
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron oxide encapsulated by copper-apatite: an efficient magnetic nanocatalyst for N-arylation of imidazole with boronic acid†
Othmane Amadine *a,
Younes Essamlalia,
Abdallah Amedlousab and
Mohamed Zahouily*ab
*a,
Younes Essamlalia,
Abdallah Amedlousab and
Mohamed Zahouily*ab
aVARENA Center, MAScIR Foundation, Rabat Design, Rue Mohamed El Jazouli, Madinat El Irfane, 10100-Rabat, Morocco. E-mail: m.zahouily@mascir.com; o.amadine@mascir.com
bLaboratoire de Matériaux, Catalyse et Valorisation des Ressources Naturelles, URAC 24, FST, Université Hassan II-Casablanca, Morocco
First published on 11th November 2019
Abstract
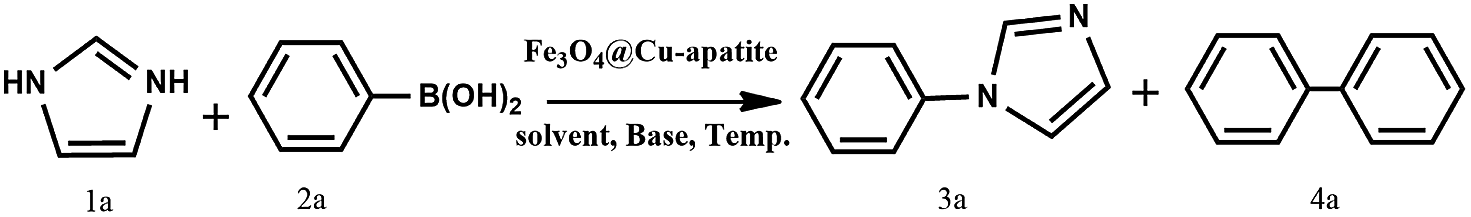
N-Arylation of imidazole was carried out with various arylboronic acids on iron oxide encapsulated by copper-apatite (Fe3O4@Cu-apatite), producing excellent yields. Firstly, the iron nanoparticles were prepared using a solvothermal method, and then they were encapsulated by copper-apatite to obtain magnetic Fe3O4@Cu-apatite nanocatalysts. Several physico-chemical analysis techniques were used to characterize the prepared nanostructured Fe3O4@Cu-apatite catalyst. The prepared Fe3O4@Cu-apatite was used as a nanocatalyst for N-arylation of imidazole with a series of arylboronic acids with different substituents to reaffirm the effectiveness of this magnetic nanocatalyst. The Fe3O4@Cu-apatite nanocatalyst can also be easily separated from the reaction mixture using an external magnet. More importantly, the as-prepared Fe3O4@Cu-apatite exhibited good reusability and stability properties in successive cycles. However, there was a notable loss of its catalytic activity after multiple cycles.
1. Introduction
Over the years, N-arylheterocycles have gained prominence due to their presence in a wide variety of natural products and bioactive compounds. They have also played an important role as building blocks in organic synthesis.1,2 There is a special emphasis on N-aryl imidazole because it exhibits antipsychotic, antiallergic, and herbicidal3,4 properties, as well as others. The development of a mild and highly efficient method for the synthesis of N-arylheterocycles over classical Ullman type,5,6 nucleophilic aromatic substitution reactions,7 or coupling with organometallic reagents has recently gained considerable attention in synthetic chemistry.8,9 But the harsh conditions of these reactions, such as very high temperature and strong bases have restricted and limited their applications. At the turn of the 21st century, recent development of N-arylation with boronic acids unleashed the power of C-heteroatom bond formation reaction due to the mild reaction conditions (room temperature, weak base and ambient atmosphere).10–13 However, the synthetic scope of this reaction is strongly limited due to some disadvantages, such as product contamination – toxic waste produced after separation of the catalyst, which cannot be easily recovered after the reaction.14 These disadvantages can be overcome by anchoring the metal on suitable supports, which can be easily recovered, and then potentially be reusable with a minimal amount of product contamination. Recently, diverse forms of heterogeneous catalysts have been developed for N-arylation, such as cellulose supported copper(0),15 polymer supported Cu(II),16 MCM-41-immobilized bidentate nitrogen copper(I),17 CuO nanoparticles18 and Cu-exchanged fluorapatite.19 Magnetic separation has also received a lot of attention as a solid catalyst separation technology, since it can be very efficient and fast. Numerous studies have focused on the immobilization of copper catalytic systems on a magnetic medium based on iron in order to separate them by the simple application of a magnet.20,21 In the same way, Alper and co-workers have immobilized Cu(I) catalyzed on the surface of Fe3O4 magnetic nanoparticle-supported L-proline as a recyclable and recoverable catalyst for N-arylation of heterocycles.Calcium phosphate, such as hydroxyapatite and its derivates appear very attractive due to their ion-exchange capability, adsorption capacity, and acid–base properties.22 Hydroxyapatite based materials have been used in the biomedical field23,24 lately and in some important chemical transformations.25–28 The latter applications are related to its well-known ability to immobilize divalent and trivalent metal ions, by partial cationic exchange with calcium.29–33 Recently, the possibility to combine the properties of apatite with other inorganic phases within core–shell nanostructures has attracted great attention. For instance, hydroxyapatite/TiO2,34 hydroxyapatite/carbon nanotubes,35 and hydroxyapatite/SiO2 (ref. 36) core–shell nanoparticles have been studied for different applications, such as photocatalytic processes, fluorescence imaging and drug delivery. Indeed, the combination of hydroxyapatite with iron oxide particles could be useful to design sorbents that combine large activity and easy recovery via magnetic separation.37,38 Hence, several iron oxide–hydroxyapatite nanocomposites have been described in the literature.39–45 However, the Fe3O4@Cu-apatite has only rarely been explored, and to the best of our knowledge, the use of Fe3O4@Cu-apatite as a heterogeneous catalyst for the N-arylation of imidazole has not been reported in literature. Fe3O4 is one of the cheaper magnetic oxide, which act in this study as a high surface area framework that is coated by the growth of Cu-apatite shell. The Fe3O4 core has to facilitate the recycle of nanocatalysts through magnetic separation. This present work was achieved within our progressive program to develop an eco-friendly and efficient approach for the synthesis of various products.46–50 This is the background upon which this work is situated upon, and the objective is based on the synthesis and characterization of Fe3O4@Cu-apatite nanoparticles, and then their use as magnetic catalysts for the N-arylation of imidazole with arylboronic acids (Scheme 1).
