DOI:
10.1039/C9RA07051F
(Paper)
RSC Adv., 2019,
9, 37232-37240
Therapeutic polymeric nanomedicine: GSH-responsive release promotes drug release for cancer synergistic chemotherapy†
Received
3rd September 2019
, Accepted 1st November 2019
First published on 15th November 2019
Abstract
To obtain an efficient dual-drug release and enhance therapeutic efficiency for combination chemotherapy, a glutathione (GSH)-responsive therapeutic amphiphilic polyprodrug copolymer (mPEG-b-PCPT) is synthesized to load doxorubicin (DOX) via hydrophobic and π–π stacking interaction. In this nanomedicine system (mPEG-b-PCPT/DOX), the ratio of the two drugs can be easily modulated by changing the loading content of DOX. The in vitro drug release curves and laser confocal images suggested that the release of CPT and DOX is induced through a “release promotes release strategy”: after internalization into tumor cells, the disulfide bonds in the nanomedicine are cleaved by glutathione (GSH) in the cytoplasm and then lead to the release of CPT. Meanwhile, the disassembly of nanomedicine immediately promotes the co-release of DOX. The optimum dose ratio of CPT and DOX is evaluated via the combination index (CI) value using HepG-2 cells. The results of cell apoptosis and cell viability prove the better synergistic efficiency of the nanomedicine than free drugs at the optimum dose ratio of 1. Consequently, this stimuli-responsive synergistic chemotherapy system provides a direction for the fabrication of nanomedicines possessing promising potential in clinical trials.
1. Introduction
Chemotherapy has been the major modality for cancer therapy. A variety of chemotherapeutic agents, such as anti-metabolites, alkylating agents, topoisomerase inhibitors, anti-microtubule agents and cytotoxic antibiotics, have been widely employed for cancer chemotherapy.1 The clinical application of traditional small molecule chemotherapeutic agents still has some obstacles, including low bioavailability, superimposed side effects and drug resistance.2,3 Over the past decades, the combination of two or more drugs has been frequently used as a therapeutic regimen benefiting from the complementary pharmacological mechanisms of multiple drugs to interrupt tumor growth.4–9 However, the bioavailability and half-life of each drug formulation can be extremely different, resulting in a problem to find the best dose combination of drugs.
Nanomedicine encapsulating multi-drugs is able to co-deliver drugs into cells to promote therapeutic performance.10–17 Among them, nanomedicines formed from amphiphlic polyprodrugs and another encapsulated drugs for combination therapy exhibit several remarkable advantages, including easy modulation of drug loading content, controllable release triggered by exogenous stimuli.18–26 The common strategy for polyprodrug preparation is to conjugate drugs to the carriers through post-modification of polymeric backbones. But the shortcoming is that the drug content of polyprodrugs can not be precisely controlled and the graft ratio of drugs is hardly controlled between different batches, which are critical for further applications and even clinical translations. Additionally, it is extremely difficult using the post-modification method to adjust the optimal ratio between conjugated drug and loaded drug to realize synergistic efficacy. Thus, an amphiphilic polyprodrug with precise conjugation number is extremely important for combination therapy. Moreover, inefficient release of drugs from the nanomedicines after internalization into cancer cells also significantly reduce the synergistic effect.27,28 The common attention is mainly paid to increasing co-release of multi-drugs in nanomedicine design, taking advantage of covalent linkage or physical interactions between chemotherapeutic drugs and functional materials.29–35 Notably, glutathione (GSH)-responsiveness is an ideal candidate for the development of controlled drug release system because there is a significant difference in GSH concentration between intracellular (2–10 mM) and extracellular environment (2–20 μM), so that the nanomedicine will be able to keep stable in extracellular environment and release the conjugated drug in the cells, thus enhancing the therapeutic efficacy while reducing the side effects.36–42
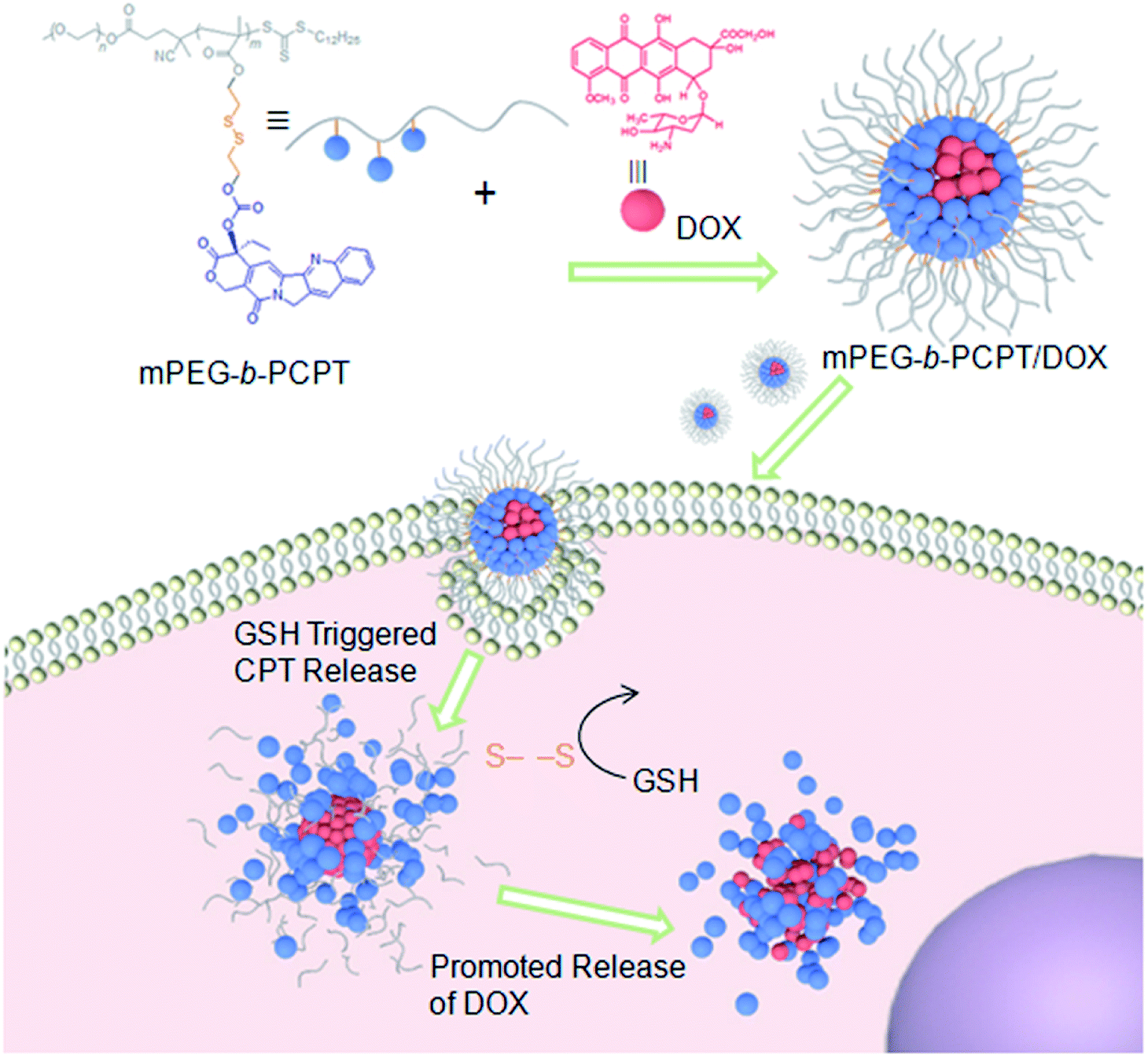
Herein, we utilize an amphiphilic polyprodrug copolymer (mPEG-b-PCPT) in which the drug (camptothecin, CPT) is linked to the backbone through a GSH-responsive disulfide bond by reversible addition–fragmentation transfer (RAFT) polymerization to encapsulate doxorubicin (DOX) via hydrophobic and π–π stacking interaction to form a nanomedicine (mPEG-b-PCPT/DOX). The topoisomerase I inhibitor (CPT) and topoisomerase II inhibitor (DOX) with a complementary anticancer mechanism is used for combination therapy.43–49 Benefiting from the RAFT living polymerization, the polymerization degree of prodrug segment can be precisely controlled, so that the optimized ratio of two drugs can be easily modulated by changing the loading content. The synergistic chemotherapy of CPT and DOX is induced by the mPEG-b-PCPT/DOX nanomedicine through a “release promote release strategy”, which is shown in Scheme 1. After the nanoparticles (NPs) were efficiently internalized by tumor cells, the disulfide bonds are rapidly cleaved under reductive cytosolic circumstance to release the active CPT. The disassembly of hydrophobic core immediately results in a boost release of DOX, promoting the intercellular drug co-release, thus achieving excellent synergistic effect. Compared to the reported nanomedicine for combination therapy, this combination therapeutic system integrating features of flexible preparation of different drug ratios, reduction-responsive co-release and good synergistic effect is crucial for potential clinical combination therapy research.
