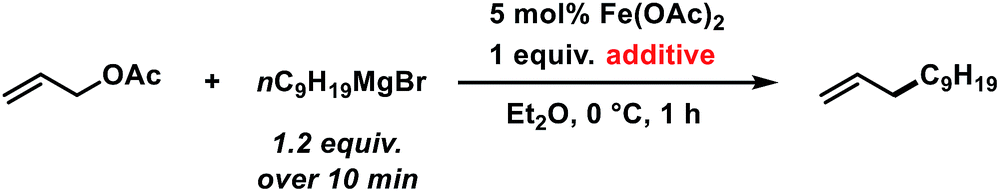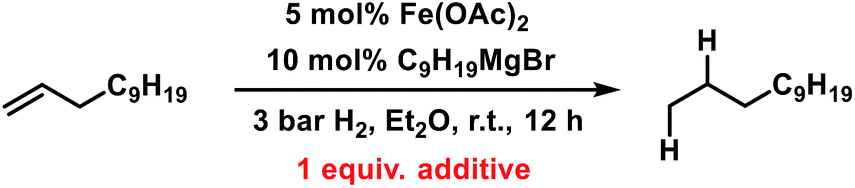DOI:
10.1039/C9RA07604B
(Paper)
RSC Adv., 2019,
9, 31217-31223
Iron-catalysed allylation–hydrogenation sequences as masked alkyl–alkyl cross-couplings†
Received
22nd August 2019
, Accepted 25th September 2019
First published on 2nd October 2019
Abstract
An iron-catalysed allylation of organomagnesium reagents (alkyl, aryl) with simple allyl acetates proceeds under mild conditions (Fe(OAc)2 or Fe(acac)2, Et2O, r.t.) to furnish various alkene and styrene derivatives. Mechanistic studies indicate the operation of a homotopic catalyst. The sequential combination of such iron-catalysed allylation with an iron-catalysed hydrogenation results in overall C(sp3)–C(sp3)-bond formation that constitutes an attractive alternative to challenging direct cross-coupling protocols with alkyl halides.
Introduction
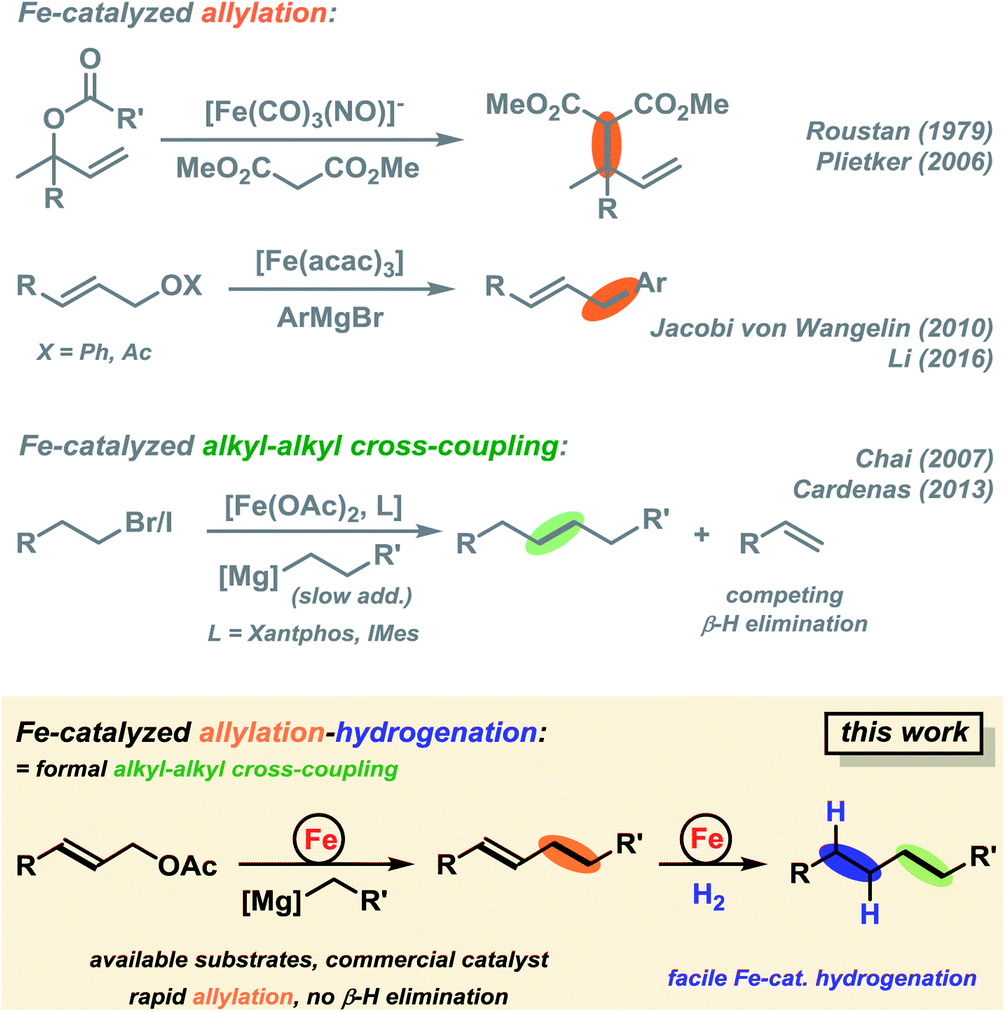
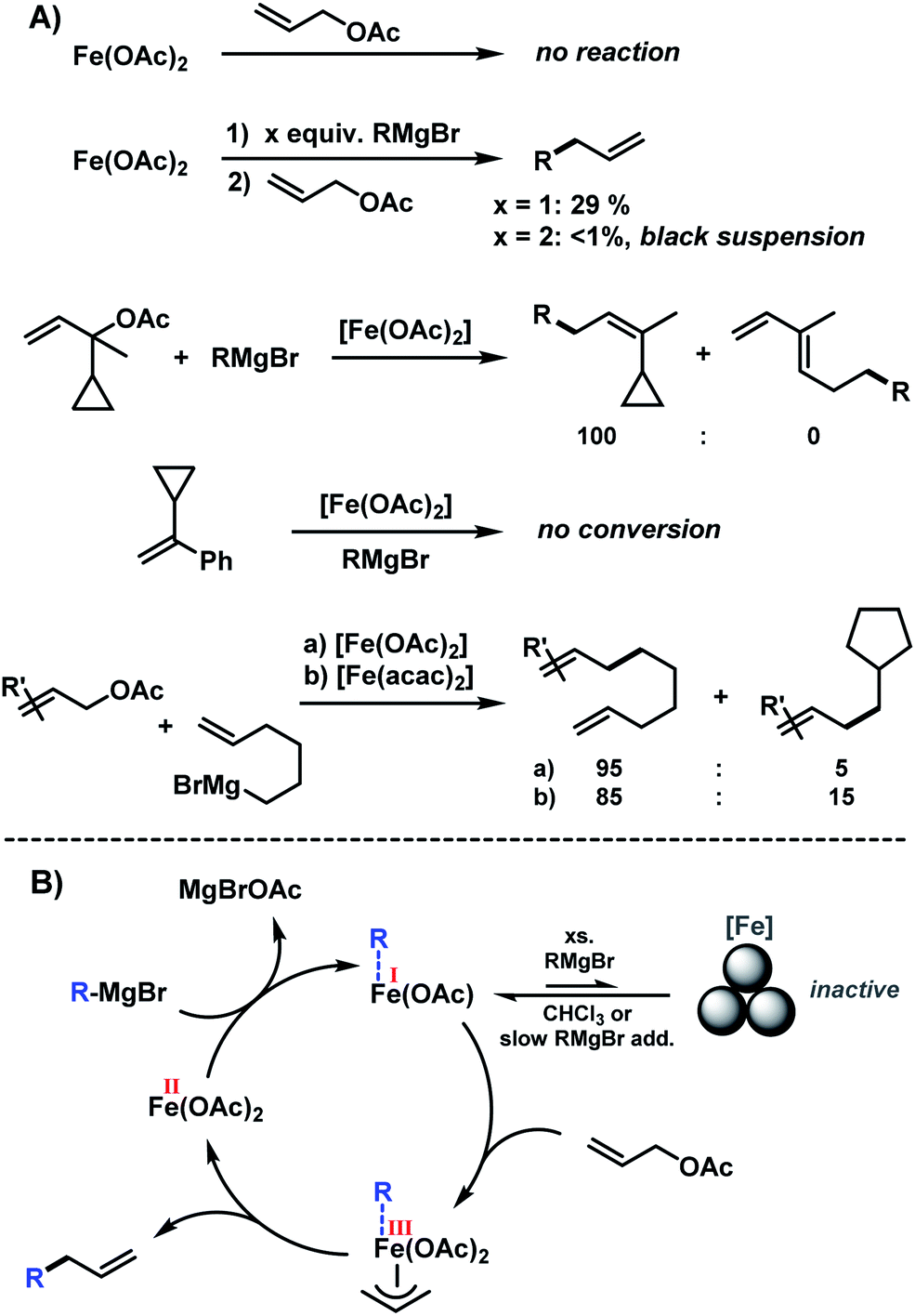
The development of transition metal-catalysed cross-coupling reactions has propelled the art of C–C bond formation like no other new methodology in the past decades.1,2 Among them, Pd and Ni catalysts have clearly dominated the field by virtue of their high versatility and chemoselectivity.3 However, noble and toxic metal catalysts (e.g. Pd, Rh, Ir, Ni and Co) have high costs and/or exhibit significant levels of toxicity which limit their general applicability under modern sustainability criteria. Iron-catalysed cross-coupling