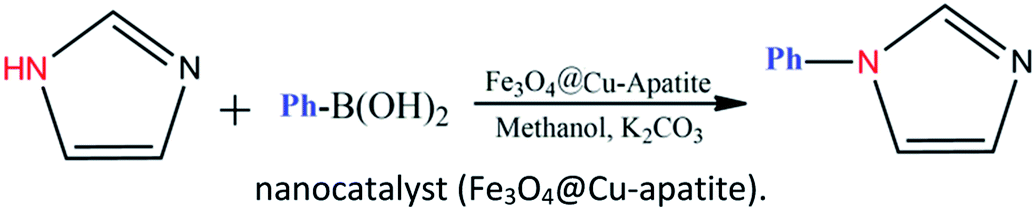 | ||
| Scheme 1 N-Arylation of imidazole with boronic acid by a nanocatalyst (Fe3O4@Cu-apatite). | ||
2. Experimental
2.1. Materials and apparatus
Copper(II) nitrate (Cu(NO3)2·3H2O, ≥98.0%), ferric chloride (FeCl3·6H2O), calcium nitrate (Ca(NO3)2·4H2O, ≥99.0%), ammonium hydrogen phosphate ((NH4)2HPO4), sodium acetate (CH3COONa), ethylene glycol (EG), sodium hydroxide (NaOH), ammonia solution (NH3·H2O, 30%), poly (ethylene glycol) (PEG, 1000) were purchased from Aldrich Chemical Company and were used as received without any further purification. Deionized water was used in all experiments. X-ray diffraction (XRD) patterns of the catalysts were obtained at room temperature on a Bruker AXS D-8 diffractometer using Cu-Kα radiation in Bragg–Brentano geometry (θ–2θ). SEM and STEM micrographs were obtained on a Tecnai G2 microscope at 120 kV. The average diameter of sample was quantified from the SEM image using ImageJ software. The gas adsorption data was collected using a Micromeritics 3Flex Surface characterization analyzer, using nitrogen. Prior to nitrogen sorption, all samples were degassed at 150 °C overnight. The specific surface areas were determined from the nitrogen adsorption/desorption isotherms (at −196 °C), using the BET (Brunauer–Emmett–Teller) method. Pore size distributions were calculated from the N2 adsorption isotherms with the “classic theory model” of Barret, Joyney and Halenda (BJH). Fourier transformation infrared (FT-IR) spectra of samples in KBr pellets were measured on a Bruker Vector 22 spectrometer. Magnetic properties of Fe3O4 and Fe3O4@Cu-apatite were investigated in a MPMS-XL-7AC superconducting quantum interference device (SQUID) magnetometer. The magnetic measurements were performed from −15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 15000 Oe at room temperature. The element content of Fe3O4@Cu-apatite materials was determined by an Elemental analysis was realized using inductively coupled plasma atomic emission spectrometry (ICP AES; Ultima 2 – Jobin Yvon). The 1H NMR spectra were recorded in CDCl3 or DMSO d6, using a Bruker Avance 600 spectrometer.
000 to 15000 Oe at room temperature. The element content of Fe3O4@Cu-apatite materials was determined by an Elemental analysis was realized using inductively coupled plasma atomic emission spectrometry (ICP AES; Ultima 2 – Jobin Yvon). The 1H NMR spectra were recorded in CDCl3 or DMSO d6, using a Bruker Avance 600 spectrometer.
2.2. Catalyst preparation
:Cu 1:1). After heating at 90 °C for 2 h, 10 mL of (NH4)2HPO4 (0.1981 g) solution was also added in drops and the pH value of the solution was tuned to 10 with ammonia water (molar ratio of Ca:P 10:6). After 0.5 h, the solution was transferred into a Teflon-lined stainless-steel autoclave, where it was sealed and heated at 165 °C for 12 h. Finally, the Fe3O4@Cu-apatite NPs were collected using a magnet bar.2.3. General procedure of imidazol's N-arylation with arylboronic acid
In a 50 mL round-bottomed flask, imidazole (1 mmol), phenylboronic acid (1.2 mmol), K2CO3 (1.5 mmol) and Fe3O4@Cu-apatite (15 mol%) were added and stirred in MeOH under air at 60 °C for the required time, monitoring by TLC. After completion, the mixture was diluted with H2O and the product was extracted with EtOAc (3 times). The combined extracts were washed with brine (3 times) and dried over Na2SO4. The product was purified using column chromatography (60–120 mesh silica gel, eluting with EtOAc–hexane). The structures of the prepared products were confirmed by 1H NMR and assigned on the basis of their spectral data in comparison with those reported in the literature.3. Results and discussion
3.1. Catalyst characterization
XRD was used to analyze the crystalline phases of the as-prepared Fe3O4 and Fe3O4@Cu-apatite samples (Fig. 1). The Fig. 1a shows the characteristic diffraction peaks of Fe3O4 nanoparticles observed clearly at 2θ = 30.2, 35.4, 43.3, 53.7, 57.3, and 62.98, which are attributed to the (220), (311), (400), (422), (511), and (440) Bragg reflections of the face-centered cubic lattice of Fe3O4 nanoparticles, respectively (JCPDS no. 19-0629). In the XRD pattern of Fe3O4@Cu-apatite (Fig. 1b), we can observe the presence of a secondary phase in which the diffraction peaks correspond to calcium copper phosphate Ca20Cu(PO4)14 (JCPDS 49-1080). The average crystallite size of Fe3O4 and Fe3O4@Cu-apatite was estimated using the Debye–Scherrer formula, which gave 21 and 33 nm for Fe3O4 and Fe3O4@Cu-apatite, respectively.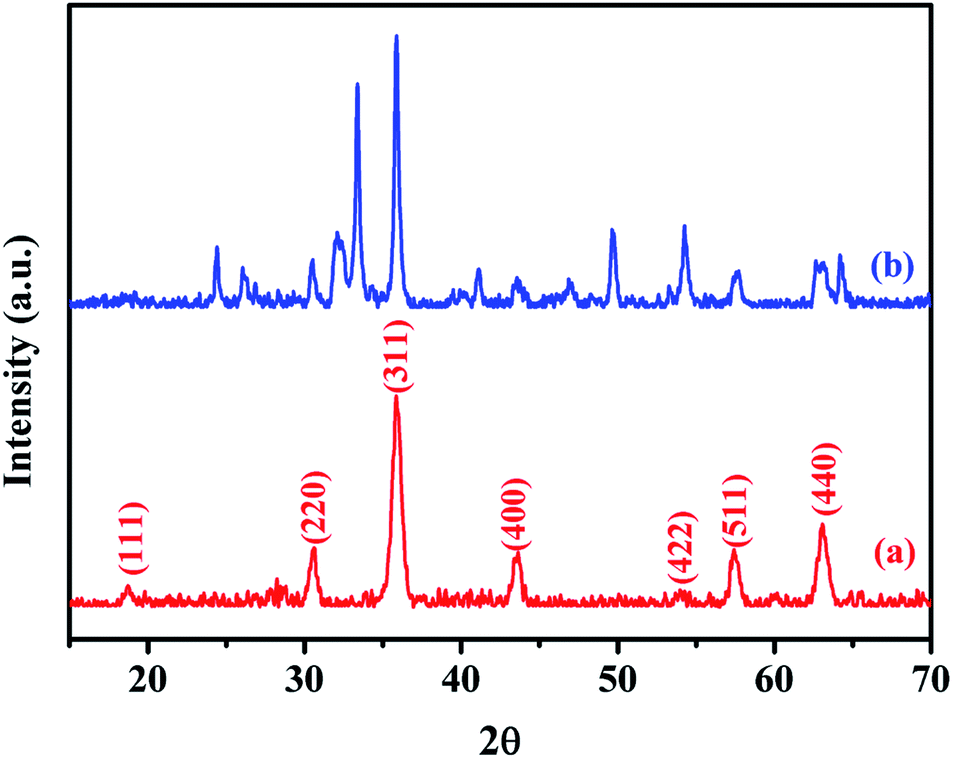 | ||
| Fig. 1 XRD patterns of the Fe3O4 (a) and Fe3O4@Cu-apatite (b). | ||
The FTIR spectra of Fe3O4 and Fe3O4@Cu-apatite are presented in Fig. 2. This figure shows the FTIR spectrum of Fe3O4@Cu-apatite nanoparticles, where there are observable peaks around 1020, and 1090, 964, 600, 561 and 470 cm−1, which are attributed to the asymmetric and symmetric stretching mode of P–O group and bending mode of O–P–O group, respectively. The characteristic bands at about 3571 and 3641 cm−1 are related to the stretching vibrations of the OH group. The weak and broad bands at around 1470 cm−1 and 873 cm−1 are most likely assigned to carbonate groups adsorbed during the synthesis process. In addition, the fingerprint of iron oxide is assigned in the FTIR spectroscopy to the Fe–O stretching vibration, and detected towards 587 cm−1.