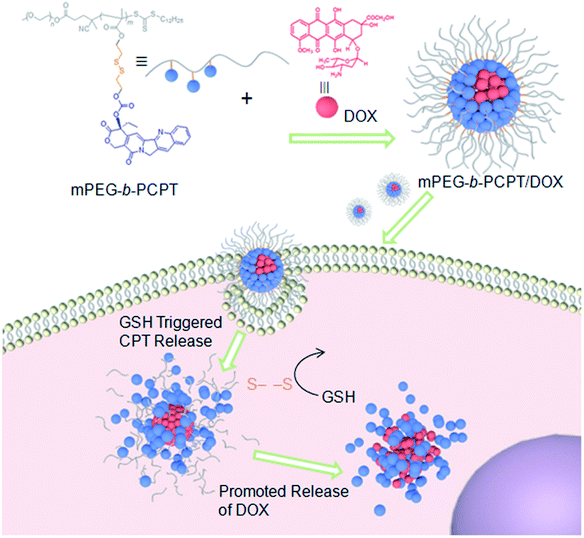 |
| | Scheme 1 Schematic illustration of the preparation of mPEG-b-PCPT/DOX nanomedicine and the “drug release promotes release strategy” of mPEG-b-PCPT/DOX for cancer synergistic chemotherapy. | |
2. Results and discussion
2.1 Synthesis and characterization of mPEG-b-PCPT
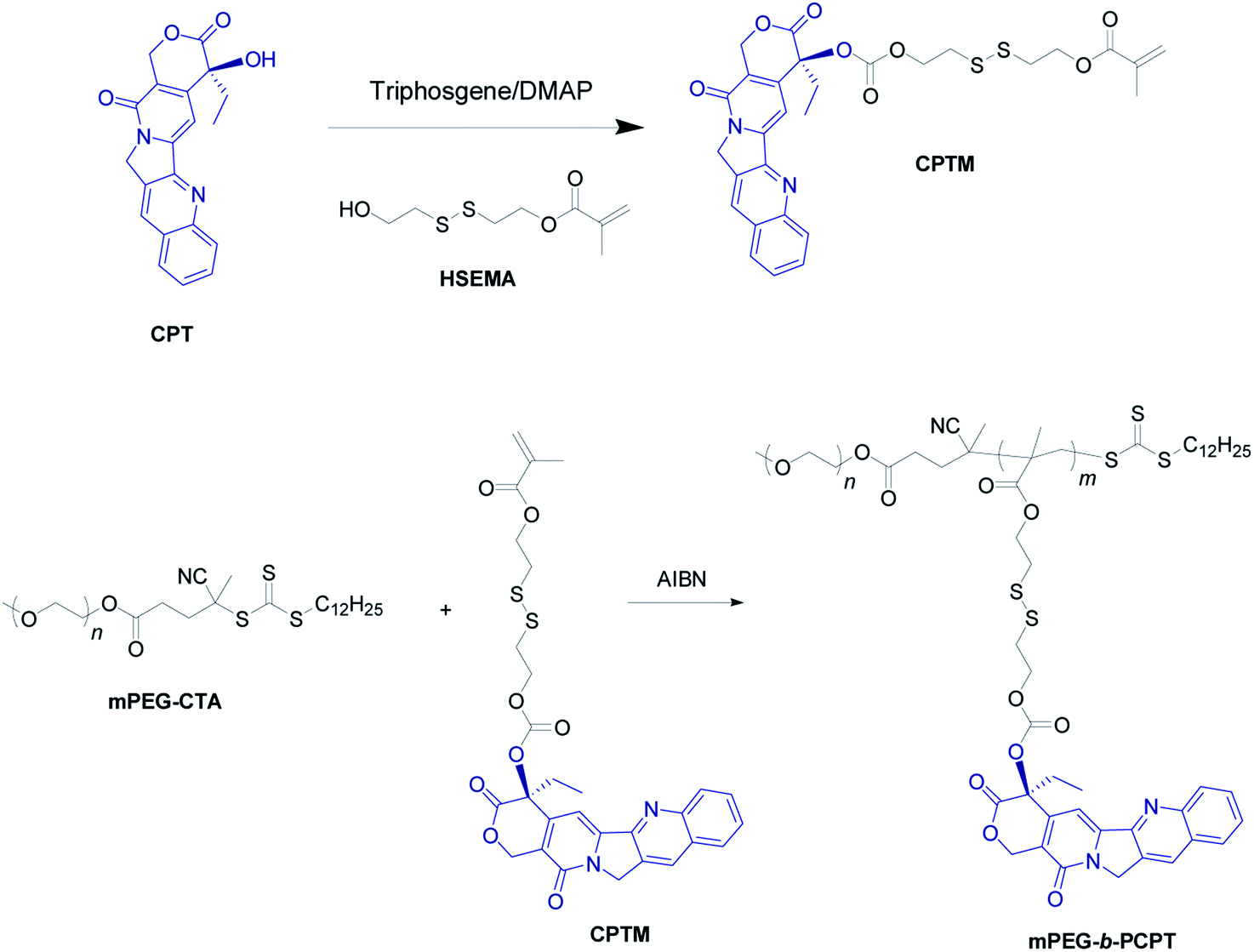
The mPEG-b-PCPT was synthesized by reversible addition–fragmentation transfer (RAFT) polymerization with a cleavable prodrug monomer (CPTM) and a PEG-based macroRAFT agent (mPEG-CTA) (Scheme 2).50 1H NMR and 13C NMR of CPTM and mPEG-b-PCPT confirmed the chemical structure and revealed the actual degree of polymerization (DP) of mPEG-b-PCPT to be 50 (Fig. S1–S3†), indicating a high drug loading content of CPT (55.1%). More importantly, the CPT loading content could be easily modulated by DP. GPC analysis suggested the Mn of this amphiphilic polydrug was 34.7 kDa and the Mw/Mn was 1.27 (Fig. S4†). The generated diblock copolymer mPEG-b-PCPT exhibiting controllable DP and narrow polydispersity index showed great potential for further application of DOX loading and combination chemotherapy.