reactions have recently been developed to great maturity and now constitute a powerful alternative to the established noble metal systems.4–10 Most protocols utilize organic halides as electrophiles (mostly I, Br); only very few reactions involve activated ester derivatives (triflates, tosylates, phosphonates).11–14 Iron-catalysed cross-couplings reactions at alkenyl acetates were only recently reported.15–17 Allyl alcohols constitute one of the most easily accessible classes of electrophiles by numerous substitution or reduction methods from abundant starting materials (allyl halides, α,β-unsaturated carbonyls). However, there is no concise report of iron-catalysed reactions of simple allyl alcohol derivatives with organo-metallic reagents. A handful of iron-catalysed allylations have been reported.18–20 The Hieber-type salt Na[Fe(CO)3(NO)] (which is iso-electronic to Pd0) was especially competent in the catalytic allylation of malonates (Scheme 1, top).21 Xu and Zhou later reported similar reactions with the stable tetrabutylammonium salt in a CO atmosphere.22 Plietker et al. performed similar reactions in the presence of phosphine ligands that prevented the formation of inactive catalyst derivatives.18 Substitutions of allyl carbonates with N-, O-, S-nucleophiles and stereoselective reactions were reported under such conditions.23–26 Arylations of selected allyl alcohol derivatives with aryl-Grignard reagents have been reported by Li and coworkers and us.15,27 We