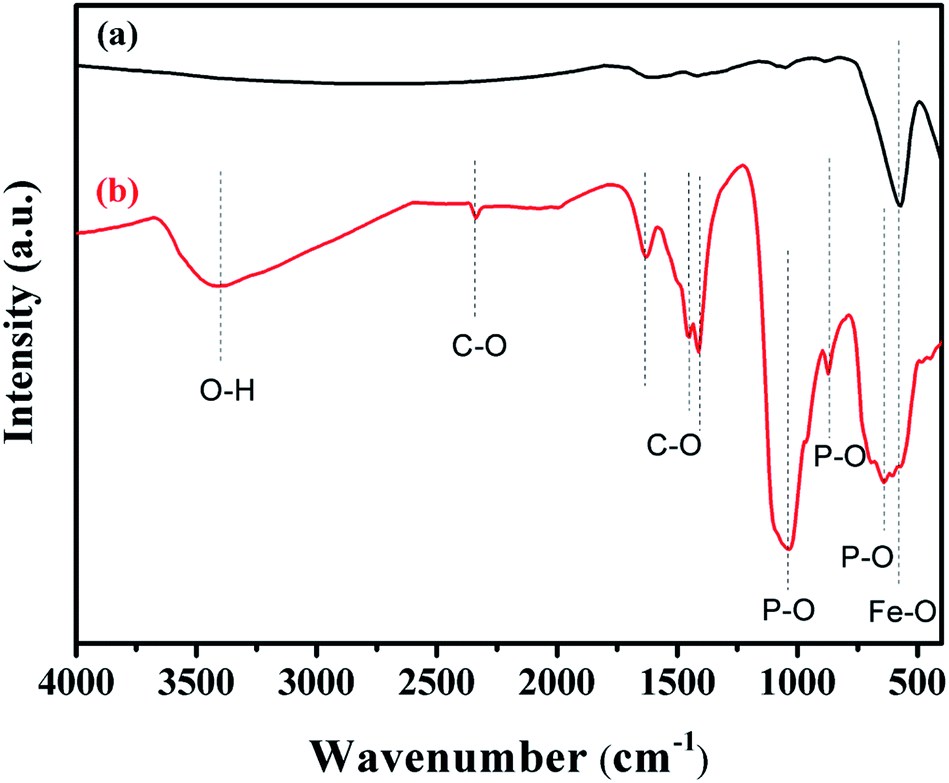 | ||
| Fig. 2 FT-IR spectra of (a) Fe3O4 and (b) Fe3O4@Cu-apatite. | ||
The calculated texture parameters of Fe3O4 and Fe3O4@Cu-apatite are summarized in Table 1. The calculation of the BET surface area for Fe3O4 is 22 m2 g−1 and the pore volume is 0.0305 cm3 g−1. The surface area and pore volume of the Fe3O4@Cu-apatite are 84 m2 g−1 and 0.1350 cm3 g−1, respectively. This change in surface properties was assumed to be due to the coating of Cu-apatite on the surface of Fe3O4 nanoparticles. The rough surface provides more pores for nitrogen adsorption. The nitrogen sorption isotherm of Fe3O4 is type IV, displaying a hysteresis loops type H3, indicating the mesoporous nature of the material (Fig. 3a). For the Fe3O4@Cu-apatite, the nitrogen sorption isotherm is type (IV) with a hysteresis loop type H3, which proves the existence of mesopores according to the IUPAC manifests (Fig. 3b).
