
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermination of microcystins in water samples by deep eutectic solvent-based vortex-assisted liquid–liquid microextraction coupled with ultrahigh-performance liquid chromatography-high resolution mass spectrometry†
Yung-Chih Chen,
Yi-Ting Ao and
Wang-Hsien Ding *
*
Department of Chemistry, National Central University, Chung-Li, 320, Taiwan. E-mail: wanghsiending@gmail.com; Fax: +886-3-4227664; Tel: +866-3-4227151 ext 65905
First published on 26th November 2019
Abstract
Rapid screening of two microcystins (i.e., microcystin-YR (MC-YR) and microcystin-LR (MC-LR)) in surface water samples was performed by a simple and eco-friendly procedure using deep eutectic solvent-based vortex-assisted liquid–liquid microextraction (DES-based VALLME) combined with ultrahigh-performance liquid chromatography and electrospray ionization (+)-quadrupole time-of-flight mass spectrometry (UHPLC-ESI(+)-qTOF-MS) detection. To obtain an efficient water-miscible DES, choline chloride and phenol at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 were used as an extractant for VALLME. To optimize factors of DES-based VALLME, response surface design alongside Box–Behnken design was used. The limits of quantitation (LOQs) were 0.5 ng mL−1 and 0.4 ng mL−1 for MC-YR and MC-LR, respectively, which is sensitive enough to meet the World Health Organization (WHO) maximum guideline level for MC-LR in water of 1.0 ng mL−1. Moreover, satisfactory precision with relative standard deviations (RSD) for both intra- and inter-day analysis lower than 11%, and trueness (also known as mean extraction recovery) ranged from 85.5 to 113% based on the ICH method validation guideline.
:2 were used as an extractant for VALLME. To optimize factors of DES-based VALLME, response surface design alongside Box–Behnken design was used. The limits of quantitation (LOQs) were 0.5 ng mL−1 and 0.4 ng mL−1 for MC-YR and MC-LR, respectively, which is sensitive enough to meet the World Health Organization (WHO) maximum guideline level for MC-LR in water of 1.0 ng mL−1. Moreover, satisfactory precision with relative standard deviations (RSD) for both intra- and inter-day analysis lower than 11%, and trueness (also known as mean extraction recovery) ranged from 85.5 to 113% based on the ICH method validation guideline.
1. Introduction
Microcystins (MCs) are a group of monocyclic heptapeptides produced by a broad range of cyanobacteria species in freshwater blooms. They are tumor promoters and possess hepatotoxic properties due to their inhibition of protein phosphatases 1 and 2A, and are health hazards, sometimes even fatal ones, to humans and animals.1–4 MC-LR and MC-YR are probably the most concerning and toxic microcystins, and are also widely distributed and detected in the freshwater system worldwide.5–7 The WHO has established a provisional guideline value of 1.0 ng mL−1 for MC-LR in drinking water.8 To prevent potential adverse effects caused by long-term exposure of these toxins and the increasing concerns for public health prompted us to develop a simple and eco-friendly method for the rapid screening of MC-YR and MC-LR in surface water samples.Solid-phase extraction (SPE) has been the most common sample pretreatment method for microcystins analysis,9–11 and also reviewed by Picardo et al.12 However, SPE is time consuming (depending on the sample volume, flow rate of SPE, and manifold setup), and SPE cartridge blocking problems further discourage its use. In recent years, sample pretreatment strategies have trended toward becoming efficient and “greener” in order to eliminate the use of hazardous organic solvents and reduce time required. Methods such as liquid–liquid microextraction (LLME), which uses less volume of both extractant and aqueous sample, as well as requires less time, has become an attractive pretreatment approach for various aqueous and liquid samples.13–15 Vortex-assisted liquid–liquid microextraction (VALLME) is one of the alternative LLME techniques developed by Yiantzi et al. at 2010.16 Since its original publication, VALLME has become a highly popular LLME technique due to the minimal time required to perform it, no dispersion solvent requirement, and excellent extraction efficiency. The process of VALLME is as follows: instead of dispersing solvents, target analytes are extracted from aqueous samples by using vortex agitation, which accelerates the mass-transfer process between two immiscible phases. Using VALLME procedure to extract various persistent organic pollutants and emerging contaminants from aqueous samples has been reviewed by Ojeda and Rojas.17 However, hazardous chlorinated solvents, such as chloroform, chlorobenzene, carbon tetrachloride, etc., are often used as extractants in LLME techniques. To overcome this problem, deep eutectic solvents (DESs), a group of novel “green” solvents, have been introduced as substituting solvents in various LLME techniques, and their applications in the extraction and separation for sample preparations have been reviewed by Tang et al.,18 Li and Row,19 and Cunha and Fernandes.20 DESs also benefit from being easily prepared at room temperature, cheap, biodegradability, good solubility for organic compounds and metal ions, and even lower toxicity compared to room-temperature ionic liquids. Therefore, DESs have become attractive alternative solvents for various scientific research, and substituted conventional volatile organic solvents, especially for hazardous chlorinated solvents.
