
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of flavonol glycoside to anthocyanin: an interpretation of the oxidation–reduction relationship of biosynthetic flavonoid-intermediates†
Kin-ichi
Oyama
a,
Yuki
Kimura
b,
Satoru
Iuchi
c,
Nobuaki
Koga
c,
Kumi
Yoshida
 *c and
Tadao
Kondo
*c
*c and
Tadao
Kondo
*c
aResearch Institute for Materials Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan
bGraduate School of Information Sciences, Nagoya University, Chikusa, Nagoya 464-8601, Japan
cGraduate School of Informatics, Nagoya University, Chikusa, Nagoya 464-8601, Japan. E-mail: yoshidak@i.nagoya-u.ac.jp; kondot@info.human.nagoya-u.ac.jp
First published on 3rd October 2019
Abstract
An efficient conversion of rutin to the corresponding anthocyanin, cyanidin 3-O-rutinoside, was established. Clemmensen-type reduction of rutin gave a mixture of flav-2-en-3-ol and two flav-3-en-3-ols, which were easily oxidised by air to give the anthocyanin. The interconversion reactions of these flavonoids provide insight into their biosynthetic pathway.
Introduction
Because anthocyanins are attractive plant pigments with beautiful colours1 and beneficial functions for human health,2 it is important to supply large amounts of the compounds, which can be problematic. Currently, almost all the anthocyanins for commercial demand and research needs are prepared from plants as a mixture, or purified by very expensive repeated column chromatography. Only simple anthocyanins, anthocyanidin mono- or di-glucosides, are synthesised using the classical Robinson's method via aldol condensation.3 The disadvantage of Robinson's route is that the alkaline reaction conditions in the last step are drastic and the yield is not very good, especially in the case of delphinidin derivatives (yield approximately below 20%).3 A Clemmensen-type reduction of flavonol glycosides to the corresponding anthocyanins by a metal or hydride reagent is another option for the synthesis of anthocyanins.3c,4,5 This route is short, does not need protecting groups, and is easy to carry out at large scale. However, it still has problems with low yield; previous attempts gave only a 20–30% yield of pigment.4,5 Another advantage to the Clemmensen-type reduction over Robinson's method is that acylated anthocyanins can be synthesised. We reported the synthesis of cyanidin 3-O-6′′-O-acetyl-β-D-glucopyranoside from kaempferol 3-O-6′′-O-acetyl-β-D-glucopyranoside according to Brouillard's Zn–Hg reduction.5 However, the yield was not satisfactory, being only 10% with a deacylated anthocyanin (15%).5 To improve the reaction, we analysed the detailed reaction procedure of the reduction, and found that non-toxic dried Zn powder can replace Zn–Hg.6 Furthermore, we found that the reaction proceeded in two steps: in the first step, flavonol was reduced to give a mixture of flav-2-en-3-ol and two flav-3-en-3-ol derivatives; in the second step, these flavenols were oxidised to the anthocyanin.6 The intermediate flavenols are highly reactive and easily oxidised under acidic conditions without any reagent other than the oxygen in the air.In the well-accepted biosynthetic pathway of Cy3G 1, one of the simplest and most abundant anthocyanins in plants, the last colourless compound is cis-leucocyanidin, and this precursor is oxidised by anthocyanidin synthase (ANS) to give a coloured cyanidin, followed by glucosylation at 3-OH to give 1 (Scheme 1).7 Inspired by this route, we previously designed a cis-leucocyanidin derivative and tried an oxidation reaction in the total synthesis of 1.8 However, this compound did not give a cyanidin chromophore. We then transformed the cis-leuco compound to a flav-3-en-3-ol 3-O-glucoside derivative by dehydration, which had the same oxidative level as the cis-leuco compound. Air oxidation of the flav-3-en-3-ol 3-O-glucoside derivative gave 1. Furthermore, flav-2-en-3-ol 3-O-glucoside (2) has been found in the seed-coat of immature black soybeans.9 Combining these findings strongly suggested that 2 is a new anthocyanin precursor, and that one should pay attention to the interconversion of the labile flavonoids involved in anthocyanin redox potentials and biosynthetic pathways. In addition, the mechanism of ANS and the oxidation–reduction relationship of flavonoids should be re-examined. Herein, we report the Zn reduction of rutin (3) followed by air oxidation to cyanidin 3-O-rutinoside (4). To determine the absolute stereostructure of the intermediates, we carried out electronic circular dichroism (ECD) spectra calculations and the synthesis of catechin derivatives. We anticipated that the obtained chemical characteristics and reactivity would add to the understanding of flavonoid biosynthesis.