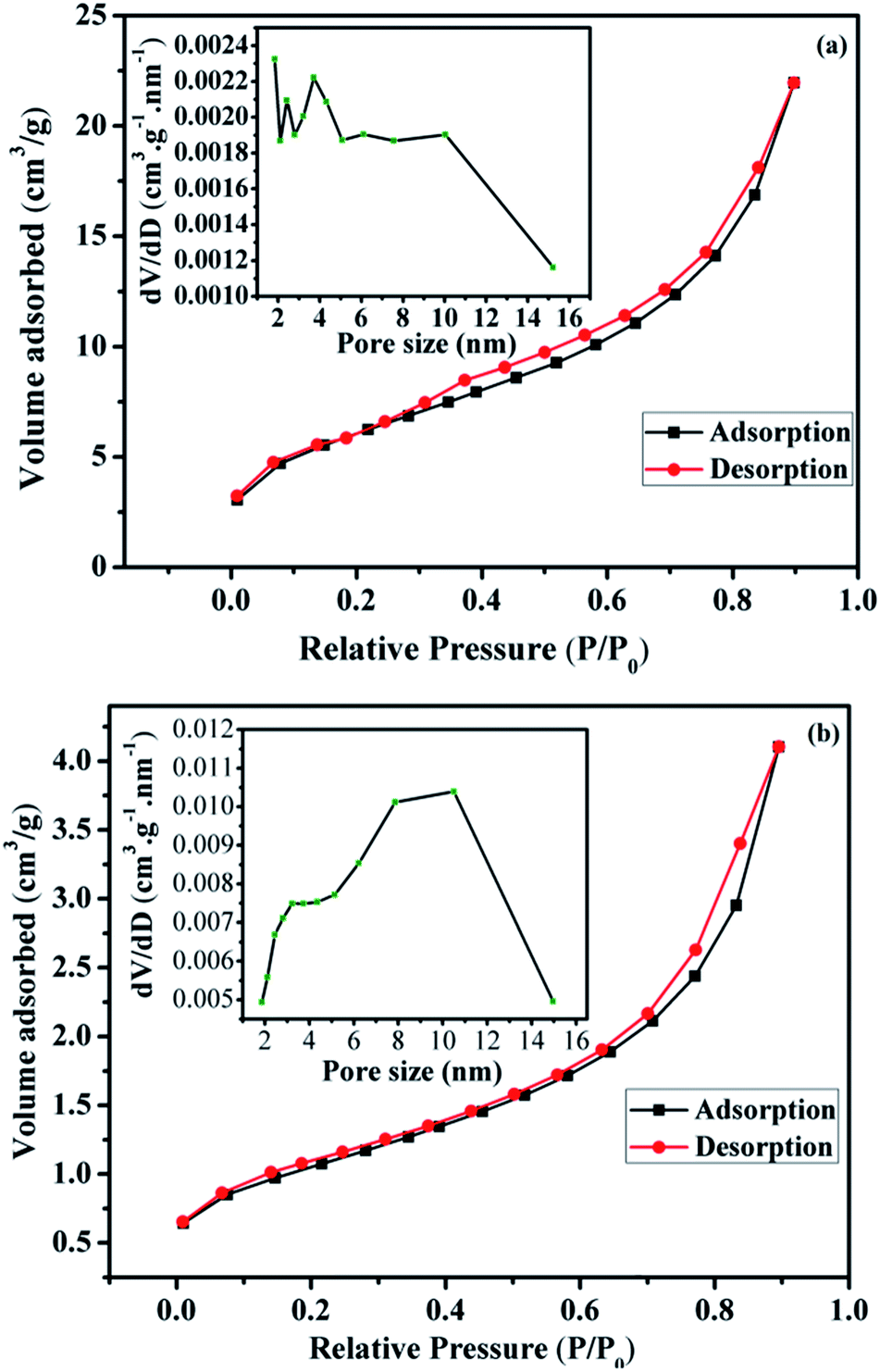 | ||
| Fig. 3 Nitrogen adsorption/desorption isotherms and BJH pore size distribution of the (a) Fe3O4 and (b) Fe3O4@Cu-apatite. | ||
The corresponding pore size distribution curve indicates that Fe3O4 and Fe3O4@Cu-apatite have a centralized pore size distribution around 4 and 11 nm, respectively, which corresponding to mesoporous materials (inset Fig. 3a and b).
SEM was used to study the surface morphology of Fe3O4 and Fe3O4@Cu-apatite (Fig. 4). The micrographs show that Fe3O4 is composed of spherical nanoparticles. Furthermore, in the case of Fe3O4@Cu-apatite, some particles were observed on the surface of Fe3O4, which may be due to the coating of the magnetic nanoparticles with Cu-apatite nanoparticles. From the SEM images of Fe3O4, we determined that the average sizes and the relative standard deviation (SD) values for the diameters of the spherical Fe3O4 MNPs was 264 ± 28 nm. Moreover, ICP analysis was performed to determine the weight of element composition of Fe3O4@Cu-apatite. From the ICP analysis, the Ca, P, Fe and Cu content were found to be 30.96, 18.63, 47, 1.83%, respectively.
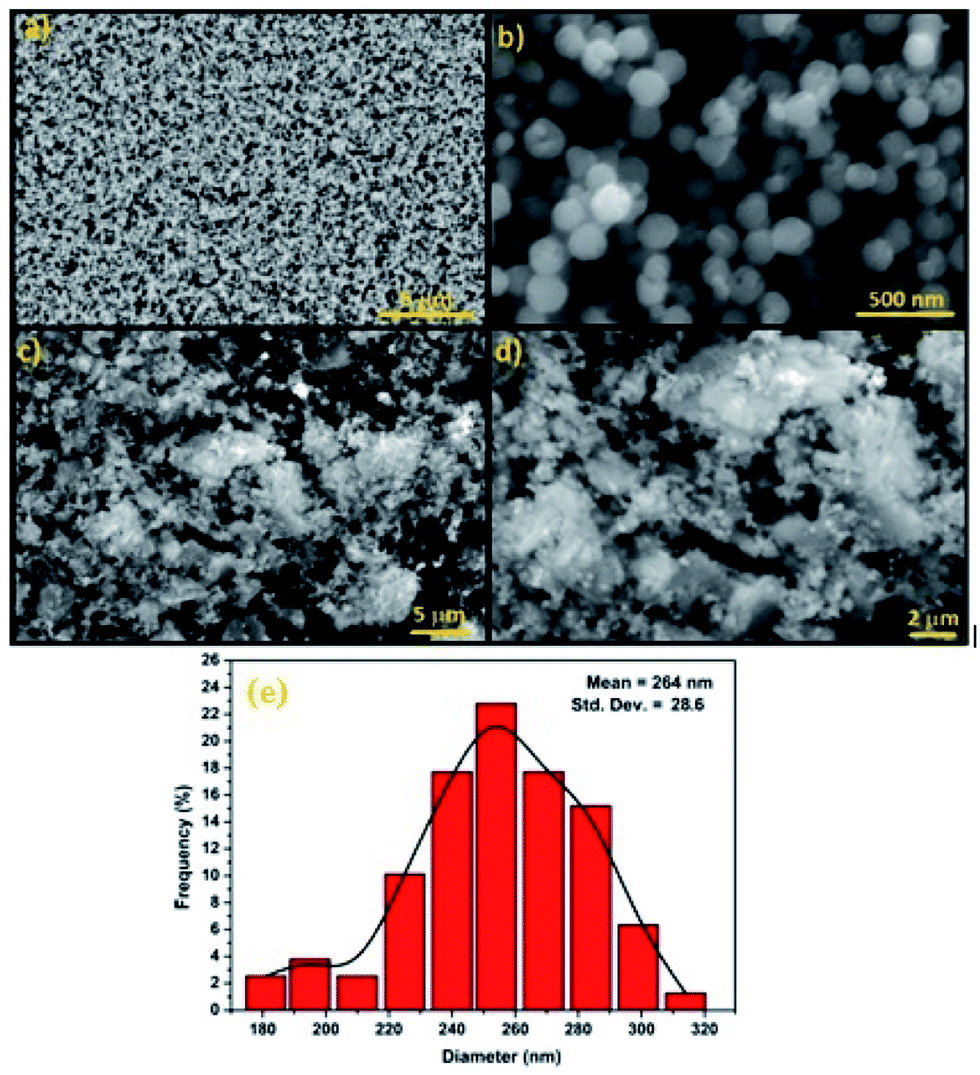 | ||
| Fig. 4 SEM images of (a and b) Fe3O4, (c and d) Fe3O4@Cu-apatite and diameter distribution of Fe3O4 nanoparticles (e). | ||
STEM was used to analyze the microstructure of the Fe3O4 and Fe3O4@Cu-apatite materials. From Fig. 5a and b, it can be observed that Fe3O4 NPS show a very good dispersion of spherical particles. These Fe3O4 spheres were further used as cores for the growth of Cu-apatite shells to obtain the Fe3O4@Cu-apatite core–shell nanostructures. Compared to the Fe3O4 cores, the outside surfaces became coarse after the growth process, as shown in Fig. 5c and d. These results indicate that the Cu-apatite nanoparticles were successfully loaded onto the Fe3O4 sphere surfaces, which clearly demonstrates the formation of core–shell nanostructures. We have also noticed that Cu-apatite was still tightly anchored on the surface of Fe3O4 even after the severe conditions under which the sample preparation for STEM analysis were conducted (a long time of mechanical stirring and sonication), suggesting the existence of strong interactions between Cu-apatite and Fe3O4.