The goal of the present study was to develop a green, simple, and sustainable extraction procedure, called deep eutectic solvent-based vortex-assisted liquid–liquid microextraction (DES-based VALLME) technique, for the rapid extraction of MC-YR and MC-LR in surface water samples, and to demonstrate the feasibility of applying DES-based VALLME plus UHPLC-ESI(+)-qTOF-MS as an effective and sensitive method for microcystins determination. Structures of MC-LR and MC-YR used for method development and validation can be found in Table S1.† To minimize the number of experiments, expenses, and reagents required, optimization of the factors affecting DES-based VALLME were done via Box–Behnken Design (BBD), one of the most commonly used response surface designs, which is more efficient than the central composite design and three-level full factorial design for the quadratic model used to optimize the analytical method studies, as previously reported.21 The developed method's precision and trueness were evaluated based on the ICH guideline, and its applicability and practicality to detect microcystins in water samples was also tested.
2. Experimental
2.1. Reagents and chemicals
Highly pure standards (all greater than 98%), viz., microcystin (MC-YR) and microcystin-LR (MC-LR) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Reagent-grade chemicals, viz., phenol, choline chloride, tetrahydrofuran (THF), methanol, acetonitrile (ACN), and formic acid were also purchased from Sigma-Aldrich.2.2. Samples collection
Four surface water samples (specific conductance: 200 to 210 μS cm−1) were collected from the Shihmen Reservoir, which is Taiwan's third largest reservoir, and provides irrigation, flood control, hydroelectricity, and domestic water supply for more than three million people in northern Taiwan. The reservoir is also a recreational lake, offering shuttle boats for passengers to visit the tourist attractions. 250 mL sample was collected in duplicate. All samples were filtered through a 0.45 μm membrane filter, and adjusted pH to 2 in order to depress microbial degradation, and then stored at 4 °C. The samples were analyzed within one week.2.3. DES preparation
The DESs were simply prepared by mixing choline chloride (ChCl, as a hydrogen-bond acceptor) with phenol (as a hydrogen-bond donor) at various molar ratios (i.e., DES1 (molar ratio 1:2), DES2 (1:3), DES3 (1:4), and DES4 (2:3)). The two components were placed in a capped flask, and then constantly stirred at 50 °C until a homogeneous clear liquid was formed (around 20 min).
2.4. DES-based VALLME procedure
The procedure of DES-based VALLME was done under optimal extraction conditions as follows: water sample 5.0 mL (pH = 2) was placed in a 15 mL screw capped centrifuge tube, and DESs 0.9 mL (as an extractant) and tetrahydrofuran (THF) 1.4 mL were added into the water sample. Herein, an aprotic solvent THF, commonly acting as an emulsifier agent in DES-based LLME techniques, was used to separate DES from the aqueous solution.20,22–25 The mixture was then vigorously shaken using a vortex agitator (Vortex-Genie 2) for 50 s at maximum speed. The turbid solution was then centrifuged at 5000 rpm for 5 min to speed up the separation of the DES-rich phase and the aqueous phase. The sedimented DES phase (0.3 mL) was collected and diluted to 0.5 mL by methanol to reduce the viscosity of DES for easily autosampler injection, as suggested by Solaesa et al.26 Then, the final extract (2.0 μL) was subsequently determined by UHPLC-ESI(+)-qTOF-MS.2.5. UHPLC-ESI-qTOF-MS analysis
Chromatography was carried out on a Dionex UltiMate 3000 UHPLC system (Thermo Fisher, Waltham MA, USA). Separation was achieved by an Agilent Poroshell 120 EC-C18 column (2.7 μm, 2.1 × 100 mm). The column temperature was set at 30 °C, and the injection volume was 2.0 μL. Elution was performed at a flow rate of 0.6 mL min−1 using (A) 0.1% formic acid aqueous solution, and (B) ACN as the mobile phases. The linear gradient for phase B was as follows: 0.0 min 10% B, 1.0 min to 30% B, 4.0 min to 60% B, 4.5 min to 90% B, and then a reversion to initial conditions. In total, gradient elution time was 4.5 min, and stabilization time was 1.5 min.Identification and quantitation of target analytes were performed on a Bruker “Compact” quadrupole time-of-flight mass spectrometry (qTOF-MS, Bremen, Germany) coupled with an Apollo II ion funnel electrospray ionization (ESI) source. Positive ion mode with the scan range from m/z 800 to 1350 was applied for ESI-qTOF-MS analysis. The ESI(+)-qTOF-MS operating parameters were as follows: dry gas flow, 10 L min; capillary voltage, −4.5 kV; dry gas temperature, 220 °C; nebulizer gas pressure, 3.5 bar. The quantitation ions of MC-YR and MC-LR were listed in Table 1. A cluster of sodium formate ions (i.e., [Na(NaCOOH)n]+, n = 12 to 18) were employed to calibrate the accurate masses for high resolution qTOF-MS.