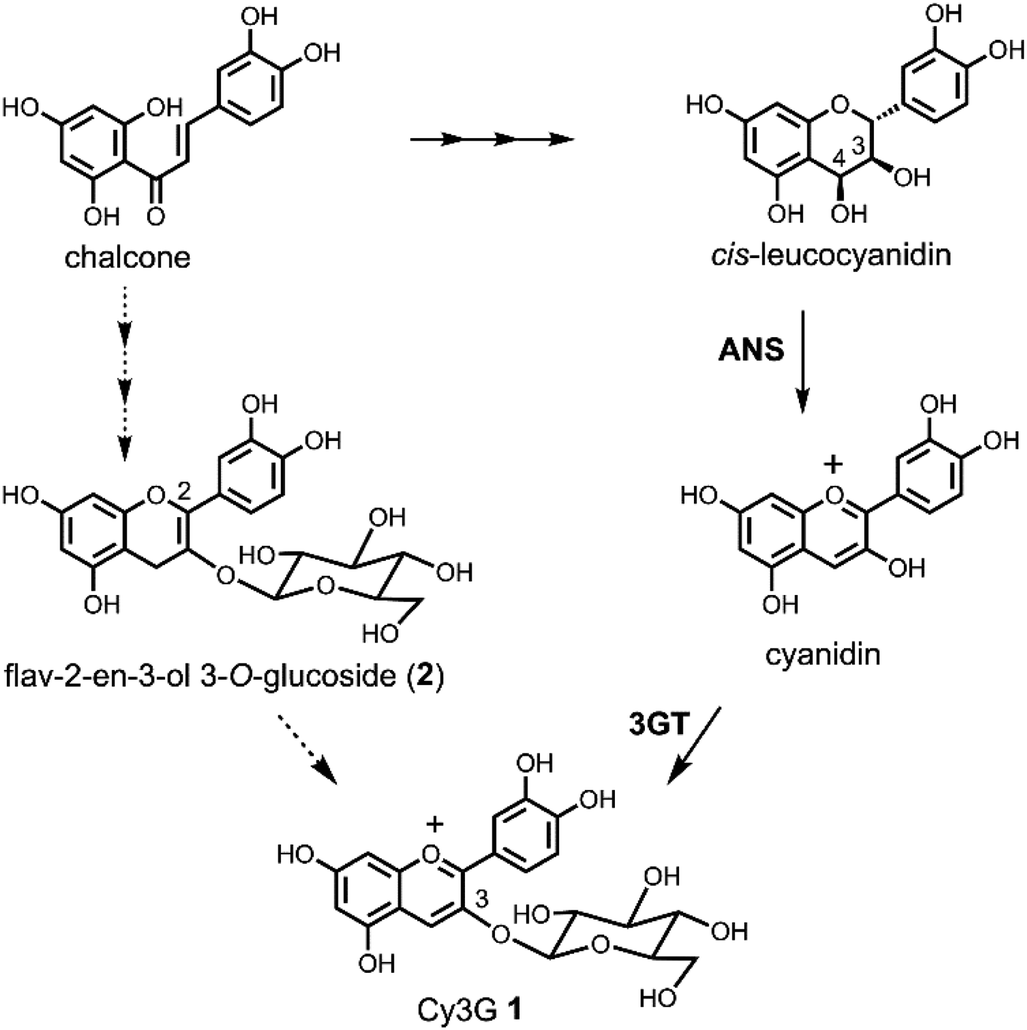 | ||
| Scheme 1 Biosynthetic pathway of anthocyanins and flav-2-en-3-ol 3-O-glucoside (2) as a plausible biosynthetic intermediate. | ||
Results and discussion
In the previously reported Clemmensen-type reduction of 3, all the reactions were carried out in air.3c,4c Therefore, we first examined the reactions under the same conditions. Rutin (3) was dissolved in dried MeOH. Zn powder and then dried HCl–MeOH were added and the mixture stirred vigorously. The mixture was red at the beginning of the reaction, but quickly became colourless. The colourless reaction mixture was filtered with a cartridge filter (pore size: 0.45 μM) to remove Zn powder and analysed by HPLC. The obtained chromatogram showed peaks for two colourless intermediates 5 and 6, and, interestingly, a sample solution for HPLC analysis without Zn turned red within a couple of minutes. The HPLC chromatogram of the red solution showed a decrease in the colourless peaks and the formation of 4. This strongly indicated that the reduction of 3 does not give 4 directly, but other colourless compounds 5 and 6 as intermediates. To analyse these intermediates in detail, we tried the Zn reduction under argon atmosphere and detected the three colourless compounds 5–7 with a small amount of 4 (Fig. S1†). Compound 7 could not be detected under air reaction conditions because 7 is very sensitive to oxidation. After removing the Zn powder by filtration, the solution was allowed to stand at room temperature under air overnight, whereupon the solution became red. At this point, compounds 5–7 had disappeared and only 4 was detected by HPLC (Fig. S1†).To determine the structure