envisioned the development of an Fe-catalysed cross-coupling between alkylmagnesium reagents and diverse allyl acetates that benefits from the intrinsic properties of allyl acetates as activated C-electrophiles and the utility of the pendant alkene moiety for further manipulation. While being a formal sp3-electrophile, allyl-X substrates exhibit strikingly different reactivity patterns than alkyl-electrophiles due to the vicinal alkenyl moiety, the absence of β-hydrogen atoms, and the ability to engage in η3-coordination to transition metals. In comparison with allyl-X electrophiles, alkyl-X electrophiles exhibit a low propensity to undergo oxidative addition to transition metal complexes, engage in rapid side reactions (β-H elimination, rearrangement), and undergo slow reductive elimination. Consequently, there are very few literature reports on iron-catalysed alkyl–alkyl cross-couplings which exhibit only moderate yields, limited substrate scope (few examples, alkyl-Br/I), and require special conditions (slow addition over 7 h) and/or expensive ligands (Xantphos, IMes; Scheme 1, center).28–30 We surmised that the combination of an effective allylation reaction with a subsequent hydrogenation reaction would constitute an attractive alternative to the challenging alkyl–alkyl cross-coupling reactions (Scheme 1, bottom).15 Such method utilizes the wide availability and easy preparation of alkyl-Grignard reagents. The success of the cross-coupling with substituted allyl acetates