3. Results and discussion
3.1. DES-based VALLME optimization
The extraction efficiency of DES-based VALLME could be affected by several factors, such as the type and volume of DES, the volume of THF (as an emulsifier agent), and the vortex-extraction time. To enhance extraction efficiency for VALLME, the DES extractant should extract microcystins in an efficient manner, and be easily separable from water after centrifugation. Based on previous studies involving DES-based LLME techniques, the DESs formed by the combination of ChCl and phenol at different molar ratios have resulted in excellent recoveries for various organic compounds and metal ions in a variety of matrices.20,22–25 Moreover, these two components are relatively cheap and widely available, and easily preparing DESs at room temperature. Therefore, ChCl and phenol at different molar ratios were initially selected to prepare as the DES-based extractants for subsequent experiments.The molar ratio of ChCl and phenol of DES is another important factor that may influence the extraction efficiency. Furthermore, THF has been commonly employed as the emulsifier agent in DES-based LLME to separate DES from the aqueous solution.20,22–25 Therefore, four DESs with different molar ratios of ChCl and phenol combined with THF, respectively, were examined to explore the extraction efficiency under the same extraction conditions. Fig. 1 shows that no significant change in the peak areas of two microcystins was observed when the molar ratios of phenol increased from 2 to 4, but when DES4 (molar ratio 2:3) was used as the extractant, relatively lower peak areas were observed. Therefore, DES1 (molar ratio 1:2) was employed in all subsequent studies because its preparation also required fewer amount of phenol.
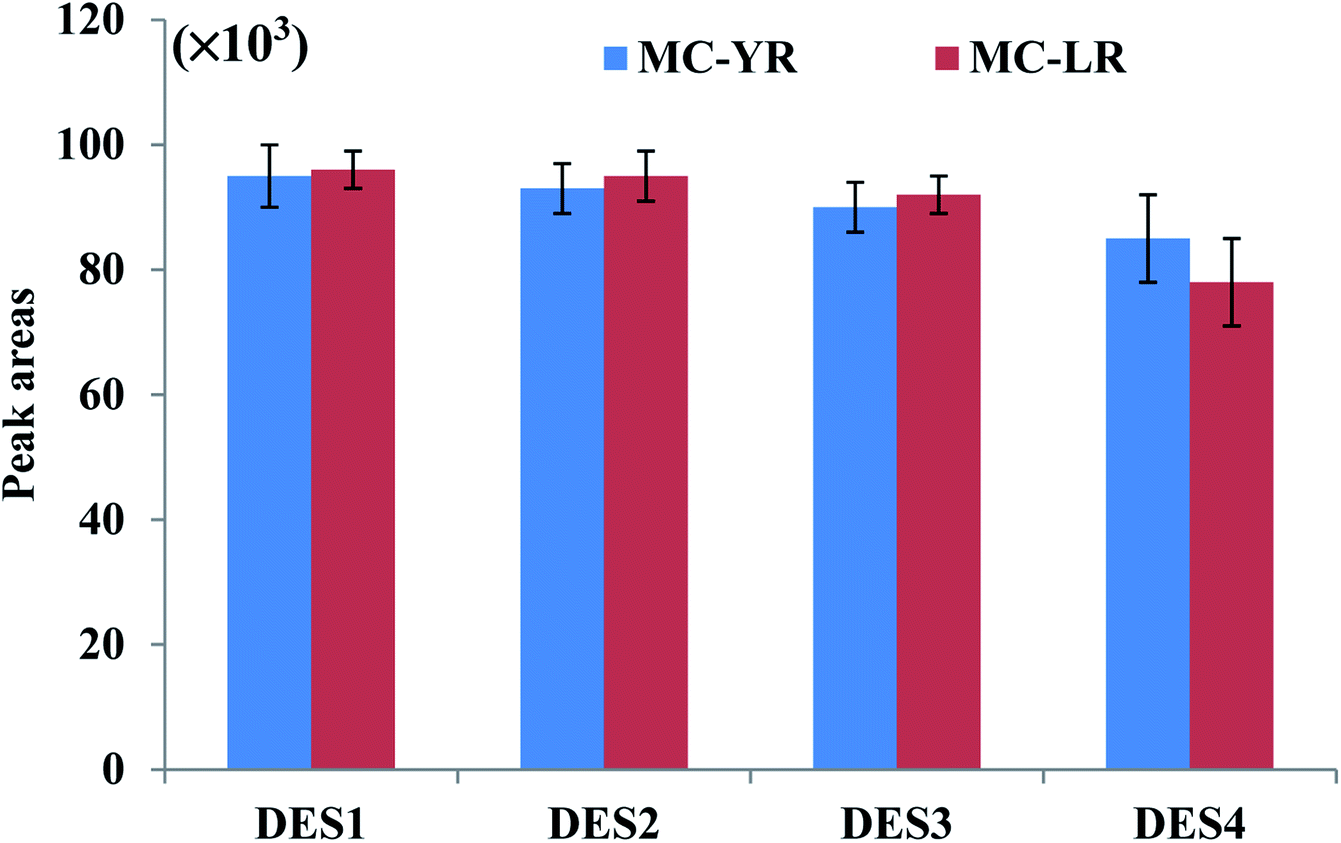 | ||
| Fig. 1 Effect of type of DESs on the extraction of MC-YR and MC-LR from water samples. Experimental conditions: 1.0 mL of DES1, DES2, DES3, or DES4 was added into water sample (5.0 mL), respectively. The solution was then mixed with 1.0 mL of THF. The mixture was vortexed for 50 s. The turbid solution was then centrifuged at 5000 rpm for 5 min. The sedimented DES phase (0.3 mL) was collected, diluted to 0.5 mL by methanol, and then subjected to UHPLC-ESI(+)-qTOF-MS analysis. | ||
Afterwards, a statistics-based Box–Behnken Design (BBD) was employed and combined with response surface methodology to evaluate the effects of each factor and to optimize the extraction efficiency of DES-based VALLME. Three major factors, considering as independent variables, were the volume of THF, the volume of DES, and the vortex-extraction time. Three levels of the factors were: the volume of THF (0.8, 1.4 and 2.0 mL), the volume of DES (0.2, 0.6 and 1.0 mL), and the vortex-extraction time (10, 50 and 90 s). Stat-Ease Design-Expert 8.0.6 software (Stat-Ease, Inc., Minneapolis, MN, USA) was performed to evaluate the design of the experiment and to analyze the data. Table 2 illustrates the experimental domain, which had seventeen