of 5–7, a Zn reduction of 3 (1 g, 1.64 mmol) in dried HCl–MeOH under argon atmosphere at −20 °C was performed (Scheme 2). After removal of Zn by filtration, the reaction mixture was diluted with water and adsorbed on an Amberlite XAD-7 column, then eluted with aq. CH3CN. The obtained fraction containing 5–7 was evaporated and purified using preparative HPLC eluted with neutral aq. CH3CN. Pure samples of 5–7 were obtained in 14%, 55%, and 3% yield, respectively. The HR-ESI-MS peaks of 5–7 were consistent with the theoretical values (Calcd for C27H31O15 [M − H]− 595.1668, found 5: 595.1665; 6: 595.1668; 7: 595.1671). After analysis using various 1D and 2D NMR techniques, 5 and 6 were both determined to be 3,5,7,3′,4′-pentahydroxylflav-3-en-3-ol 3-O-rutinoside with differences in the absolute configuration of C2. The structure of 7 was determined to be 3,5,7,3′,4′-pentahydroxylflav-2-en-3-ol 3-O-rutinoside (Scheme 2).
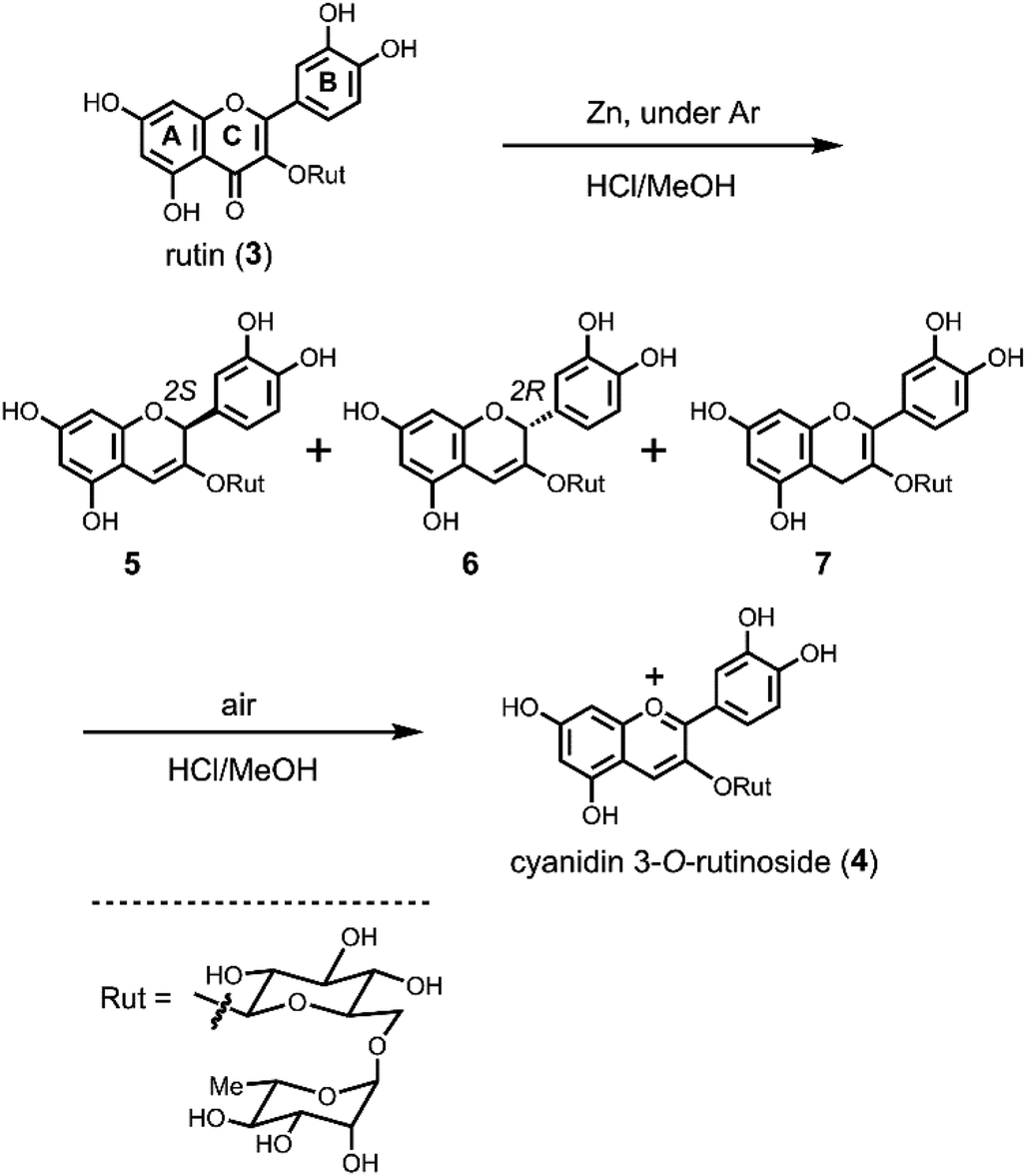 | ||
| Scheme 2 Conversion of rutin (3) to cyanidin 3-O-rutinoside (4) by Zn reduction and air oxidation. | ||
To confirm the role of these intermediates, the air oxidation of isolated 5–7 to cyanidin 3-O-rutinoside (4) was examined (Scheme 2). Compounds 5–7 were each dissolved in dried HCl–MeOH and stirred in air at room temperature. All the solutions became red and were converted to 4, although the reaction rate was different for each compound; the conversion rate of 7 was significantly higher than that of 5 and 6. If water was present in the oxidation reaction, the yield of 4 was lower. Finally, starting with 2 g of rutin (3), pure cyanidin 3-O-rutinoside (4) as the TFA salt was obtained in 50% yield (1.175 g).