relies on the strict control of chemoselectivity as the nucleophilic Grignard reagent may readily undergo direct attack at the carboxyl function under thermodynamic control. Iron-catalysed hydrogenations have been reported for a variety of olefins.31 There are a handful of powerful homogeneous iron catalysts based on pincer-type ligands32,33 and heterogeneous catalysts derived from the reduction of iron salts with organometallic or hydride reagents.34–37 With regard to the latter catalyst class, we surmised that the iron catalyst that formed under the conditions of the cross-coupling reaction with the organomagnesium halide might also be competent in the subsequent alkene hydrogenation (Scheme 1, bottom right).15
Results and discussion
Initial optimizations
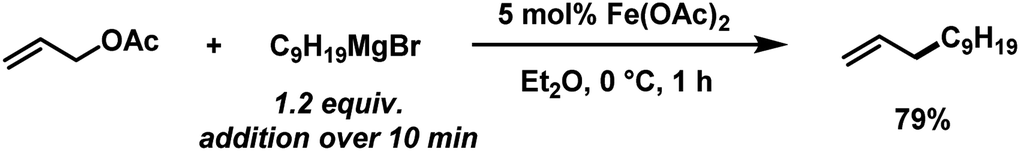
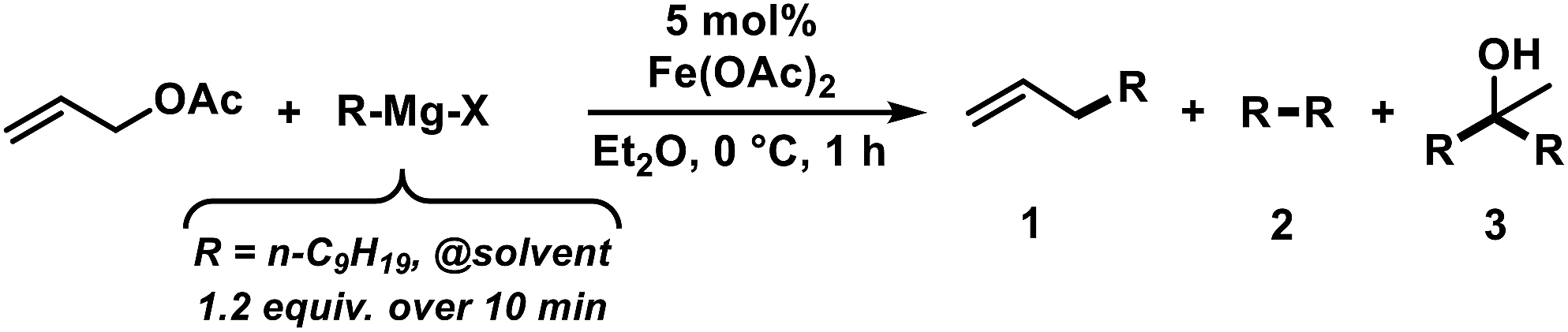
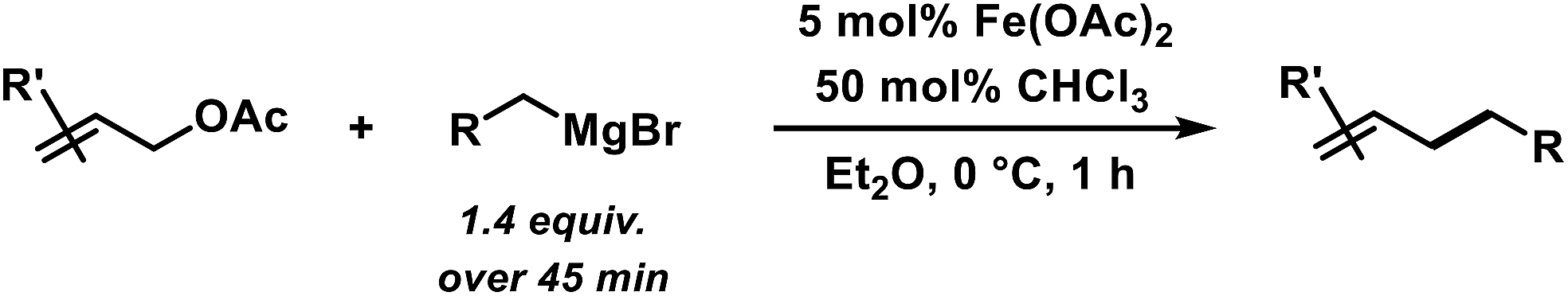
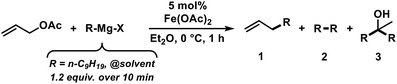
The envisaged sequence of an iron-catalysed allylation of organomagnesium halides and an iron-catalysed hydrogenation required the development of a robust allylation reaction with alkylmagnesium halides. From a rapid survey of various catalyst precursors, additives, solvents, and conditions, reactions between allyl acetate and a very low excess of the n-alkylmagnesium bromide (1.2 equiv., addition over 10 min) with the commercial pre-catalyst iron(II) acetate (5 mol% Fe(OAc)2) in diethylether were identified as being most effective (Scheme 2).38 Importantly, Fe(OAc)2 