experiments in random order (the central point containing five replicates) plus the corresponding experimental results (represented as total peak areas and peak areas for each analyte). ANOVA was also applied to evaluate the significance of the factors. The ANOVA results for total peak areas were summarized in Table 3, which shows that the model and the volume of DES (B) were statistically significant at the 95% confidence level. The F-value of the “Lack-of-Fit” was insignificant which confirmed that the model could perfectly fit the responses variables with good prediction at the 95% confidence level. Moreover, good quality of the fit of the second order polynomial model (quadratic model) was explained by the coefficient of determination (R2 = 0.9510 and adjusted-R2 = 0.9081), which indicated that the response was in reasonable agreement with the predicted-R2 (0.8879). Table 3 also lists the estimated coefficients for the second order polynomial equation of each independent factor to represent the total peak areas adequately for each response. The ANOVA results of peak areas for individual MC-LR and MC-YR were summarized in Tables S2 and S3,† respectively, which shows that the model and the volume of DES were statistically significant at the 95% confidence level. The values of predicted-R2 show in good agreement with the predicted-R2 for individual MC-LR and MC-YR. Tables S2 and S3† also display the estimated coefficients for the second order polynomial equation of each independent factor to represent the peak areas of individual MC-LR and MC-YR adequately for each response. The following values: 1.4 mL of the volume of THF, 0.9 mL of the volume of DES, and 50 s of the vortex time, were selected to achieve the optimal experimental results after the desirability function predication (the value of desirability is 0.949).22
| Run | A-THF volume (mL) | B-DES volume (mL) | C-vortex time (s) | Total peak areas | Peak areas (MC-LR) | Peak areas (MC-YR) |
|---|---|---|---|---|---|---|
| 1 | 2 | 0.2 | 50 | 98632 |
48281 |
50351 |
| 2 | 2 | 0.6 | 90 | 169632 |
84123 |
85509 |
| 3 | 1.4 | 0.6 | 50 | 242138 |
120024 |
122114 |
| 4 | 0.8 | 0.2 | 50 | 140046 |
69843 |
70203 |
| 5 | 2 | 0.6 | 10 | 188182 |
94395 |
93787 |
| 6 | 0.8 | 1 | 50 | 197811 |
98703 |
99108 |
| 7 | 1.4 | 0.2 | 90 | 139879 |
69758 |
70121 |
| 8 | 2 | 1 | 50 | 206314 |
104086 |
102228 |
| 9 | 1.4 | 0.2 | 10 | 115090 |
57312 |
57778 |
| 10 | 1.4 | 0.6 | 50 | 229263 |
114730 |
114533 |
| 11 | 1.4 | 0.6 | 50 | 226147 |
113531 |
112616 |
| 12 | 0.8 | 0.6 | 10 | 170223 |
85022 |
85201 |
| 13 | 1.4 | 1 | 90 | 211015 |
105517 |
105498 |
| 14 | 1.4 | 1 | 10 | 192249 |
95943 |
96306 |
| 15 | 0.8 | 0.6 | 90 | 184273 |
91763 |
92510 |
| 16 | 1.4 | 0.6 | 50 | 213479 |
106855 |
106624 |
| 17 | 1.4 | 0.6 | 50 | 205791 |
102221 |
103570 |
| Source | Sum of squares | df | Mean Square | F Value | p-value Prob > F | Coefficientb (estimated) |
|---|---|---|---|---|---|---|
| a Significant.b The estimated coefficients for the second order polynomial equation. | ||||||