To determine the absolute configuration at C2 in intermediates 5 and 6, quantum chemical calculations for ECD spectra were performed,10 as explained in detail in the ESI.† Optimised conformations of model compounds with 2S and 2R configurations derived from 5 and 6, respectively, were obtained by density functional theory (DFT) calculations. Then, the ECD curve, Δε, of each conformer was computed by time-dependent DFT calculations. In these calculations, we simplified the structure by replacing the rutinoside moiety with OMe to reduce the number of plausible conformations and hence to reduce the computational cost. Eight conformers were considered for both the 2S and 2R structures of the model compounds (Fig. S2†). As shown in Fig. 1, the computed UV absorption spectra reasonably agree with the experimental spectra of 5 and 6 (Fig. 1), verifying that the replacement of ORut with OMe should not significantly affect the electronic character in the main skeleton of 5 and 6. To highlight the absolute configurations at C2, the computed Δε values at ∼273 nm were examined in detail. As explained in the ESI,† the excited state at this region is attributed to the π–π* transition. The calculated ECDs were compared with those of the experimental data (Fig. 1). This comparison between the theoretical and experimental results shows that the positive and negative Cotton effects around 280 nm are assigned to the S and R absolute configurations at C2, respectively. Note that, as seen in Fig. S4† in the ESI,† all the 2R conformers show a negative Cotton effect in the Δε and the 2S conformers show a positive Cotton effect. This result further supports the above assignment.