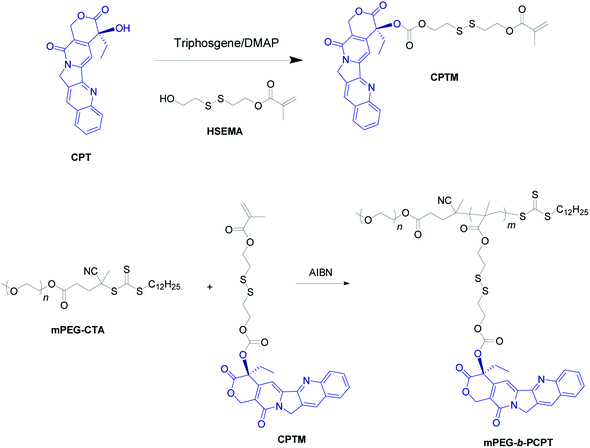 |
| | Scheme 2 Synthetic route of mPEG-b-PCPT. | |
2.2 Preparation and characterization of mPEG-b-PCPT/DOX
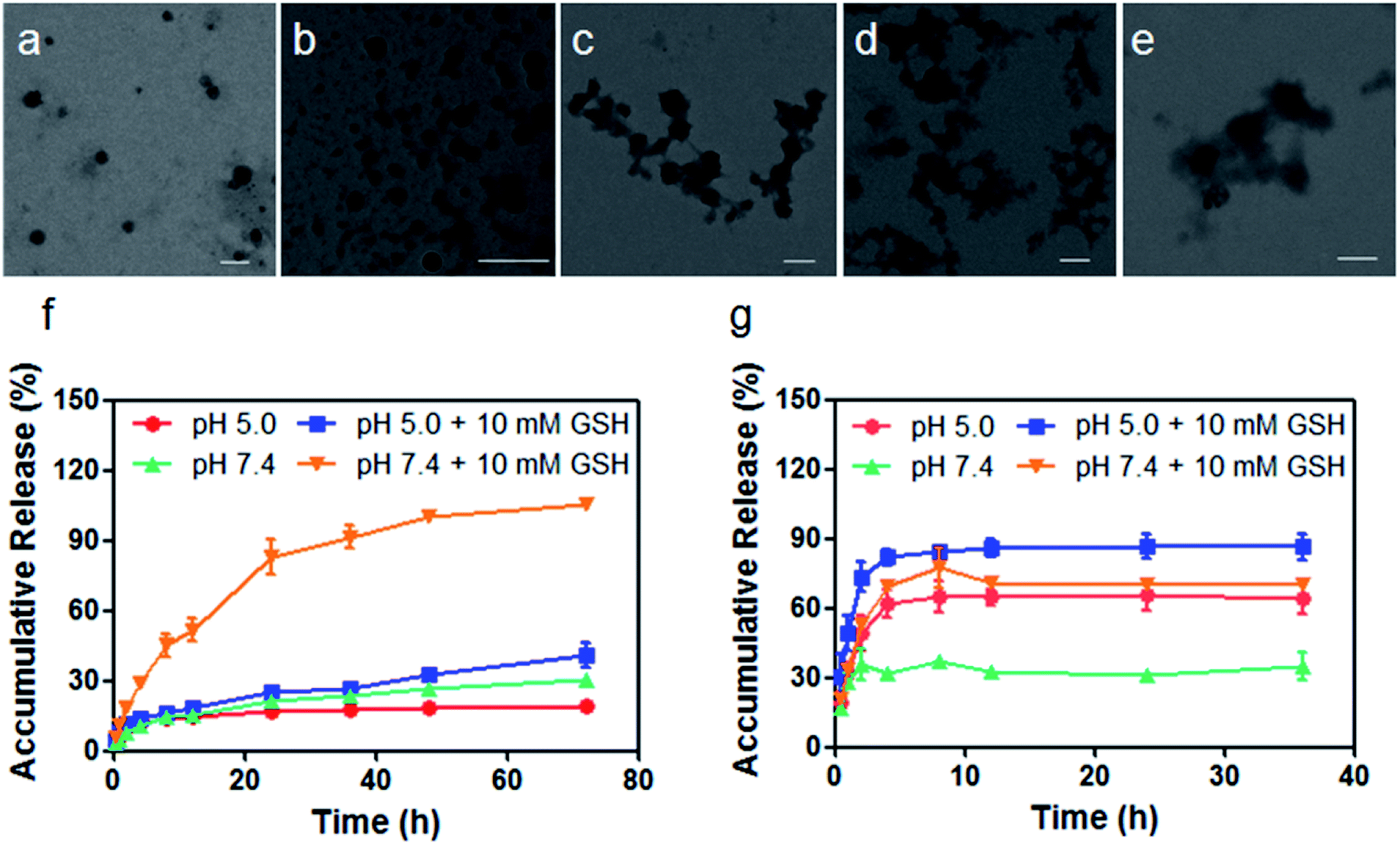
The therapeutic diblock copolymer mPEG-b-PCPT was utilized to encapsulate DOX through hydrophobic and π–π stacking interactions to fabricate a nanomedicine (mPEG-b-PCPT/DOX). Transmission electron microscopy (TEM) was used to investigate the self-assembly morphology of copolymer. As can be seen in Fig. 1a, mPEG-b-PCPT self-assembled into spherical nanoparticles with a diameter of around 150 nm. Compared with polyprodrug mPEG-b-PCPT, negligible diameter change was monitored for the dual-therapeutic mPEG-b-PCPT/DOX (Fig. 1b). After incubation with aqueous solution containing 10 mM GSH for different time, the morphology of mPEG-b-PCPT/DOX was also studied by TEM. As shown in Fig. 1c–e, the NPs disassembled into irregular aggregates in the presence of GSH, indicating the GSH-responsiveness of mPEG-b-PCPT/DOX. To reveal the drug loading capacity of mPEG-b-PCTP, loading content (LC) and encapsulation efficiency (EE) were measured at different feeding ratio. As shown in Table 1, mPEG-b-PCTP showed an excellent drug loading capacity with the EE as high as above 80%. With this nanomedicine in hand, the drug release behaviors of CPT and DOX were then carefully studied under different conditions (Fig. 1f and g). In the absence of GSH, mPEG-b-PCPT/DOX NPs were stable at pH 7.4 and only a small portion of CPT (21.4 ± 0.1%) was released after 24 h. The release rate was significantly boosted in the presence of GSH (10.0 mM) and 82.9 ± 7.3% CPT was released from the NPs. The activity of GSH could be greatly weakened in the acidic environment, which was confirmed by the low release rate of CPT (24.9 ± 0.7%) at pH 5.0 after 24 h. In the case of DOX, only 31.0 ± 0.9% of DOX was released at pH 7.4 after 24 h. However, the release efficiency increased up to 65.6 ± 6.8% at pH 5.0, suggesting a pH-responsive drug release attributing to the protonation of the amine group. More interestingly, the release efficiency of DOX reached to 70.3 ± 2.3% in the presence of GSH at pH 7.4, indicating the GSH-responsive CPT release significantly promoted the release of DOX because of the disassembly of the NPs by changing the amphiphilic property of the polymeric carrier, thus achieving amplify effect to improve their synergistic anticancer efficacy. Based on the above results, the nanomedicine could be stable in bloodstream and extracellular matrix due to the low level of GSH and high pH value, remarkably decreasing the side effects towards normal tissues, while after cellular internalization, partial DOX is release in endo/lysosome triggered by the low pH environment. Additionally, the disulfide bonds are cleaved by the GSH in cytoplasm, further promoting the release of loaded DOX.