effectively inhibited the competing formal β-H elimination of the Grignard reagent.39 Other solvents like THF, toluene, N-methyl-2-pyrrolidinone (NMP), and 1,2-dimethoxyethane (DME) afforded much lower yields. Toluene/Et2O mixtures and ethyl acetate gave similar results. The addition of ligands (amines, N-heterocyclic carbenes) showed no significant effect on the reaction outcome, whereas phosphines led to complete catalyst inhibition.38 The major side reactions observed were the formation of homocoupling products from the Grignard reagent29 and the nucleophilic attack at the carbonyl group. The composition of the Grignard reagent (RMgX, X = Cl, Br, I; RMgX vs. R2Mg) and the choice of solvent exerted a strong influence on the reaction selectivities, most likely as a direct consequence of the Schlenk equilibrium (Table 1).40 The alkyl-magnesium chloride and the dialkylmagnesium in Et2O predominantly afforded the carboxylate substitution product from the uncatalyzed background reaction (entries 1 and 8). The alkyl-magnesium bromide (in Et2O) and its LiCl-adduct (in THF) gave highest conversions (entries 4–6).
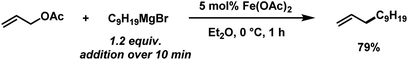 |
| | Scheme 2 Optimized conditions for iron-catalysed alkylation of allyl acetate. | |
Table 1 Variation of Grignard reagentsa
|
|
| Entry |
X |
Solvent (RMgX) |
1 [%] |
2 [%] |
3 [%] |
|
Yields were determined by quantitative GC-FID vs. internal n-pentadecane.
MgBr2 was filtered off prior to Grignard reagent addition.