 | ||
| Fig. 5 STEM images of (a and b) Fe3O4 and (c and d) Fe3O4@Cu-apatite. | ||
The magnetic properties of Fe3O4 and Fe3O4@Cu-apatite were investigated using SQUID from −15000 to 15000 Oe at room temperature. As shown in Fig. 6 the saturation magnetization (Ms) of Fe3O4 is close to 57 emu g−1, a higher value than that obtained for the corresponding Fe3O4@Cu-apatite (30.66 emu g−1). These results indicate that Fe3O4 core was successfully coated with a Cu-apatite shell. Moreover, the field-dependent magnetization curves of Fe3O4 and Fe3O4@Cu-apatite show negligible remanence (Mr) and coercivity (Hc), indicating superparamagnetic behavior for both samples at room temperature.
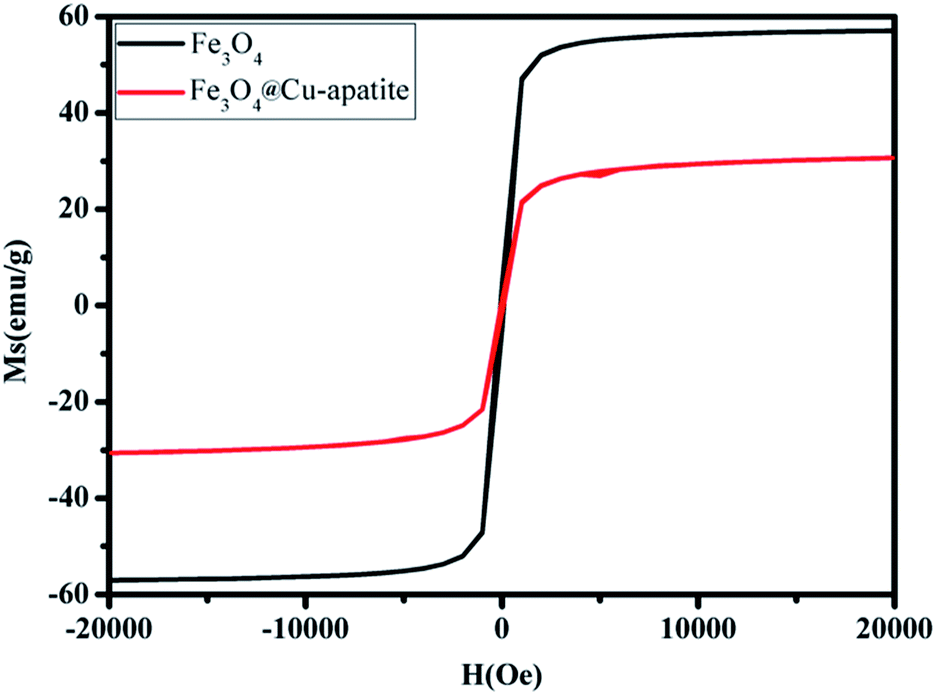 | ||
| Fig. 6 Magnetization curves of Fe3O4 and Fe3O4@Cu-apatite. | ||
This superparamagnetism can make the magnetic nanoparticles dispersible in the solution with negligible magnetic interactions between each other, which avoids magnetic clustering. As shown in the Fig. 7, Fe3O4@Cu-apatite can be dispersed in deionized water to form a stable brown suspension before magnetic separation. However, when a magnet was placed close to the reaction vessel for a while, it could be observed that the synthesized samples were rapidly attracted to the magnet side, and a nearly colorless solution was obtained.