| Model | 2.51 × 1010 | 9 | 2.79 × 109 | 15.104 | 0.0008a | |
| Intercept | 2.23 × 105 | |||||
| A-THF volume | 1.09 × 108 | 1 | 1.09 × 108 | 0.594 | 0.4663 | −3699 |
| B-DES volume | 1.23 × 1010 | 1 | 1.23 × 1010 | 66.713 | < 0.0001a | 39218 |
| C-vortex time | 1.91 × 108 | 1 | 1.91 × 108 | 1.034 | 0.3431 | 4882 |
| AB | 6.23 × 108 | 1 | 6.23 × 108 | 3.377 | 0.1087 | 12479 |
| AC | 2.66 × 108 | 1 | 2.66 × 108 | 1.440 | 0.2691 | −8150 |
| BC | 9068011 |
1 | 9068011 |
0.049 | 0.8309 | −1506 |
| A2 | 2.54 × 109 | 1 | 2.54 × 109 | 13.783 | 0.0075 | −24572 |
| B2 | 6.11 × 109 | 1 | 6.11 × 109 | 33.123 | 0.0007 | −38091 |
| C2 | 1.81 × 109 | 1 | 1.81 × 109 | 9.795 | 0.0166 | −20714 |
| Residual | 1.29 × 109 | 7 | 1.84 × 108 | |||
| Lack of fit | 4.89 × 108 | 3 | 1.63 × 108 | 0.814 | 0.5491 | |
| R2 | 0.9510 | |||||
| Adjusted R2 | 0.9081 | |||||
| Predicted R2 | 0.8879 | |||||
In Fig. 2, three-dimensional response surface plots display the interaction between two independent variables calculated by the BBD in order to examine the interactive effects of each pair of factors on the extraction efficiency. Accordingly, Fig. 2(a) shows the response surface obtaining as a function of the volume of THF and the volume of DES with a fixed vortex time at 50 s, Fig. 2(b) displays the response surface developed for the volume of THF and the vortex time, whilst fixed the volume of DES at 0.9 mL, and Fig. 2(c) shows the response surface obtained for the volume of DES and the vortex time with a fixed volume of THF at 1.4 mL. As can be seen, medium volume of THF (1.4 mL) and vortex time (50 s), as well as relatively higher volume of DES (0.9 mL) provided the highest total peak areas (represented as extraction efficiency) for the two target microcystins. The maximal spiked recovery, as estimated under the optimized conditions, ranged from 85.5 to 106% with an average of 96.1 ± 8.8% (as illustrated in part of Table 4: mean spiked recovery of intra-day at three spiked concentrations). Three-dimensional response surface plots for individual MC-LR and MC-YR display the similar response trends in all studied conditions, as shown in Fig. S1 and S2,† respectively. Collectively, these results indicate that the optimal conditions for extracting the MC-YR and MC-LR from water sample, using the DES-based VALLME, are as follows: water sample 5.0 mL mixed with DES (0.9 mL, as an extractant) and THF (1.4 mL, as an emulsifier agent). The mixture was vortexed for 50 s, and the turbid solution was then centrifuged at 5000 rpm for 5 min. The sedimented DES phase (0.3 mL) was collected, diluted to 0.5 mL by methanol, and then subsequently applied for UHPLC-ESI(+)-qTOF-MS analysis.
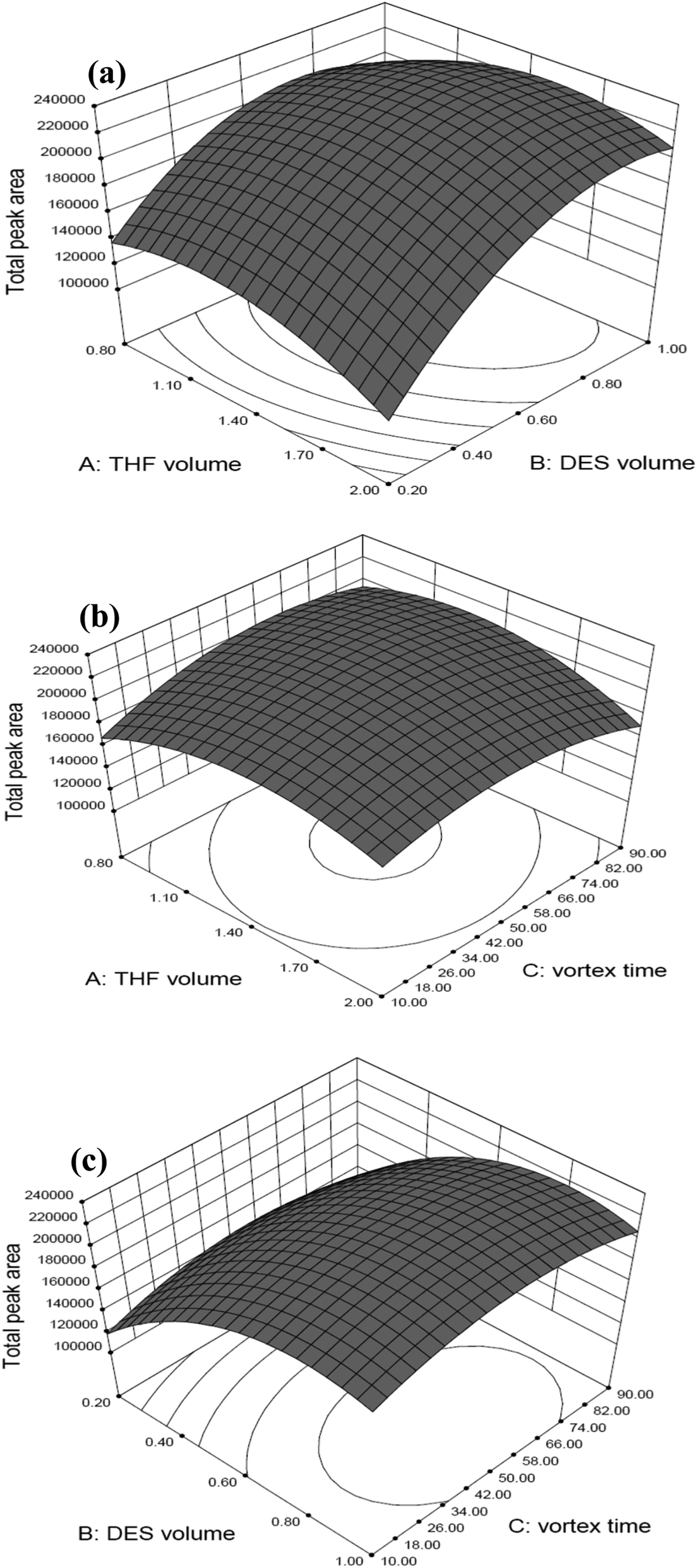 | ||
| Fig. 2 3D response surface plots for the total peak area of the target analytes estimated from the BBD on each pair of independent variables: (a) volume of THF vs. volume of DES; (b) volume of THF vs. vortex-time; (c) volume of DES vs. vortex-time. | ||
| Analytes | Intra-day | Inter-day | ||||
|---|---|---|---|---|---|---|