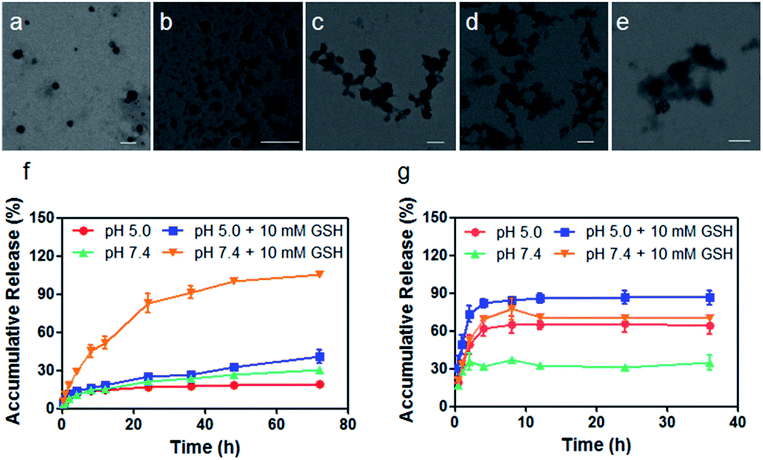 |
| | Fig. 1 Characterization of mPEG-b-PCPT and mPEG-b-PCPT/DOX. TEM images of (a) mPEG-b-PCPT and (b) mPEG-b-PCPT/DOX. TEM images of mPEG-b-PCPT/DOX after the treatment of GSH (10 mM) for (c) 2 h, (d) 6 h and (e) 24 h. Scale bar = 200 nm. Drug release curves of (f) CPT and (g) DOX from mPEG-b-PCPT/DOX NPs under different conditions. Data represent as mean ± SD (n = 3). | |
Table 1 Drug loading capacity of mPEG-b-PCPT
| mPEG-b-PCPT (mg) |
DOX (mg) |
Encapsulation efficiencya (EE, %) |
Loading contentb (LC, %) |
| Encapsulation efficiency (EE) = mload/madd × 100%, where madd and mload represent the drug mass added during the preparation of drug-loaded nanomedicine and loaded by the nanomedicine, respectively. Loading content (LC) = mload/(mload + m) × 100%, where m represents the mass of polyprodrug used during the preparation of drug-loaded nanomedicine. |
| 5.0 |
0.8 |
86.8 ± 4.4 |
12.2 ± 0.5 |
| 5.0 |
1.0 |
80.6 ± 3.3 |
13.9 ± 0.5 |
| 5.0 |
2.0 |
90.3 ± 1.7 |
26.5 ± 0.4 |
| 5.0 |
3.0 |
85.3 ± 1.3 |
33.9 ± 0.4 |
| 5.0 |
4.0 |
85.3 ± 1.5 |
40.6 ± 0.4 |
| 5.0 |
5.0 |
81.6 ± 1.6 |
44.9 ± 0.5 |
2.3 Cellular uptake of mPEG-b-PCPT/DOX
Confocal laser scanning microscopy (CLSM) was performed to assess the cellular uptake and intracellular drug release of mPEG-b-PCPT/DOX NPs. Glutathione reduced ethyl ester (GSH-OEt) was pretreated for 2 h to increase the intracellular GSH concentration. As shown in Fig. 2, at the time point of 2 h, 6 h and 12 h, in sharp contrast, the fluorescence intensity of free CPT and DOX was much weaker than the one after incubation of mPEG-b-PCPT/DOX NPs in HepG-2 (hepatocellular carcinoma) cells, demonstrating that the drugs were uptaken into the cells via different pathway, endocytosis of mPEG-b-PCPT/DOX NPs and diffusion of free drugs, and endocytosis was more efficient. The fluorescence of CPT and DOX displayed high co-localization in the cells cultured with mPEG-b-PCPT/DOX NPs in both cytoplasm and nucleus at 2 h, 6 h, and 12 h, revealing the co-delivery of two drugs. Notably, the GSH-OEt pretreated cells showed higher fluorescence intensity of DOX and CPT than the cells without pre-treatment of GSH-OEt, confirming the reductive-responsive enhancement of drug co-release by the increased intracellular GSH.
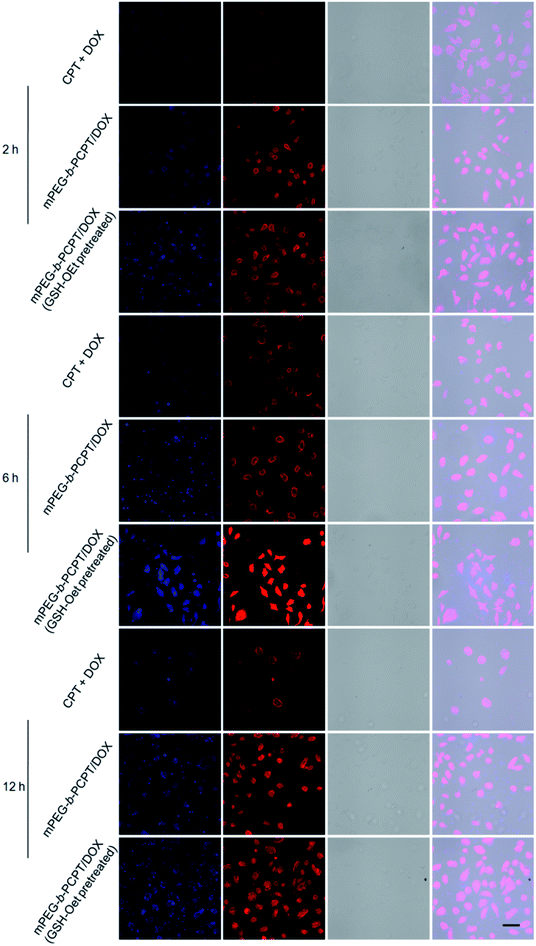 |
| | Fig. 2 CLSM images of HepG-2 cells treated with the mixture of free CPT and DOX, mPEG-b-PCPT/DOX NPs for 2 h, 6 h and 12 h, respectively. The cells for GSH pretreated group were pretreated with glutathione reduced ethyl ester (GSH-OEt) (concentration of 10 mM) for 2 h. Scale bar = 25 μm. | |
2.4 Synergistic effect of mPEG-b-PCPT/DOX
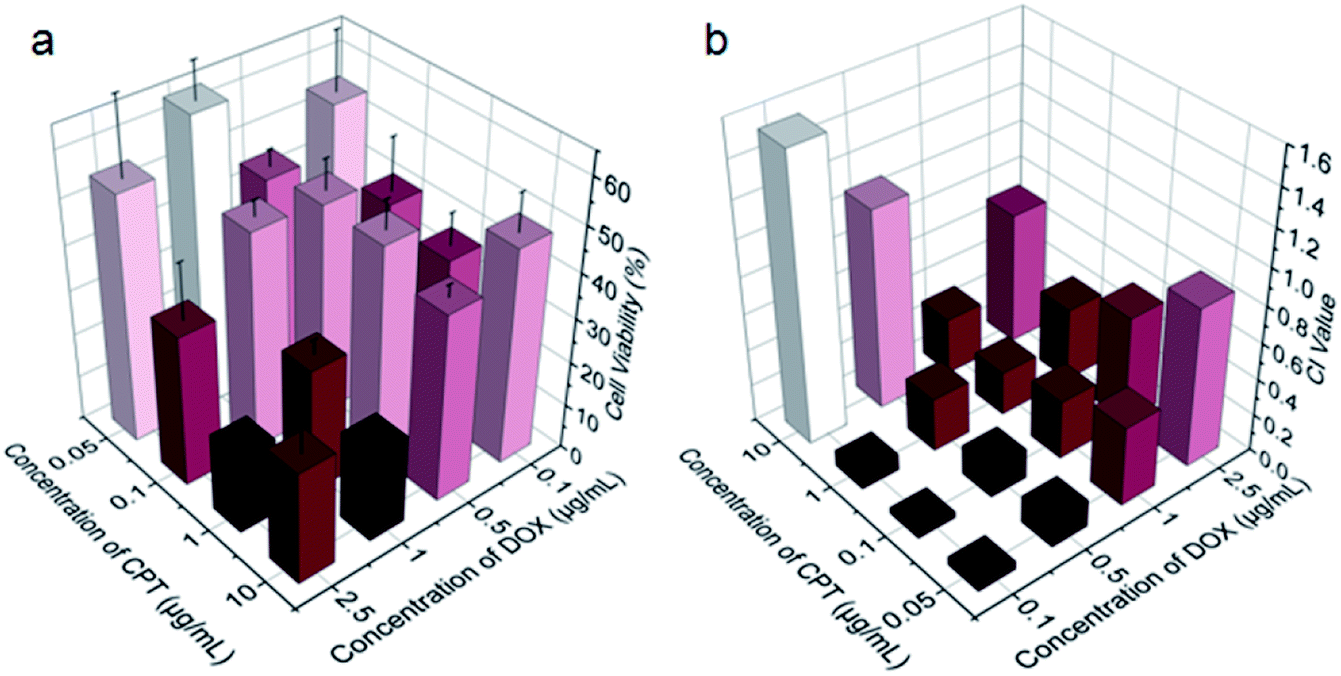