 | ||
| Fig. 7 Photographs of the separation processes Fe3O4@Cu-apatite: (a) without external magnetic field, and (b and c) with external magnetic field. | ||
3.2. N-Arylation of imidazole over Fe3O4@Cu-apatite catalyst
Firstly, the activities of some samples were screened, such as: Fe3O4, Cu-apatite, Fe3O4@Cu-apatite and Cu-apatite@Fe3O4 catalyzing imidazole N-arylation using phenyl boronic acid as a model reaction. The resultant obtained results are summarized in Table 2. First of all, the reaction doesn't occur without the addition of a catalyst (Table 2, entry 1), and the iron oxide is also inactive in this reaction (Table 2, entry 2). However, the reaction of N-arylation of imidazole was occurring when Cu-apatite was used as catalyst with a yield of 98% (Table 2, entry 3). These results can be explained by the presence of the copper, which play an important role as catalytic element for enhancing the cross-coupling reaction. Additionally, coating iron oxide by copper-apatite (Fe3O4@Cu-apatite) in order to separate them by the simple application of a magnet has no significant effect on its catalytic activity (Table 2, entry 4) with a yield of 97%. On the other hand, coating Cu-apatite by Fe3O4 decrease their catalytic activity (Table 2, entry 5), this can be explain by the decrease of the contact surface of the catalytic element and the reactants. This result shows the importance of the catalytic system developed in this work (Fe3O4@Cu-apatite) for the N-arylation of imidazole.The influence of various reaction parameters, such as type of solvent, nature of the base, reaction temperature, and catalyst loading was investigated. The results obtained are summarized in Table 3. Initially, with 20 mol% of the catalyst and K2CO3 as a base, the screening of the effect of different solvents showed that H2O may not be the optimal solvent, when the reaction was carried out in EtOH or MeOH, the yield of the product 1-phenyl-1H-imidazole was isolated in 87 and 97%, respectively (Table 3, entry 1 and 3). After optimizing the solvent, the effect of the base was studied (Table 3, entries 4–6), and it was found that the best yields were obtained by using K2CO3 as base. Otherwise, it was noted that the presence of the base was necessary for the achievement of the reaction as reported in the literature. This is explained by the role of the base to activate the boronic acid in the mechanism of the reaction.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base | Catalyst (mol%) | Temp. (°C) | Yield of 3ab (%) | Yield of 4ab (%) |
| a Reaction conditions: arylboronic acid (1 mmol), imidazole (0.5 mmol), base (1 mmol), solvent (2 mL) for 6 h.b Isolated yield. | ||||||
| 1 | Ethanol | K2CO3 | 20 | Reflux | 87 | — |
| 2 | Water | K2CO3 | 20 | Reflux | 66 | — |
| 3 | Methanol | K2CO3 | 20 | Reflux | 97 | — |
| 4 | Methanol | Na2CO3 | 20 | Reflux | 94 | — |
| 5 | Methanol | KOH | 20 | Reflux | 88 | — |
| 6 | Methanol | NaOH | 20 | Reflux | 84 | — |
| 7 | Methanol | K2CO3 | 20 | 70 | 95 | — |
| 8 | Methanol | K2CO3 | 20 | 60 | 93 | — |
| 9 | Methanol | K2CO3 | 20 | 50 | 89 | — |
| 10 | Methanol | K2CO3 | 20 | 40 | 75 | — |
| 11 | Methanol | K2CO3 | 15 | 60 | 93 | — |
| 12 | Methanol | K2CO3 | 10 | 60 | 84 | — |
| 13 | Methanol | K2CO3 | 5 | 60 | 72 | — |
The study of the influence of temperature, in the case of using MeOH, indicated that this reaction is very sensitive to changes in temperature (Table 3, entries 7–10). According to these studies we can conclude that temperature is a key parameter in this type of reaction, and the optimum yield was obtained at 60 °C. Furthermore, the decreasing the catalyst loading to half results in a yield of 84% (Table 3, entry 12), which demonstrate the influence of the catalyst amount in this type of reactions. Thus, the optimum conditions for this reaction are at 60 °C in MeOH in the presence of 15 mol% of nanocatalyst with respect to imidazole and K2CO3 (1 mmol) as the base (Table 3, entry 11).
With the optimized reaction conditions in hand, a variety of arylboronic acids were chosen as the substrates in this N-imidazole cross-coupling reaction and the results are shown in Table 4. The substitution effects of arylboronic acid were investigated. It was found that the reaction with phenylboronic acids with an electron donating group afforded better yields (Table 4, entries 1–5) than with electron withdrawing groups (Table 4, entries 6). Similar observation was made when indole was used in place of imidazoles to obtain the corresponding N-arylindole (Table 4, entries 8–10).
|
|||
|---|---|---|---|
| Samples | N(H)-heterocycle | Aryl | Yieldb (%) |
| a Reaction conditions: Fe3O4@Cu-apatite (15 mol%), arylboronic acid (1 mmol), N(H)-heterocycles (0.5 mmol), K2CO3 (1 mmol), methanol (2 mL), 60 °C for 6 h.b Isolated yield. | |||
| 1 | Imidazole | C6H5 | 89 |
| 2 | Imidazole | 4-CH3–C6H4 | 87 |
| 3 | Imidazole | 3-CH3–C6H4 | 85 |
| 4 | Imidazole | 2-CH3–C6H4 | 83 |
| 5 | Imidazole | 4-MeO–C6H4 | 92 |
| 6 | Imidazole | 4-NO2–C6H4 | 78 |
| 7 | Indole | C6H5 | 91 |
| 8 | Indole | 4-CH3–C6H4 | 86 |
| 9 | Indole | 3-CH3–C6H4 | 88 |
| 10 | Indole | 2-CH3–C6H4 | 85 |
| 11 | Indole | 4-MeO–C6H4 | 96 |
| 12 | Indole | 4-NO2–C6H4 | 72 |
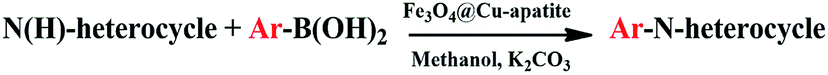
Another benefit of this nanocatalyst system consists of its ease of recyclability. The coupling of imidazole with arylboronic acid was chosen as a model reaction for the reusability study (Fig. 8). After the cross-coupling reaction, the recovered catalyst was washed twice by dichloromethane and water, dried at room temperature before being reused under similar conditions for the next run. It has to be noted that the catalytic behavior of the Fe3O4@Cu-apatite catalyst remains nearly the same for the five successive runs. After the 5th cycle, a notable loss of the catalytic activity was observed. The N-arylation of N-arylheterocycles over Fe3O4@Cu-apatite was compared with other catalysts reported in the literature as tabulated in Table 5. From this, we can show clearly that our Fe3O4@Cu-apatite catalyst exhibited a best catalytic activity of N-arylation of imidazole comparing with other catalytic systems.
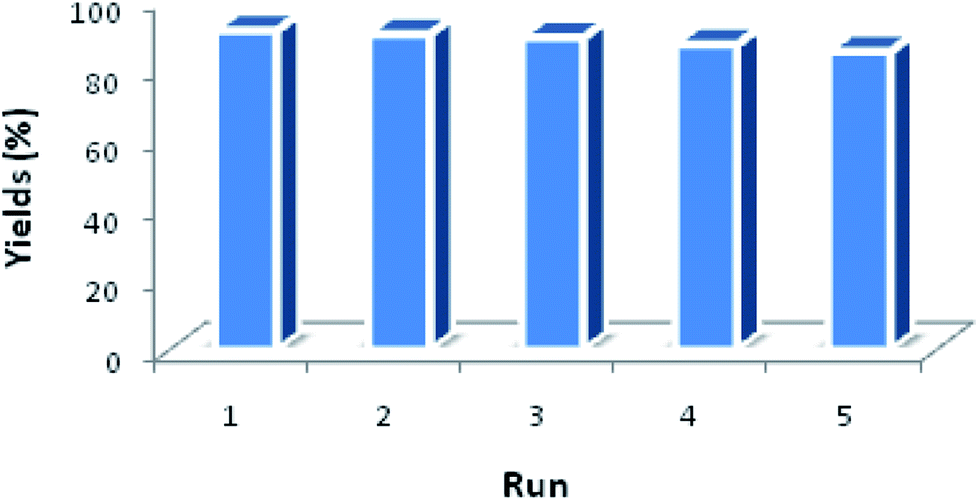 | ||
| Fig. 8 Reuse performance of Fe3O4@Cu-apatite catalyst in the N-arylation of imidazole. Reaction conditions: Fe3O4@Cu-apatite (10 mol%), arylboronic acid (1 mmol), imidazole (0.5 mmol), K2CO3 (1 mmol), methanol (2 mL), 60 °C for 8 h. | ||
In order to investigate the behavior of the Fe3O4@Cu-apatite catalyst during recycling experiments, XRD and STEM analysis were performed (Fig. 9). It was observed from the XRD pattern of the Fe3O4@Cu-apatite catalyst that, after one catalytic cycle, the crystalline composition of the catalyst remained unchanged. Also, from STEM analysis, Cu-apatite was still tightly anchored on the surface of Fe3O4 after one catalytic cycle. The catalytic effect of copper ions leached from the Fe3O4@Cu-apatite was studied in the N-arylation reaction, which was carried out under the optimum conditions. After 2 hours of heating, the catalyst was removed and the remaining reaction mixture was reheated. It was observed that the concentration of the desired product didn't increase even after heating for an additional 8 h. To further evaluate the catalytic effect of copper ions leached from the Fe3O4@Cu-apatite, ICP analysis was used against the catalyst before and after reaction. The copper concentration of the catalyst was found to be 1.83 wt% for the fresh catalyst and 1.82 wt% after one catalytic cycle in N-arylation of imidazole, which confirms negligible copper leaching.