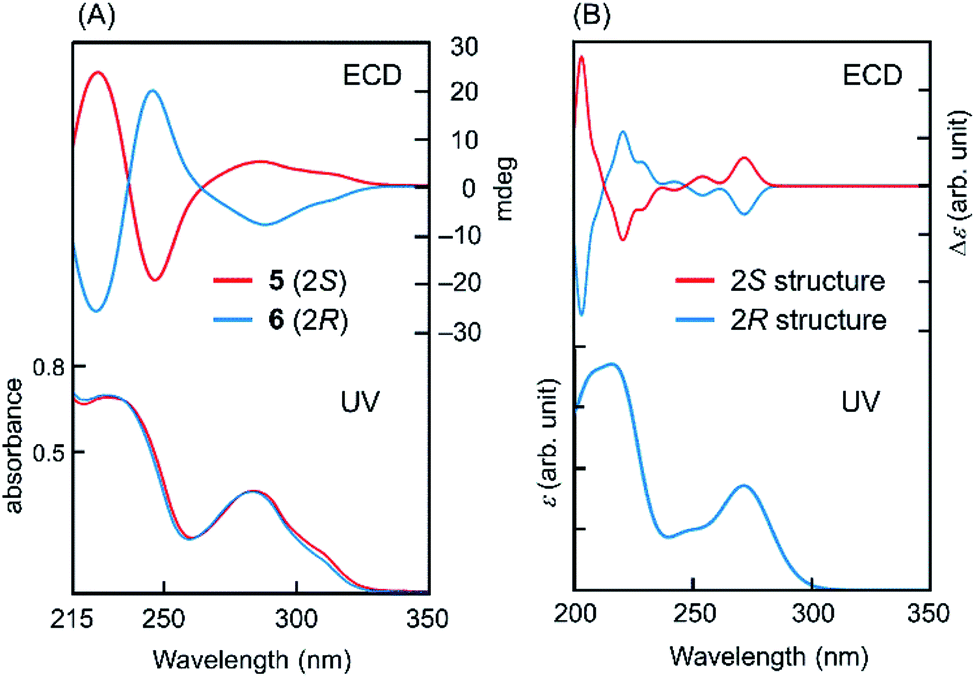 | ||
| Fig. 1 UV and ECD for (A) 5, 6 and (B) model compounds (computation). The computed UV spectra for 2S and 2R structures are virtually identical. | ||
The chirality at C2 of 5 and 6 was also confirmed by derivatization to the corresponding flavanols by hydrogenation (Scheme 3) and comparison with authentic compounds which were synthesised from (+)-catechin (11) and (−)-epicatechin (12). Compound 5 was dissolved in MeOH and reduced by H2/Pd–C at room temperature to give a single product 8 in 52% yield with a 2,3-cis-configuration (H2: broad singlet). The same reduction of 6 gave 9 and 10 in 16% and 23% yield, respectively. NMR analysis indicated that 9 has a 2,3-cis-configuration (J2,3 = 2.0 Hz) and 10 has a 2,3-trans-configuration (J2,3 = 6.5 Hz). Hydrogenation of 7 gave only 8 in 36% yield. Flavan-3-ol 3-rutinosides (10) were synthesised as shown in Scheme S1.† After selective benzylation of (+)-catechin (11),11 glycosylation of 3-OH with rutinosyl immediately followed by deprotection gave 10 in 21% overall yield. All the physicochemical data of this product were consistent with the reductant 10 obtained from 6. Using a similar procedure, (−)-epicatechin (12) was transformed to 9 and all characterisation data of this product were also consistent with the reductant 9 obtained from 6 (Scheme S1†). From these results, the chirality at C2 of 6 was determined to be the R configuration, whereupon the other 2,3-cis-flavan-3-ol 3-rutinoside 8 must have an ent–epi structure, and therefore C2 of 5 was determined to have an S configuration. Thus, ECD computational calculations for determining the chiral centre of flav-3-ene compounds were proven to be correct. This methodology is promising to the structure determination of chiral natural compounds.