Benefiting from the efficient endocytosis of mPEG-b-PCPT/DOX NPs and the co-release of CPT and DOX triggered by reductive GSH, the synergistic therapeutic effect could be higher than free drug pairs. The synergistic effect of this nanomedicine was investigated by a 3-(4′,5′-dimethylthiazol-2′-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Fig. S5 and S6† showed the cell viability after incubation with different concentrations of drugs for 24 h against HepG-2 and human umbilical vein endothelial (HUVEC) cells. Obviously, the healthy HUVEC cells, which were used as control, were not as sensitive as the carcinoma cells to the chemotherapeutic drugs. For the free drugs, the half maximal inhibitory concentration (IC50) of CPT and DOX was calculated to be 1.81 and 0.106 μg mL−1, respectively. Interestingly, the ratio between CPT and DOX in the NPs could be easily controlled by changing the loading content of DOX due to the satisfactory drug loading capacity of mPEG-b-PCPT. In order to figure out the optimum combination value, the cell viability of HepG-2 cells at different mass ratio of DOX and CPT were carried out (Fig. 3a). Then combination index (CI) values were fully investigated and utilized to quantify synergy of CPT and DOX (CI value below 1 implies synergy whereas above 1 implies antagonism). As shown in Fig. 3b and S7,† the CI value was related to the drug mass ratio and drug dose. It was increased by increasing the fraction of DOX, when the concentration of DOX and CPT reached 0.1 μg mL−1, the optimum synergy was observed (CI = 0.036, mass ratio = 1).
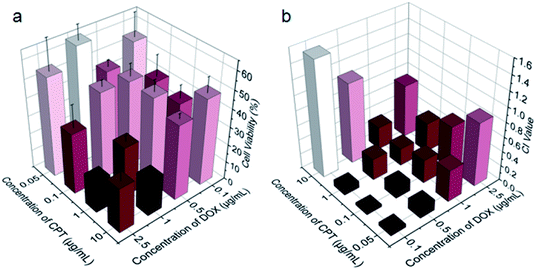 |
| | Fig. 3 (a) Viability of HepG-2 cells treated with the mixtures of CPT and DOX at indicated concentrations for 24 h. Data represent as mean ± SD (n = 4). (b) Calculated CI values for the CPT and DOX combinations for HepG-2 cells at indicated concentrations. | |
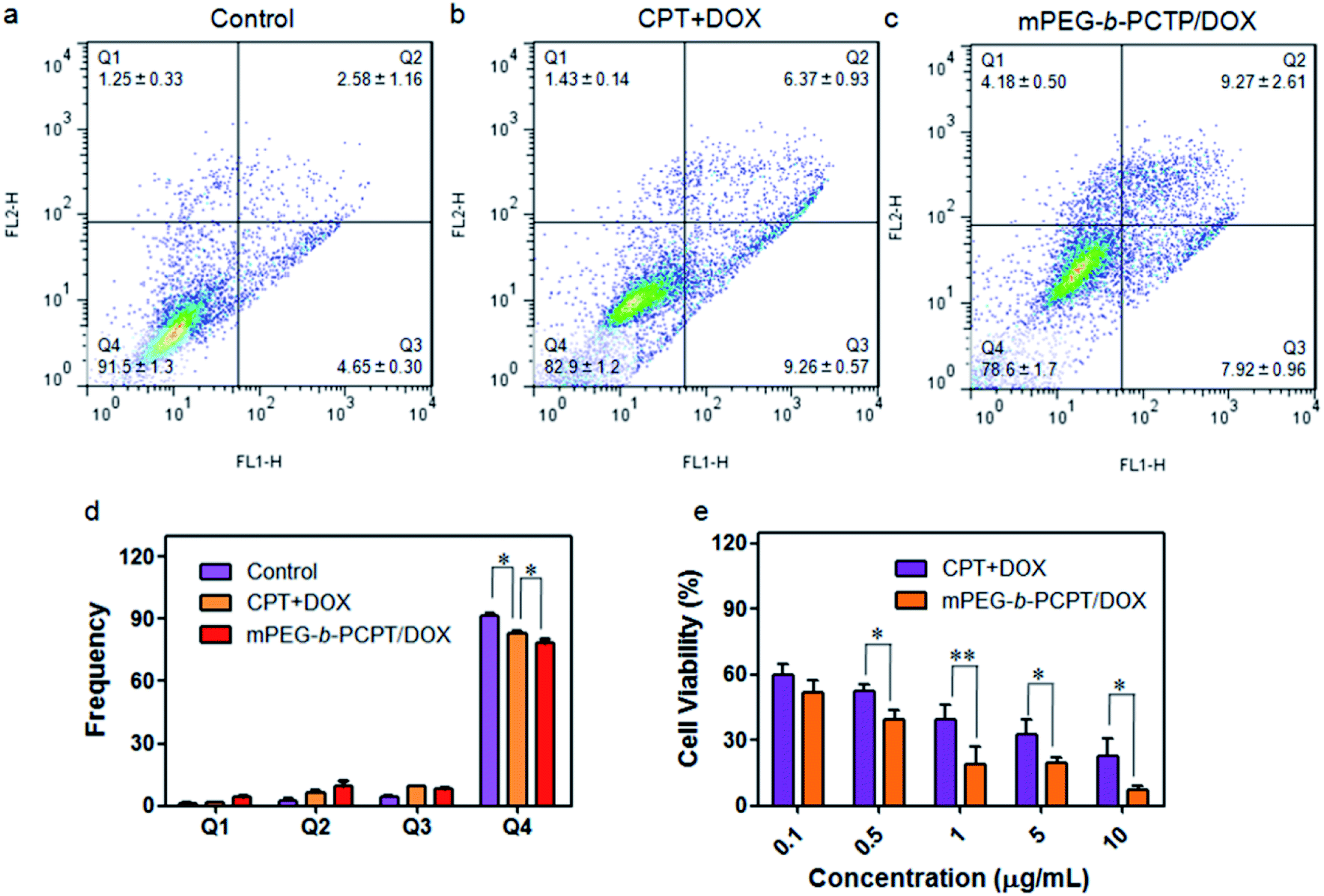
As CPT and DOX inhibit topoisomerase I and topoisomerase II, respectively, which both induce the DNA damage, so we studied the cell apoptosis induced by mPEG-b-PCPT/DOX NPs using Annexin V-FITC/PI kit by flow cytometry at the optimum mass ratio. We incubated HepG-2 cells with low-dose (equivalent drug dose of 0.1 μg mL−1) of mPEG-b-PCPT/DOX or free drugs to avoid the influence of red fluorescence from DOX. Living cells were represented in Q4 phase, whereas apoptosis cells and necrotic cells were shown in Q2, Q3 and Q1 phases. Fig. 4a–d indicated that the frequency of living cells were significantly decreased from 82.9 ± 1.2 to 78.6 ± 1.7 (p < 0.05) when the CPT/DOX combination was utilized as mPEG-b-PCPT/DOX NPs. The efficacy of mPEG-b-PCPT/DOX co-release system was higher than free drug combination, demonstrating the advantage of this nanomedicine formulation benefiting from the GSH-triggered “release promote release strategy”, which led to the co-localization and co-release of synergistic drugs in the cells. Furthermore, the combination effect was assessed by MTT assay at the optimum mass ratio of 1 mentioned above. As shown in Fig. 4e, the cell viabilities of HepG-2 cells treated with mPEG-b-PCPT/DOX NPs were significantly lower than free drug pairs. According to these results, we consequently concluded that mPEG-b-PCPT/DOX NPs could effectively co-deliver two drugs into cancer cells via endocytosis pathway, induced GSH-triggered co-release at an optimum ratio, resulting in an enhancement of synergistic effect to kill the cancer cells.