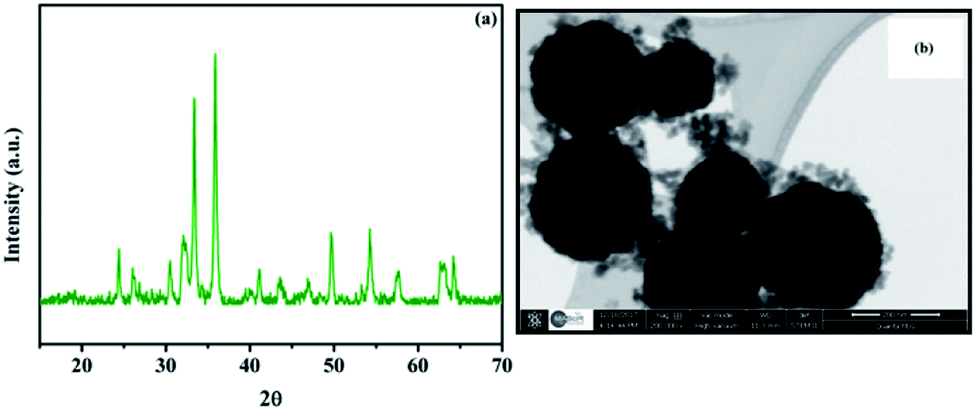 | ||
| Fig. 9 XRD (a) and STEM (b) of Fe3O4@Cu-apatite after the first catalytic test. | ||
4. Conclusion
In summary, magnetic Fe3O4@Cu-apatite core–shell nanocatalysts have been successfully prepared using a hydrothermal method. The physico-chemical properties of these materials were evaluated by FT-IR, XRD, SEM, STEM as well as the adsorption–desorption of nitrogen. The prepared catalysts showed potential capability for the preparation of N-arylimidazoles through the N-arylation reaction under mild conditions. Thereafter, the optimal reaction conditions were explored, and methanol was identified as the optimum solvent of the reaction, which is environmentally friendly. Furthermore, the Fe3O4@Cu-apatite can also be easily separated by an external magnet and reused for five runs with only a slight decrease in its catalytic activity.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The financial assistance of the MAScIR Foundation, towards this research is hereby acknowledged.References
- P. Cozzi, G. Carganico, D. Fusar, M. Grossoni, M. Menichincheri, V. Pinciroli, R. Tonani, F. Vaghi and P. Salvati, J. Med. Chem., 1993, 36, 2964–2972 CrossRef CAS.
- T. Gungor, A. Fouquet, J. M. Teulon, D. Provost, M. Cazes and A. Cloarec, J. Med. Chem., 1992, 35, 4455–4463 CrossRef CAS.
- K. M. Meyers, N. Kim, J. L. Méndez-Andino, X. E. Hu, R. N. Mumin, S. R. Klopfenstein, J. A. Wos, M. C. Mitchell, J. L. Paris, D. C. Ackley, J. K. Holbert, S. W. Mittelstadt and O. Reizes, Bioorg. Med. Chem. Lett., 2007, 17, 814–818 CrossRef CAS.
- I. Salama, C. Hocke, W. Utz, O. Prante, F. Boeckler, H. Hübner, A. Torsten Kuwert and P. Gmeine, J. Med. Chem., 2007, 489–500 CrossRef CAS.
- P. E. Fanta, Synthesis, 1974, 1974, 9–21 CrossRef.
- K. Matsuo, Y. Shichida, H. Nishida, S. Nakata and M. Okubo, J. Phys. Org. Chem., 1994, 7, 9–17 CrossRef CAS.
- A. Kiyomori, J.-F. Marcoux and S. L. Buchwald, Tetrahedron Lett., 1999, 40, 2657–2660 CrossRef CAS.
- P. Y. S. Lam, S. Deudon, K. M. Averill, R. Li, M. Y. He, A. Philip DeShong and C. G. Clark, J. Am. Chem. Soc., 2000, 122, 7600–7601 CrossRef CAS.
- G. I. Elliott and J. P. Konopelski, Org. Lett., 2000, 2, 3055–3057 CrossRef CAS.
- J. Baruah, K. Gogoi, A. Dewan, G. Borah and U. Bora, Bull. Korean Chem. Soc., 2017, 38, 1203–1208 CrossRef CAS.
- A. Gogoi, G. Sarmah, A. Dewan and U. Bora, Tetrahedron Lett., 2014, 55, 31–35 CrossRef CAS.
- I. Munir, A. F. Zahoor, N. Rasool, S. A. R. Naqvi, K. M. Zia and R. Ahmad, Mol. Diversity, 2019, 23, 215–259 CrossRef CAS.
- A. Siva Reddy, K. Ranjith Reddy, D. Nageswar Rao, C. K. Jaladanki, P. V. Bharatam, P. Y. S. Lam and P. Das, Org. Biomol. Chem., 2017, 15, 801–806 RSC.
- Y. Zou, H. Lin, P. A. Maggard and A. Deiters, Eur. J. Org. Chem., 2011, 2011, 4154–4159 CrossRef CAS.
- K. R. Reddy, N. S. Kumar, B. Sreedhar and M. L. Kantam, J. Mol. Catal. A: Chem., 2006, 252, 136–141 CrossRef CAS.
- S. M. Islam, S. Mondal, P. Mondal, A. S. Roy, K. Tuhina, N. Salam and M. Mobarak, J. Organomet. Chem., 2012, 696, 4264–4274 CrossRef CAS.
- R. Xiao, H. Zhao and M. Cai, Tetrahedron, 2013, 69, 5444–5450 CrossRef CAS.