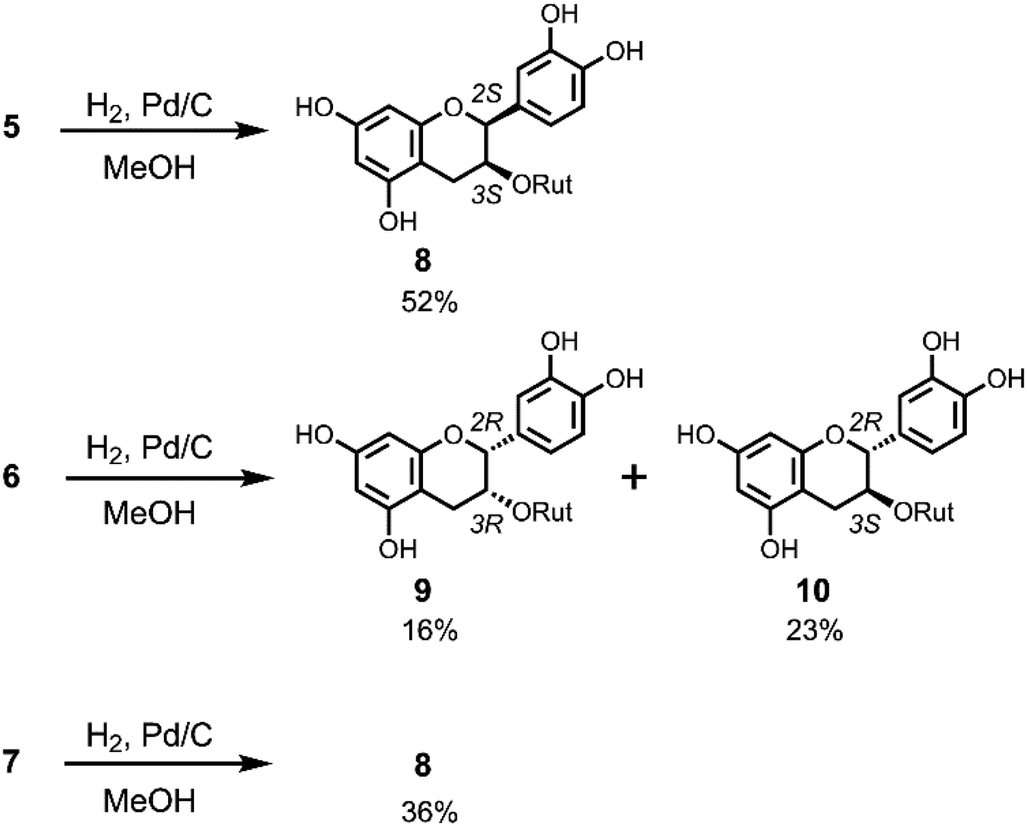 | ||
| Scheme 3 Hydrogenation of 5–7. | ||
To further confirm the interconversion reaction in flavonoids intermediates, we reduced the anthocyanin. At first, we attempted the Zn reduction of 4 in HCl–MeOH. HPLC analysis showed that 4 gave a mixture of 5–7 with a similar ratio as that obtained by the Zn reduction of rutin (3). However, hydrogenation of 4 by H2/Pd–C in MeOH at room temperature gave a different result (Scheme 4). Two flavanols were obtained and identified as the major product 8 (55%) and the minor product 9 (2%). This indicated that hydrogenation occurs selectively, possibly a result of the steric hinderance of rutinoside. A similar stereoselective reduction was found in the hydrogenation of 5 and 7; therefore, hydrogenation of 4 might involve 5, 7, or both as an intermediate. We also attempted the reduction of 4 with a hydride reagent, NaBH3CN, which can be used in acidic conditions (Scheme 4). Compound 4 was dissolved in MeOH, 3 eq. of NaBH3CN was added, and the mixture stirred at −20 °C for 30 min. The result was different from that of the Zn reduction; the major product was 2-flavenol 7 with small amounts of 5 and 6. Purification by preparative ODS-HPLC with 10% aqueous CH3CN solution gave pure 7 in 70% yield.