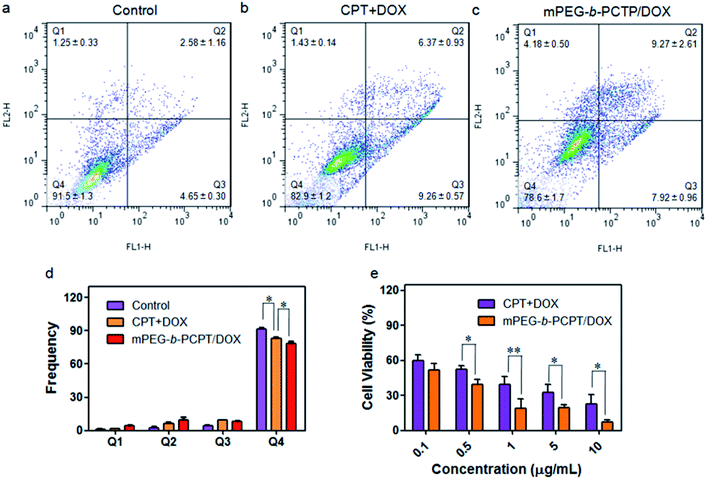 |
| | Fig. 4 Apoptosis analysis of HepG-2 cells treated with medium (a), mixture of free CPT and DOX (b) and mPEG-b-PCPT/DOX NPs (c) at the concentration of 0.1 μg mL−1 CPT and DOX for 24 h. (d) Frequencies of necrotic cells (Q1), late apoptotic cells (Q2), early apoptotic cells (Q3), and living cells (Q4) analyzed by flow cytometry after treatments of indicated formulations. Data represent mean ± SD (n = 3). *p < 0.05, Student's t-test. (e) Viability of HepG-2 cells treated with various concentrations of the mixture of free CPT and DOX and mPEG-b-PCPT/DOX NPs at a drug mass ratio of 1 for 24 h. Data represent mean ± SD (n = 4). **p < 0.01, *p < 0.05, Student's t-test. | |
3. Experimental
3.1 Materials and methods
All reagents were commercially available and used as supplied without further purification. 1H NMR spectra were recorded on a Bruker Avance III-300 spectrometer with internal standard TMS. Transmission electron microscopy (TEM) images were taken by a HT-7700 transmission electron microscopy (Hitachi, Japan). The particle size was performed by dynamic light scattering (DLS) on the Zetasizer (Nano ZS, Malvern Instruments, Worcestershire, UK).
3.2 Cell cultures
HepG-2 cells were cultured in RPMI 1640 culture medium containing 10% calf serum and 1% penicillin/streptomycin. The cells were harvested from the cell culture medium by incubating in a trypsin solution. The cells were centrifuged, and the supernatant was discarded. The cells were resuspended in serum-supplemented 1640 at a concentration of 1 × 104 cells per mL. Cells were cultured at 37 °C and 5% CO2.
3.3 Preparation of mPEG-b-PCPT/DOX
The mPEG-b-PCPT/DOX nanoparticle was prepared by the solvent evaporation method. In brief, DOX was firstly dehydrochlorinated using dichloromethane (DCM). mPEG-b-PCPT and dehydrochlorinated DOX were separately dissolved in DCM and then mixed. The mixture was slowly injected into an aqueous solution and stirred at room temperature overnight to ensure complete evaporation of DCM. To measure the drug loading capacity, the nanomedicine was prepared and centrifugated at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The supernatant was measured by a fluorescence spectrophotometry (RF-5301PC, Shimadzu, Japan) at an emission wavelength of 558 nm and excitation wavelength of 502 nm. LC and EE was determined by DOX concentration in the nanomedicine.
000 rpm for 10 min. The supernatant was measured by a fluorescence spectrophotometry (RF-5301PC, Shimadzu, Japan) at an emission wavelength of 558 nm and excitation wavelength of 502 nm. LC and EE was determined by DOX concentration in the nanomedicine.
3.4 TEM studies
The morphology of the mPEG-b-PCPT and mPEG-b-PCPT/DOX nanoparticles were measured by TEM. Samples were prepared by drop-coating the solution of nanoparticles onto a carbon-coated copper grid. The corresponding solution was left to stand for 10 min and eliminated by a microporous membrane.