|
| 1 |
Cl |
Et2O |
0 |
<1 |
52 |
| 2 |
Cl |
THF |
37 |
12 |
11 |
| 3 |
Cl·LiCl |
THF |
54 |
5 |
2 |
| 4 |
Br |
Et2O |
76 |
11 |
<1 |
| 5 |
Br |
THF |
69 |
6 |
2 |
| 6 |
Br·LiCl |
THF |
77 |
5 |
2 |
| 7 |
I |
Et2O |
32 |
35 |
0 |
| 8b |
n-C9H19 |
Dioxane |
1 |
<1 |
48 |
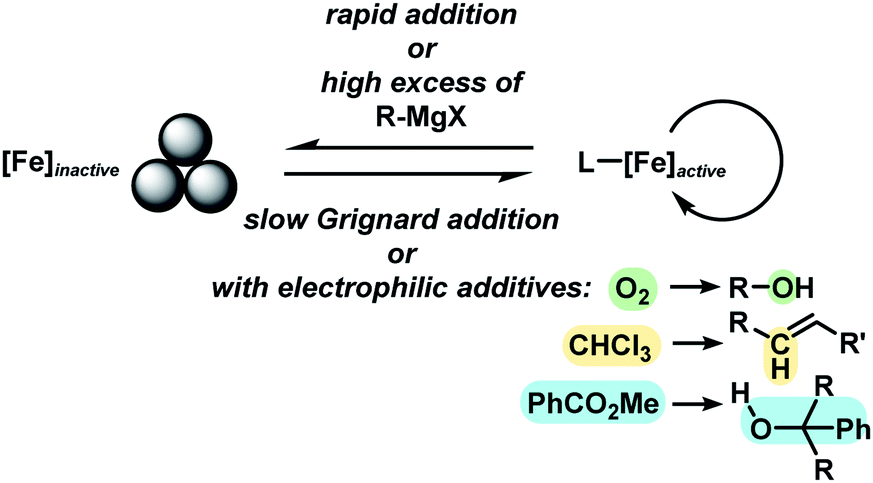
With substituted allyl acetates bearing alkyl groups, the iron-catalysed allylation was slowed and the formation of the tertiary alcohol was observed as main product. High cross-coupling yields were re-established by slow addition of the Grignard reagent (over 1–6 h). An alternative procedure involved addition of 50 mol% chloroform to the reaction which allowed Grignard addition over only 45 min. A similar protocol was reported but the role of chloroform remained unclear.12 We speculate that chloroform buffers high concentrations of Grignard reagent and thereby prevents over-reduction of the catalyst to naked Fe(0) species which would rapidly aggregate to inactive particles.41,42 A similar effect should be observed with electrophilic additives that react with the Grignard reagent in slower rates than the desired cross-coupling but sufficiently rapid to prohibit catalyst reduction. Consistent with this hypothesis, a brief evaluation of electrophiles revealed beneficial effects of the presence of esters, organochlorides, and air under the reaction conditions (Scheme 3; see also Scheme 5B).38
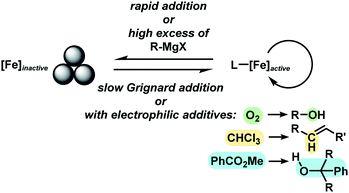 |
| | Scheme 3 Catalyst activity and effect of reaction additives. | |
Substrate scope
We then explored the substrate scope of the iron-catalysed allylation of alkylmagnesium bromides under the optimized conditions (condition A, see Table 2). The reaction proceeds with very high regiocontrol most likely through a π-allyliron intermediate as alkylation selectively occurred at the less hindered allyl termini. This is exemplified by the identical product (and yield) that was obtained from prenyl acetate and 2-methylbut-3-en-2-yl acetate, respectively (entries 4 and 5). Bulky allyl acetate derivatives required slow Grignard addition and higher catalyst loading to give moderate yields. Primary, secondary, and tertiary allyl acetates underwent alkylation under the same conditions. The (E)-alkene isomers were formed in all cases with E/Z stereoselectivities of >50/1. Halide substituents were not tolerated in the 2-allyl position (entries 7 and 8). A screening of functional additives documented moderate compatibility with nitriles and good tolerance of esters and amines (Table 3). Alcohols required the addition of an extra equivalent Grignard reagent.
Table 2 Iron-catalysed alkylations of allyl acetates (condition A)
Table 3 External functional group test
|
|
| Entry |
Additive |
Additive conversiona [%] |
Yielda [%] |
|
Yields were determined by quantitative GC-FID vs. internal n-pentadecane.