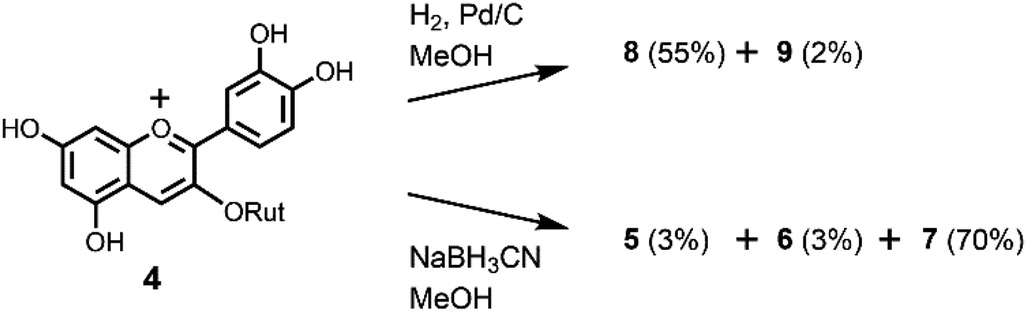 | ||
| Scheme 4 Reduction of 4. | ||
In the above conversion of flavonols to their corresponding anthocyanins, protection of the 3-OH was essential. Treatment of quercetin 3-O-glucoside gave 1; however, quercetin did not give cyanidin.6,12 It is not necessary for the 3-OH to be glycosylated; an alkyl-protected 3-O-methylquercetin was transformed to 3-O-methylcyanidin using the same reaction.13 These findings can explain the existence of 2 in immature black soybeans and raises a new hypothesis that glucosylation at 3-OH occurs first, following which oxidation by ANS gives 1. Recent reports have proposed a role and mechanism of the enzymatic reaction of ANS as oxidation at C3, and predicted 3,3-diol as an intermediate.7c,f However, according to our experimental results, oxidation to the anthocyanidin chromophore is not possible from these intermediates.
Conclusion
In summary, we studied the conversion of rutin (3) to cyanidin 3-O-rutinoside (4) by a Clemmensen-type reduction using Zn in dried HCl–MeOH. The reaction proceeded in two steps: the first step was reduction to a mixture of flavenol compounds 5–7, and in the second step, 5–7 were oxidised to anthocyanin 4. Removal of Zn before the second step improved the reaction yield and reproducibility. This reaction can be easily scaled up to gram-scale anthocyanin synthesis. For this conversion reaction, protection of 3-OH was essential.6,12,13 We also studied the reduction of 4 under various conditions and obtained chemical information about the anthocyanin–flavenols–flavanols interconversion. In the biosynthetic pathway of these flavonoids, sequential oxidation–reduction reactions were observed, and our findings should aid in the understanding of those reactivities and conversions in plants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Dr Ørjan Bjorøy, Mr Kazunari Tokuno, Ms. Emi Hayashi, Mr Yusuke Nakane, and Mr Reo Sawaguchi for their technical assistance. This research was financially supported by Grants-in-Aid for Scientific Research (No 18H02146) by MEXT. The quantum chemical calculations were performed at the Information Technology Center, Nagoya University.Notes and references
- (a) K. Yoshida, M. Mori and T. Kondo, Nat. Prod. Rep., 2009, 26, 884–915 RSC; (b) K. Yoshida, K.-I. Oyama and T. Kondo, in Polyphenol Research, ed. V. Cheynier, P. Sarni-Manchado, and S. Quideau, Wiley-Blackwell Publishing, Chichester, vol. 3, pp. 99–129 Search PubMed; (c) T. Goto and T. Kondo, Angew. Chem., Int. Ed. Engl., 1991, 30, 17–33 CrossRef.
- (a) H. E. Khoo, A. Azlan, S. T. Tang and S. M. Lim, Food Nutr. Res., 2017, 61, 1361779 CrossRef; (b) T. Tsuda, Food Sci. Technol. Res., 2012, 18, 315–324 CrossRef CAS; (c) M. A. Lila, J. Biomed. Biotechnol., 2004, 2004, 306–313 CrossRef; (d) T. C. Wallace and M. M. Giusti, in Anthocyanins in Health and Disease, CRC Press Search PubMed.
- (a) L. Cruz, N. Mateus and V. de Freitas, Tetrahedron Lett., 2013, 54, 2865–2869 CrossRef CAS; (b) G. A. Iacobucci and J. G. Sweeny, Tetrahedron, 1983, 39, 3005–3038 CrossRef CAS; (c) A. Robertson and R. Robinson, J. Chem. Soc., 1927, 242–247 RSC; (d) S. Murakami, A. Robertson and R. Robinson, J. Chem. Soc., 1931, 2665–2671 RSC; (e) R. Robinson, Ber. Dtsch. Chem. Ges. A, 1934, 67, 85–105 CrossRef.