- S. K. Das, P. Deka, M. Chetia, R. C. Deka, P. Bharali and U. Bora, Catal. Lett., 2018, 148, 547–554 CrossRef CAS.
- B. M. Choudary, C. Sridhar, M. L. Kantam, A. G. T. Venkanna and B. Sreedhar, J. Am. Chem. Soc., 2005, 127, 9948–9949 CrossRef CAS.
- N. Panda, A. K. Jena, S. Mohapatra and S. R. Rout, Tetrahedron Lett., 2011, 52, 1924–1927 CrossRef CAS.
- R. Zhang, C. Miao, Z. Shen, S. Wang, C. Xia and W. Sun, ChemCatChem, 2012, 4, 824–830 CrossRef CAS.
- S. Sebti, M. Zahouily, H. Lazrek, J. Mayoral and D. Macquarrie, Curr. Org. Chem., 2008, 12, 203–232 CrossRef CAS.
- R. Z. LeGeros, Calcium phosphates in oral biology and medicine, 1991, vol. 15 Search PubMed.
- M. Epple, K. Ganesan, R. Heumann, J. Klesing, A. Kovtun, S. Neumann and V. Sokolova, J. Mater. Chem., 2010, 20, 18–23 RSC.
- A. Solhy, R. Tahir, S. Sebti, R. Skouta, M. Bousmina, M. Zahouily and M. Larzek, Appl. Catal., A, 2010, 374, 189–193 CrossRef CAS.
- A. Smahi, A. Solhy, H. El Badaoui, A. Amoukal, A. Tikad, M. Maizi and S. Sebti, Appl. Catal., A, 2003, 250, 151–159 CrossRef CAS.
- R. Tahir, K. Banert, A. Solhy and S. Sebti, J. Mol. Catal. A: Chem., 2006, 246, 39–42 CrossRef CAS.
- A. Solhy, J. H. Clark, R. Tahir, S. Sebti and M. Larzek, Green Chem., 2006, 8, 871–874 RSC.
- Q. Y. Ma, S. J. Traina, T. J. Logan and J. A. Ryan, Environ. Sci. Technol., 1993, 27, 1803–1810 CrossRef CAS.
- X. Cao, L. Q. Ma, D. R. Rhue and C. S. Appel, Environ. Pollut., 2004, 131, 435–444 CrossRef CAS.
- M. Mouflih, A. Aklil and S. Sebti, J. Hazard. Mater., 2005, 119, 183–188 CrossRef CAS.
- Z. Elouear, J. Bouzid, N. Boujelben, M. Feki, F. Jamoussi and A. Montiel, J. Hazard. Mater., 2008, 156, 412–420 CrossRef CAS.
- S. Saoiabi, S. El Asri, A. Laghzizil, A. Saoiabi, J. L. Ackerman and T. Coradin, Chem. Eng. J., 2012, 211–212, 233–239 CrossRef CAS.
- F. Z. Shi, Y. G. Li, H. Z. Wang and Q. H. Zhang, Adv. Mater. Res., 2012, 465, 66–71 CAS.
- A. A. White, S. M. Best and I. A. Kinloch, Int. J. Appl. Ceram. Technol., 2007, 4, 1–13 CrossRef CAS.
- S. Blindow, M. Pulkin, D. Koch, G. Grathwohl and K. Rezwan, Adv. Eng. Mater., 2009, 11, 875–884 CrossRef CAS.
- L. Dong, Z. Zhu, Y. Qiu and J. Zhao, Chem. Eng. J., 2010, 165, 827–834 CrossRef CAS.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang, G. X. Xie and Z. F. Liu, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- C.-H. Hou, S.-M. Hou, Y.-S. Hsueh, J. Lin, H.-C. Wu and F.-H. Lin, Biomaterials, 2009, 30, 3956–3960 CrossRef CAS.
- D. Wang, X. Duan, J. Zhang, A. Yao, L. Zhou and W. Huang, J. Mater. Sci., 2009, 44, 4020–4025 CrossRef CAS.
- Z. Yang, X. Gong and C. Zhang, Chem. Eng. J., 2010, 165, 117–121 CrossRef CAS.
- A. Mir, D. Mallik, S. Bhattacharyya, D. Mahata, A. Sinha and S. Nayar, J. Mater. Sci.: Mater. Med., 2010, 21, 2365–2369 CrossRef CAS.
- E. B. Ansar, M. Ajeesh, Y. Yokogawa, W. Wunderlich and H. Varma, J. Am. Ceram. Soc., 2012, 95, 2695–2699 CrossRef CAS.
- R. K. Singh, A. M. El-Fiqi, K. D. Patel and H.-W. Kim, Mater. Lett., 2012, 75, 130–133 CrossRef CAS.
- E. M. Múzquiz-Ramos, D. A. Cortés-Hernández, J. C. Escobedo-Bocardo, A. Zugasti-Cruz, X. S. Ramírez-Gómez and J. G. Osuna-Alarcón, J. Mater. Sci.: Mater. Med., 2013, 24, 1035–1041 CrossRef.
- A. Amedlous, O. Amadine, Y. Essamlali, K. Daanoun, M. Aadil and M. Zahouily, RSC Adv., 2019, 9, 14132–14142 RSC.
- Y. Essamlali, O. Amadine, H. Maati, K. Abdelouahdi, A. Fihri, M. Zahouily, R. S. Varma and A. Solhy, ACS Sustainable Chem. Eng., 2013, 1, 1154–1159 CrossRef CAS.
- O. Amadine, Y. Essamlali, A. Fihri, M. Larzek and M. Zahouily, RSC Adv., 2017, 7, 12586–12597 RSC.
- O. Amadine, H. Maati, K. Abdelouhadi, A. Fihri, S. El Kazzouli, C. Len, A. El Bouari and A. Solhy, J. Mol. Catal. A: Chem., 2014, 395, 409–419 CrossRef CAS.
- Y. Essamlali, O. Amadine, M. Larzek, C. Len and M. Zahouily, Energy Convers. Manage., 2017, 149, 355–367 CrossRef CAS.
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef CAS.
- P. Li, L. Li, Y. Zhao, L. Sun and Y. Zhang, J. Inorg. Biochem., 2016, 156, 49–54 CrossRef CAS.
- T. Garnier, R. Sakly, M. Danel, S. Chassaing and P. Pale, Synthesis, 2016, 49, 1223–1230 CrossRef.
- L.-Y. Zhang and L. Wang, Chin. J. Chem., 2006, 24, 1605–1608 CrossRef CAS.
- M. L. Kantam, G. T. Venkanna, C. Sridhar, A. Bojja Sreedhar and B. M. Choudary, J. Org. Chem., 2006, 71, 9522–9524 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and spectral data the compounds listed in Table 4. See DOI: 10.1039/c9ra06991g |
| This journal is © The Royal Society of Chemistry 2019 |