3.5 In vitro release study
The in vitro drug release manner of mPEG-b-PCPT/DOX was determined using dialysis bag method in phosphate-buffered saline (PBS) in various conditions (pH = 5.0, pH = 5.0 + 10 mM GSH, pH = 7.4 and pH = 7.4 + 10 mM GSH). Briefly, 2 mL of mPEG-b-PCPT/DOX (equivalent to 20 μg mL−1 of DOX and 4 μg mL−1 of CPT) was sealed in dialysis bags (MCWO 6000–8000). The dialysis bags were added to the 50 mL conical tube containing 28 mL of drug release medium. The test conditions were a shaking speed of 100 rpm and a temperature of 37 °C ± 0.5 °C. Two milliliter of each sample was withdrawn at 0.5, 1, 2, 4, 8, 12, 24, 36, 48 and 72 hours. Free DOX and CPT was used as controls. The fluorescence intensity of DOX in the medium was measured by a fluorescence spectrophotometry at an emission wavelength of 558 nm and excitation wavelength of 502 nm. The one of CPT was measured at an emission wavelength of 435 nm and excitation wavelength of 365 nm. The standard curves were also established.
3.6 Cellular uptake study
The cellular uptake of mPEG-b-PCPT/DOX was studied by the confocal laser scanning microscopy (CLSM, IX83-FV3000-OSR, Olympus, Japan). The HepG-2 cells were seeded on 24-well plates with cell slide and grown for 18 h. The cells for GSH pretreated group were pretreated with glutathione reduced ethyl ester (GSH-OEt) (concentration of 10 mM) for 2 h. The medium was changed to serum-free 1640 medium containing mPEG-b-PCPT/DOX (at drug concentrations of 0.5 μg mL−1) and incubated for 2 h, 6 h and 12 h, respectively. After incubation, the cells were rinsed with PBS, fixed with fresh 4% paraformaldehyde, and imaged on a confocal scanning laser microscopy.
3.7 Evaluation of cytotoxicity
The cytotoxicity was evaluated by the 3-(4′,5′-dimethylthiazol-2′-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay using HepG-2 cells and HUVEC cells. Generally, the cells were seeded on a 96-well tissue culture plate at a density of 1 × 104 cells per well in 200 μL of 1640 medium containing 10% serum for 18 h. The medium was replaced with 200 μL of serum-free one containing serial dilutions of indicated concentration of drugs in various formulations for 24 h. The media were then replaced with 100 μL of the serum-free medium containing 0.5 mg mL−1 MTT and incubated for 4 h. Finally, each well was replaced with 100 μL of DMSO and measured spectrophotometrically on an ELISA plate reader (Model 550, Bio-Rad) at a wavelength 570 nm. The relative cell growth (%) related to control cells cultured in the media without the polymer was calculated by the following formula:
| V% = ([A]experimental − [A]blank)/([A]control − [A]blank) × 100%. |
The CI values were calculated by Compu Syn software.
3.8 Cell apoptosis study
The HepG-2 cells were seeded on a 12-well tissue culture plate at a density of 1.6 × 105 cells per well in 1.0 mL of 1640 medium containing 10% serum for 18 h. The media were then replaced by 1 mL of serum free 1640 containing free drug combination or mPEG-b-PCPT/DOX at drug concentrations of 0.1 μg mL−1 and incubated for 24 h. Then the cells were washed by PBS, trypsinized, stained using an Annexin V-FITC/PI apoptosis detection kit (Beyotime Biotechnology). The cells were evaluated by the flow cytometer (FACS Calibur, BD, USA).
4. Conclusions
In summary, we synthesized an amphiphilic copolymer (mPEG-b-PCPT) constructed with hydrophobic chemotherapeutic drug CPT, reductive sensitive disulfide bond and hydrophilic PEG chain by precisely controlled RATF living polymerization. By forming spherical NPs, the copolymer was utilized to load another chemotherapeutic drug DOX for synergistic chemotherapy. The mPEG-b-PCPT/DOX NPs behaved a reductive-triggered “release promote release strategy” of CPT and DOX, demonstrating the possibility of efficient co-release in the intracellular environment. The result of cellular uptake confirmed the successful internalization and reductive-responsive co-release of mPEG-b-PCPT/DOX NPs into HepG-2 cells. MTT assay and cell apoptosis study demonstrated much higher anticancer efficacy of mPEG-b-PCPT/DOX NPs than that of the free drugs combination, showing excellent synergistic effect. The present studies provided a promising GSH-responsive fast co-release system with easy controlled drug ratio for more effective combination cancer chemotherapy, providing a direction for the fabrication of nanomedicines possessing promising potentials in clinical trials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation for Young Scientists of China (Grant No. 51603184 and 81800442).
Notes and references
- S. S. Qi, J. H. Sun, H. H. Yu and S. Q. Yu, Drug Delivery, 2017, 24, 1909–1926 CrossRef CAS PubMed.
- Y. Matsumura, Jpn. J. Clin. Oncol., 2008, 38, 793–802 CrossRef PubMed.
- Y. Matsumura, Jpn. J. Clin. Oncol., 2014, 44, 515–525 CrossRef PubMed.
- Q. Fu, P. Lv, Z. Chen, D. Ni, L. Zhang, H. Yue, Z. Yue, W. Wei and G. Ma, Nanoscale, 2015, 7, 4020–4030 RSC.
- K. Hu, H. Zhou, Y. Liu, Z. Liu, J. Liu, J. Tang, J. Li, J. Zhang, W. Sheng, Y. Zhao, Y. Wu and C. Chen, Nanoscale, 2015, 7, 8607–8618 RSC.
- H. Meng, M. Wang, H. Liu, X. Liu, A. Situ, B. Wu, Z. Ji, C. H. Chang and A. E. Nel, ACS Nano, 2015, 9, 3540–3557 CrossRef CAS PubMed.
- C. Wang, G. Y. Zhang, G. H. Liu, J. M. Hu and S. Y. Liu, J. Controlled Release, 2017, 259, 149–159 CrossRef CAS PubMed.
- J. Mo, L. Wang, X. Huang, B. Lu, C. Zou, L. Wei, J. Chu, P. K. Eggers, S. Chen, C. L. Raston, J. Wu, L. Y. Lim and W. Zhao, Nanoscale, 2017, 9, 13142–13152 RSC.
- M. Yin, S. Tan, Y. Bao and Z. Zhang, J. Controlled Release, 2017, 258, 108–120 CrossRef CAS PubMed.
- L. Liao, J. Liu, E. C. Dreaden, S. W. Morton, K. E. Shopsowitz, P. T. Hammond and J. A. Johnson, J. Am. Chem. Soc., 2014, 136, 5896–5899 CrossRef CAS PubMed.
- T. Ramasamy, H. B. Ruttala, J. Y. Choi, T. H. Tran, J. H. Kim, S. K. Ku, H. G. Choi, C. S. Yong and J. O. Kim, Chem. Commun., 2015, 51, 5758–5761 RSC.
- S. S. Desale, K. S. Soni, S. Romanova, S. M. Cohen and T. K. Bronich, J. Controlled Release, 2015, 220, 651–659 CrossRef CAS PubMed.