-
(a) K. Shibata, Y. Shibata and I. Kasiwagi, J. Am. Chem. Soc., 1919, 41, 208–220 CrossRef CAS;
(b) H. G. Krishnamutry, V. Krishnamoorthy and T. R. Seshadri, Phytochemistry, 1963, 2, 47–60 CrossRef;
(c) M. Elhabiri, P. Figueiredo, A. Fougerousse and R. R/Brouillard, Tetrahedron Lett., 1995, 36, 4611–4614 CrossRef CAS : Elhabiri et al. reported the yield was 60%. However, we re-examined the reduction of rutin under HCl–MeOH with zinc amalgam, zinc powder, or magnesium powder and obtained cyanidin 3-O-rutinoside in less than 30% yield under the optimised conditions. This might be because the value of the molar absorption coefficients for cyanidin 3-O-rutinoside reported by Elhabiri et al. (7000 at 510 nm) is too low compared with the theoretical value (around 20
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in our results); therefore, they miscalculated the yield..
000 in our results); therefore, they miscalculated the yield.. - The deacetylated anthocyanin as a by-product was obtained in 15% yield because the acid catalyzed deacetylation was occurred in the presence of Zn–Hg and hydrogen chloride methanol solution: K.-I. Oyama, S. Kawaguchi, K. Yoshida and T. Kondo, Tetrahedron Lett., 2007, 48, 6005–6009 CrossRef CAS.
- K. Yoshida, T. Kondo and K.-I. Oyama, JPN Patent No. 5382676, 2014.
- (a) W. Heller and G. Forkmann, in The Flavonoids Advances in Research since 1986, ed. J. B. Harborne, Chapman & Hall, London, 1994, pp. 499–535 Search PubMed; (b) K. Saito, M. Kobayashi, Z. Z. Gong, M. Yamazaki and Y. Tanaka, Plant J., 1999, 17, 181–189 CrossRef CAS; (c) J. Nakajima, Y. Tanaka, M. Yamazaki and K. Saito, J. Biol. Chem., 2001, 276, 25797–25803 CrossRef CAS; (d) J. J. turnbull, W. J. Sobey, R. T. Aplin, A. Hassan, J. L. Firmin, C. J. Schofiled and A. G. Prescott, Chem. Commun., 2000, 2473–2474 RSC; (e) K. Springob, J. Nakajima, M. Yamazaki and K. Saito, Nat. Prod. Rep., 2003, 20, 288–303 RSC; (f) J.-r Zhang, C. Trossat-Magnin, K. Bathany, S. Delrot and J. chaudiere, J. Agric. Food Chem., 2019, 67, 3595–3604 CrossRef CAS.
- T. Kondo, K.-I. Oyama, S. Nakamura, D. Yamakawa, K. Tokuno and K. Yoshida, Org. Lett., 2006, 8, 3609–3612 CrossRef CAS PubMed.
- H. Fukami, Y. Yano and T. Iwashita, J. Oleo Sci., 2013, 62, 623–629 CrossRef CAS.
- (a) K. Yoshida, N. Nagai, Y. Ichikawa, M. Goto, K. Kazuma, K.-I. Oyama, K. Koga, M. Hashimoto, S. Iuchi, Y. Takaya and T. Kondo, Sci. Rep., 2019, 9, 1484 CrossRef; (b) I. Warnke and F. Furche, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 150–166 CAS; (c) G. Bringmann, T. Bruhn, K. Maksimenka and Y. Hemberger, Eur. J. Org. Chem., 2009, 2717–2727 CrossRef CAS.
- (a) H. Kawamoto, F. Nakatsubo and K. Murakami, Mokuzai Gakkaishi, 1991, 37, 488–493 CAS; (b) S. Nakamura, K.-I. Oyama, T. Kondo and K. Yoshida, Heterocycles, 2007, 73, 451–460 CrossRef CAS; (c) K.-I. Oyama, M. Kuwano, M. Ito, K. Yoshida and T. Kondo, Tetrahedron Lett., 2008, 49, 3176–3180 CrossRef CAS.
- When quercetin was subjected to the same reaction, a colourless compound with a five-membered ring (aurone-type compound) was obtained instead of cyanidin..
- (a) Y. Kimura, K.-I. Oyama, Y. Murata, A. Wakamiya and K. Yoshida, Int. J. Mol. Sci., 2017, 18, 427 CrossRef; (b) A. B. El-Meligy, T. Ishihara, K.-I. Oyama, A. M. El-Nahas and K. Yoshida, Heterocycles, 2018, 97, 946–959 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06986k |
| This journal is © The Royal Society of Chemistry 2019 |