| 1.0 ng mL−1 | 10 ng mL−1 | 50 ng mL−1 | 1.0 ng mL−1 | 10 ng mL−1 | 50 ng mL−1 | |
| a Average spiked recovery (trueness, %, n = 5).b Relative standard deviation (RSD) of spiked recovery (precision, %, n = 5). | ||||||
| MC-YR | 106a (4)b | 96.6a (8)b | 105a (2)b | 113a (2)b | 111a (7)b | 109a (2)b |
| MC-LR | 85.5 (7) | 86.3 (6) | 97.0 (3) | 103 (11) | 97.3 (5) | 99.2 (2) |
3.2. Method validation and applications
The developed method was validated through evaluating its linearity, selectivity, limits of detection (LODs), LOQ, precision and trueness, which based on ICH Harmonised tripartite Guideline.27To investigate the influence of DES in final extract and the feasibility of quantitation by ESI(+)-qTOF-MS, we firstly compared the peak areas of analytes obtained from final extract solution (i.e., DES: methanol (3:2, v/v); n = 3) to those of obtained from standards solution (i.e., methanol). Table S4† shows that the peak areas for both target analytes in the final extract (containing DES) was enhanced by more than 11%. The differences of the peak areas from these two kinds of determinations were then evaluated by a t-test (with confidence intervals at 95%). After calculation, the t-calculated value was 11.7, which was higher than the t-tabulated value (t(95%, df = 5) = 2.571), indicating that the peak areas obtained from these two solutions were significantly different. Therefore, to obtain satisfactory quantitative results, the calibration standards were prepared in final extract solutions, and five-point “matrix-matched” calibration curves were employed to quantitate the target analytes. Each curve covered the range from 1 to 100 ng mL−1 (i.e., 1, 2, 5, 20, and 100 ng mL−1). As shown in Table 1, the coefficients of determination (r2) were 0.9998 to 0.9999 for MC-YR and MC-LR, respectively, demonstrating the excellent linearity. Furthermore, by analyzing through the Mandel's fitting test, the linear regression was proven to be a better fit with the experimental data than that of quadratic regression, since the calculated F-values were lower than that of the tabulated limit at the 95% confidence level (Table 1).
For selectivity, the accurate masses of the protonated molecules ([M + H]+, as listed in Table 1) of the MC-LR and MC-YR were used for both quantitation and confirmation. Moreover, the traces of extracted ion chromatogram (EIC) with narrow mass window (i.e., ±10 mDa mass interval) were applied to increase the selectivity and sensitivity for qTOF-MS high resolution measurements. Fig. S3† depicts that no interfering peaks were observed at or around the retention times of the target analytes, which exhibits excellent selectivity by easily identifying target analytes via their retention times.
The LODs and LOQs were defined at a signal to noise (S/N) ratio of 3 and 10, respectively. The LODs, estimated through DES-based VALLME plus UHPLC-ESI(+)-qTOF-MS procedures of the spiked reservoir water samples, have values 0.16 and 0.14 ng mL−1 for MC-YR and MC-LR, respectively, and the LOQs were 0.5 and 0.4 ng mL−1.