- X. Wu, L. Zhou, Y. Su and C. M. Dong, Biomacromolecules, 2016, 17, 2489–2501 CrossRef CAS PubMed.
- W. P. Fan, B. Yung, P. Huang and X. Y. Chen, Chem. Rev., 2017, 117, 13566–13638 CrossRef CAS PubMed.
- M. Rahimi, K. D. Safa and R. Salehi, Polym. Chem., 2017, 8, 7333–7350 RSC.
- C. Cheng, X. L. Zhang, Y. B. Meng, L. Chen and Q. Q. Zhang, J. Mater. Chem. B, 2017, 5, 8487–8497 RSC.
- C. Q. Lan and S. L. Zhao, J. Mater. Chem. B, 2018, 6, 6685–6704 RSC.
- W. Cao, L. Wang and H. P. Xu, Chem. Commun., 2015, 51, 5520–5522 RSC.
- H. Z. Chen, H. P. Tham, C. Y. Ang, Q. Y. Qu, L. Z. Zhao, P. Y. Xing, L. Y. Bai, S. Y. Tan and Y. L. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 24319–24324 CrossRef CAS PubMed.
- D. L. Cao, J. L. He, J. Y. Xu, M. Z. Zhang, L. Zhao, G. X. Duan, Y. W. Cao, R. H. Zhou and P. H. Ni, Polym. Chem., 2016, 7, 4198–4212 RSC.
- P. Sun, N. Wang, X. Jin and X. Y. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 36675–36687 CrossRef CAS PubMed.
- K. N. Zhu, G. H. Liu, J. M. Hu and S. Y. Liu, Biomacromolecules, 2017, 18, 2571–2582 CrossRef CAS PubMed.
- Y. L. Dai, C. Xu, X. L. Sun and X. Y. Chen, Chem. Soc. Rev., 2017, 46, 3830–3852 RSC.
- W. Yin, W. Ke, W. Chen, L. Xi, Q. Zhou, J. F. Mukerabigwi and Z. Ge, Biomaterials, 2019, 195, 63–74 CrossRef CAS.
- S. Z. F. Phua, C. C. Xue, W. Q. Lim, G. B. Yang, H. Z. Chen, Y. Y. Zhang, C. F. Wijaya, Z. Luo and Y. L. Zhao, Chem. Mater., 2019, 31, 3349–3358 CrossRef CAS.
- B. Yang, K. Y. Wang, D. Zhang, B. Ji, D. Y. Zhao, X. Wang, H. T. Zhang, Q. M. Kan, Z. G. He and J. Sun, RSC Adv., 2019, 9, 9260–9269 RSC.
- K. M. Camacho, S. Kumar, S. Menegatti, D. R. Vogus, A. C. Anselmo and S. Mitragotri, J. Controlled Release, 2015, 210, 198–207 CrossRef CAS PubMed.
- K. M. Camacho, S. Menegatti and S. Mitragotri, Nanomedicine, 2016, 11, 1139–1151 CrossRef CAS PubMed.
- Y. Wang, L. Zhu, L. Li, Y. Lu, L. Shen and L. W. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 35106–35113 CrossRef CAS PubMed.
- F. W. Zhang, S. Khan, R. C. Li, J. A. Smolen, S. Y. Zhang, G. Z. Zhu, L. Su, A. A. Jahnke, M. Elsabahy, X. Y. Chen and K. L. Wooley, Nanoscale, 2017, 9, 15773–15777 RSC.
- F. Li, T. Y. Li, W. Cao, L. Wang and H. P. Xu, Biomaterials, 2017, 133, 208–218 CrossRef CAS PubMed.
- N. He, Z. Chen, J. Yuan, L. Zhao, M. Chen, T. Wang and X. Li, ACS Appl. Mater. Interfaces, 2017, 9, 32534–32544 CrossRef CAS PubMed.
- W. Q. Lim, S. Z. F. Phua, H. Z. Chen and Y. L. Zhao, Chem. Commun., 2018, 54, 12762–12765 RSC.
- Z. J. Chen, Z. L. Zhang, M. H. Chen, S. Z. Xie, T. Wang and X. H. Li, J. Mater. Chem. B, 2019, 7, 1415–1426 RSC.
- M. Lei, S. J. Sha, X. Y. Wang, J. Wang, X. Du, H. Miao, H. Zhou, E. H. Bai, J. M. Shi and Y. Q. Zhu, RSC Adv., 2019, 9, 5512–5520 RSC.
- Q. L. Li, S. H. Xu, H. Zhou, X. Wang, B. A. Dong, H. Gao, J. Tang and Y. W. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 28656–28664 CrossRef CAS PubMed.
- H. K. Yang, M. Qi, L. Mo, R. M. Yang, X. D. Xu, J. F. Bao, W. J. Tang, J. T. Lin, L. M. Zhang and X. Q. Jiang, RSC Adv., 2016, 6, 114519–114531 RSC.
- H. Xiang, H. Chen, H. P. Tham, S. Z. F. Phua, J. G. Liu and Y. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 27553–27562 CrossRef CAS PubMed.
- M. R. Aji Alex, C. Nehate, S. Veeranarayanan, D. S. Kumar, R. Kulshreshtha and V. Koul, Biomaterials, 2017, 133, 94–106 CrossRef CAS PubMed.
- J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2017, 8, 97–126 RSC.
- D. Wu, Y. Li, J. Shen, Z. Tong, Q. Hu, L. Li and G. Yu, Chem. Commun., 2018, 54, 8198–8201 RSC.
- W. Zhang and C. H. Tung, Chem. Commun., 2018, 54, 8367–8370 RSC.
- M. X. Chen, S. Lin and H. S. Wang, J. Controlled Release, 2015, 213, e76 CrossRef PubMed.
- K. M. Camacho, S. Menegatti and S. Mitragotri, Nanomedicine, 2016, 11, 1139–1151 CrossRef CAS PubMed.
- R. Aluri and M. Jayakannan, Biomacromolecules, 2017, 18, 189–200 CrossRef CAS.
- X. L. Hu, S. D. Zhai, G. H. Liu, D. Xing, H. J. Liang and S. Y. Liu, Adv. Mater., 2018, 30, e1706307 CrossRef.
- Z. Xu, D. Wang, S. Xu, X. Liu, X. Zhang and H. Zhang, Chem.–Asian J., 2014, 9, 199–205 CrossRef CAS PubMed.
- Y. Li, H. Yang, J. Yao, H. Yu, X. Chen, P. Zhang and C. Xiao, Colloids Surf., B, 2018, 169, 273–279 CrossRef CAS PubMed.
- J. Li, Z. E. Hu, X. L. Yang, W. X. Wu, X. Xing, B. Gu, Y. H. Liu, N. Wang and X. Q. Yu, Biomater. Sci., 2019, 7, 3277–3286 RSC.
- X. L. Hu, J. M. Hu, J. Tian, Z. S. Ge, G. Y. Zhang, K. F. Luo and S. Y. Liu, J. Am. Chem. Soc., 2013, 135, 17617–17629 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR spectra and GPC curve. See DOI: 10.1039/c9ra07051f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Qiwen Wangb,
Jie Fanga,
Wangxing Shena,
Dan Wu*c,
Guping Tang
*a,
Qiwen Wangb,
Jie Fanga,
Wangxing Shena,
Dan Wu*c,
Guping Tang