Precision was evaluated through intra-day and inter-day analyses expressed as the relative standard deviation in percentage (%RSDs) at three concentration levels. Spiked reservoir water samples (n = 5) were analyzed from the same day to determine repeatability, also known as intra-day precision. Inter-day precision was obtained by analyzing through five successive days (n = 5). Trueness can be further obtained by calculating the mean recovery of these spiked water samples. In Table 4, for low-, medium- and high-level spiked samples, their intra- and inter-day precision and trueness have values from 2 to 11% and 85.5 to 113%, respectively. Robustness was evaluated through the daily fresh prepared DES1 extractant during the inter-day analyses. The excellent average spiked recovery, as estimated under the optimized conditions, ranged from 99.2 to 113% with an average of 105 ± 6% (as illustrated in part of Table 4: mean spiked recovery of inter-day at three spiked concentrations). Such satisfactory precision, trueness, and robustness demonstrate that the DES-based VALLME coupled with UHPLC-ESI(+)-qTOF-MS can achieve satisfactory selectivity and sensitivity, as well as high reproducibility for the quantitation of microcystins in tested surface water samples. However, no MC-YR and MC-LR were detected in the surface water samples collected from the Shihmen Reservoir (as shown in Table S5†), perhaps due to the samples' dilution caused values to drop below their LOQs. The average recovery of the spiked extractions of the two target microcystins ranged from 94.0 to 109% with a RSD of less than 12.2%.
3.3. “Greenness” and method comparisons
The greenness of our developed method was assessed through an “analytical Eco-scale” as proposed by Gałuszka et al.28 This scale is based on the assigning “penalty points” for each parameter in the analytical protocol, if which departs from the twelve principles of green chemistry. As shown in Table S6,† after assessment, our developed method can be classified as “an excellent green analysis” since the score was greater than 75 on the “analytical Eco-scale”. Therefore, the developed method can be used for reliable monitoring studies of the microcystins in analytical laboratories with a minimal detrimental impact on human health and the environment.Table 5 lists the comparison among our DES-based VALLME method and previously published techniques for sample preparation. Compared with DES-based VALLME, various SPE procedures required more than 10 min plus elution time for each sample (based on the sample volume, flow rate of SPE, and manifold setup).9–11 In addition, for the microextraction methods, such as ionic liquid-based dispersive liquid–liquid microextraction required less than 6 min,29 and magnetic solid-phase extraction needed at least 50 min,30 but both of these techniques required more than four days to synthesize ionic liquids as extractant and magnetic γ-cyclodextrin polymer as adsorbent, respectively. Although, the results of LOQs obtained by our developed method were higher than those obtained by SPE procedures and microextraction methods.9–11,29,30 our values meet the WHO recommended health-based level of 1.0 ng mL−1 for MC-LR in drinking water.8 Moreover, the outstanding features of our procedure was the use of a green extractant, less sample volume, and the fully exploitation of the benefits of VALLME technique (i.e., speed, simplicity, and efficiency). Furthermore, the recoveries and the precisions of the spiked samples of our results are comparable to those of other published methods.
| Extraction method | Sample volume | Time required | Detection method | Spiked recovery (%) | Precision (%RSD) | LOQ/MQL (ng mL−1) | Ref. |
|---|---|---|---|---|---|---|---|
| a DLLME: dispersive liquid–liquid microextraction; MSPE: magnetic solid-phase extraction. | |||||||
| DES-VALLME | 5 mL | <6 min | UHPLC-qTOF-MS | 85.5–113.5% | <11% | LOQ: MC-LR: 0.4, MC-YR: 0.5 | This study |
| SPE | 100 mL | 10 min + elution time | HPLC-orbitrap-MS | 97.1–100.9% | ≤5.0% | LOQ: 0.0006 | 9 |
| Dual-SPE | 400 mL | 50 min + elution time | HPLC-ESI-MS/MS | 85–95% | <15.5% | LOD: 0.004 | 10 |
| SPE | 1000 mL | 100 min + elution time | HPLC-ESI-MS/MS | MC-LR: 94% | <3.9% | MDL: MC-LR: 0.0004 | 11 |
| MC-YR: 93% | MC-YR: 0.0008 | ||||||
| Ionic liquid-based DLLME | 10 mL | < 6 min | HPLC-UV | 45–109.7% | <10.9% | HPLC-UV: 0.7 | 29 |
| HPLC-ESI-MS | HPLC-MS: < 0.005 | ||||||
| MSPE | 20 mL | < 50 min | HPLC-MS/MS | 85–100.3% | <6.2% | LOQ: 0.002–0.005 | 30 |
4. Conclusions
In this study, a developed method that combined DES-based VALLME with UHPLC-ESI(+)-qTOF-MS was applied to determine MC-YR and MC-LR in surface water samples. The DES-based VALLME was developed and fully validated, as shown by satisfactory trueness and precision. DES-based VALLME is a simple and straightforward technique, which needs only small amounts of DES for each extraction. This implied that our developed method is a low-cost and eco-friendly one with the elimination of the requirement of needing or generating hazardous substances, and could achieve excellent ranking on the analytical Eco-Scale. Many advantages of DES-based VALLME could greatly aid future routine analysis and monitoring programs on the occurrence of microcystins in our aquatic environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a grant from the Ministry of Science and Technology of Taiwan under contract no. MOST 108-2113-M-008-002. We thank the Instrumental Center of National Central University for instrumentation support. We also thank Mr James Ding, who checked over the English and grammar usage within this manuscript.References
- W. W. Carmichael, J. Appl. Bacteriol., 1992, 72, 445–459 CrossRef CAS PubMed.
- G. A. Codd, Ecol. Eng., 2000, 16, 51–60 CrossRef.
- D. R. de Figueiredo, U. M. Azeiteiro, S. M. Esteves, F. J. Goncalves and M. J. Pereira, Ecotoxicol. Environ. Saf., 2004, 59, 151–163 CrossRef CAS PubMed.
- S. Merel, D. Walker, R. Chicana, S. Snyder, E. Baurès and O. Thomas, Environ. Int., 2013, 59, 303–327 CrossRef CAS PubMed.
- E. C. Aguete, A. Gago-Martínez, J. M. Leão, J. A. Rodríguez-Vázquez, C. Menàrd and J. F. Lawrence, Talanta, 2003, 59, 697–705 CrossRef CAS.
- P. Babica, J. Kohoutek, L. Bláha, O. Adamovský and B. Maršálek, Anal. Bioanal. Chem., 2006, 385, 1545–1551 CrossRef CAS PubMed.
- E. P. Preece, F. J. Hardy, B. C. Moore and M. Bryan, Harmful Algae, 2017, 61, 31–45 CrossRef CAS.
- WHO, Guidelines for Drinking Water Quality, World Health Organisation, 2nd edn, Geneva, 1998, pp. 95–110 Search PubMed.
- D. S. W. Palagama, R. E. West III and D. Isailovic, Anal. Methods, 2017, 9, 2021–2030 RSC.
- S. K. Zervou, C. Christophoridis, T. Kaloudis, T. M. Triantis and A. Hiskia, J. Hazard. Mater., 2017, 323, 56–66 CrossRef CAS PubMed.
- S. P. Haddad, J. M. Bobbitt, R. B. Taylor, L. M. Lovin, J. L. Conkle, C. K. Chambliss and B. W. Brooks, J. Chromatogr. A, 2019, 1599, 66–74 CrossRef CAS PubMed.
- M. Picardo, D. Filatova, O. Nuñez and M. Farré, Trends Anal. Chem., 2019, 112, 75–86 CrossRef CAS.
- M. Rezaee, Y. Yamini and M. Faraji, J. Chromatogr. A, 2010, 1217, 2342–2357 CrossRef CAS PubMed.
- M. I. Leong, M. R. Fuh and S. D. Huang, J. Chromatogr. A, 2014, 1335, 2–14 CrossRef CAS PubMed.
- A. Spietelun, Ł. Marcinkowski, M. de la Guardia and J. Namieśnik, Talanta, 2014, 119, 34–45 CrossRef CAS PubMed.
- E. Yiantzi, E. Psillakis, K. Tyrovola and N. Kalogerakis, Talanta, 2010, 80, 2057–2062 CrossRef CAS PubMed.
- C. B. Ojeda and F. S. Rojas, Chromatographia, 2014, 77, 745–754 CrossRef.
- B. K. Tang, H. Zhang and K. H. Row, J. Sep. Sci., 2015, 38, 1053–1064 CrossRef CAS PubMed.
- X. X. Li and K. H. Row, J. Sep. Sci., 2016, 39, 3505–3520 CrossRef CAS PubMed.
- S. C. Cunha and J. O. Fernandes, Trends Anal. Chem., 2018, 105, 225–239 CrossRef CAS.
- S. L. C. Ferreira, R. E. Bruns, H. S. Ferreira, G. D. Matos, J. M. David, G. C. Brandão, E. G. P. da Silva, L. A. Portugal, P. S. dos Reis, A. S. Souza and W. N. L. dos Santos, Anal. Chim. Acta, 2007, 597, 179–186 CrossRef CAS PubMed.
- T. Khezeli, A. Daneshfar and R. Sahraei, J. Chromatogr. A, 2015, 1425, 25–33 CrossRef CAS PubMed.
- N. Lamei, M. Ezoddin and K. Abdi, Talanta, 2017, 165, 176–181 CrossRef CAS PubMed.
- L. L. Liu and T. Zhu, Anal. Methods, 2017, 9, 4747–4753 RSC.
- F. Aydin, E. Yilmaz and M. Soylak, Food Chem., 2018, 243, 442–447 CrossRef CAS PubMed.
- A. G. Solaesa, J. O. Fernandes, M. T. Sanz, O. Benito-Roman and S. C. Cunha, Talanta, 2019, 195, 251–257 CrossRef CAS PubMed.
- Harmonised tripartite guideline Q2 (R1), ICH, Geneva, 2005, https://database.ich.org/sites/default/files/Q2_R1__Guideline.pdf Search PubMed.
- A. Gałuszka, Z. M. Migaszewski, P. Konieczka and J. Namieśnik, Trends Anal. Chem., 2012, 37, 61–72 CrossRef.
- H. L. Yu, K. D. Clark and J. L. Anderson, J. Chromatogr. A, 2015, 1406, 10–18 CrossRef CAS PubMed.
- C. H. Huang, Y. J. Wang, Q. Huang, Y. He and L. Zhang, Anal. Chim. Acta, 2019, 1054, 38–46 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07544e |
| This journal is © The Royal Society of Chemistry 2019 |