2.2 equiv. n-C9H19MgBr.
|
| 1 |
— |
— |
77 |
| 2 |
PhCN |
0 |
21 |
| 3 |
n-C3H7CN |
9 |
49 |
| 4 |
PhNO2 |
90 |
<1 |
| 5 |
EtNO2 |
87 |
8 |
| 6 |
PhNH2 |
77 |
9 |
| 7 |
n-C6H13NH2 |
0 |
80 |
| 8 |
PhCHO |
100 |
16 |
| 9 |
PhCO2Me |
0 |
82 |
| 10 |
n-C3H7CO2Me |
4 |
72 |
| 11 |
PhOH |
0 |
75b |
| 12 |
n-C5H11OH |
6 |
79b |
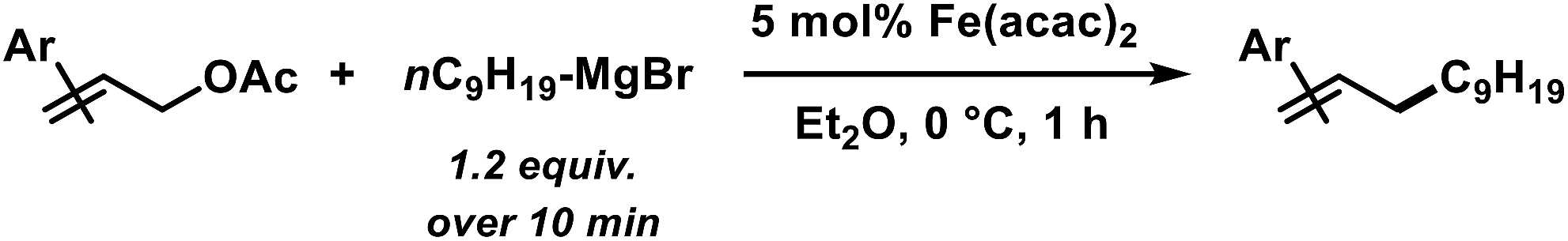
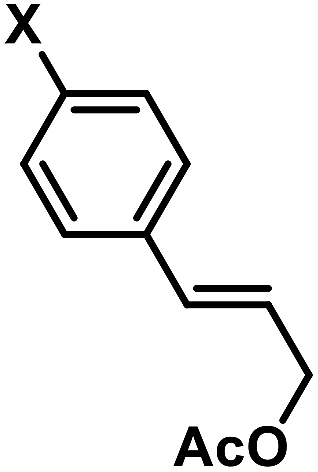
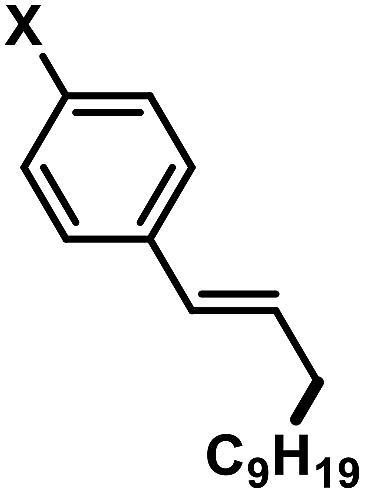
In an effort to further expand the scope of this allylation reaction, we employed aryl-substituted allyl acetates (Table 4). Enhanced selectivities were obtained from the use of Fe(acac)2 as pre-catalyst (acac = acetylacetonato). The presence of CHCl3 or slow addition of the Grignard reagent did not show any improvements (condition B). Again, increased steric hindrance of the substrates led to lower reactivities.
Table 4 Iron-catalysed alkylations of aryl-substituted allyl acetates (condition B)
Finally, we employed different Grignard reagents in reactions with various allyl acetate derivatives (Table 5). Higher yields were obtained with catalytic Fe(acac)2 when aryl groups were present in the allyl acetate or Grignard reagents. Primary and secondary alkyl-magnesium bromides and arylmagnesium bromides afforded good to very good yields of the cross-coupling products. It is important to note that the conditions A and B exhibited distinct reactivities: condition A, in the presence of CHCl3 and with catalytic Fe(OAc)2, facilitated reactions with sterically demanding allyl acetate derivatives. Condition B (no CHCl3, with Fe(acac)2) gave higher selectivities for aryl-containing substrates. Allylmagnesium bromide underwent carboxylate substitution (entry 2). 1,3-Dioxolane-2-methylmagnesium bromide was unreactive (entry 15).
Table 5 Variation of the Grignard reagents (conditions A and B)
Mechanistic studies
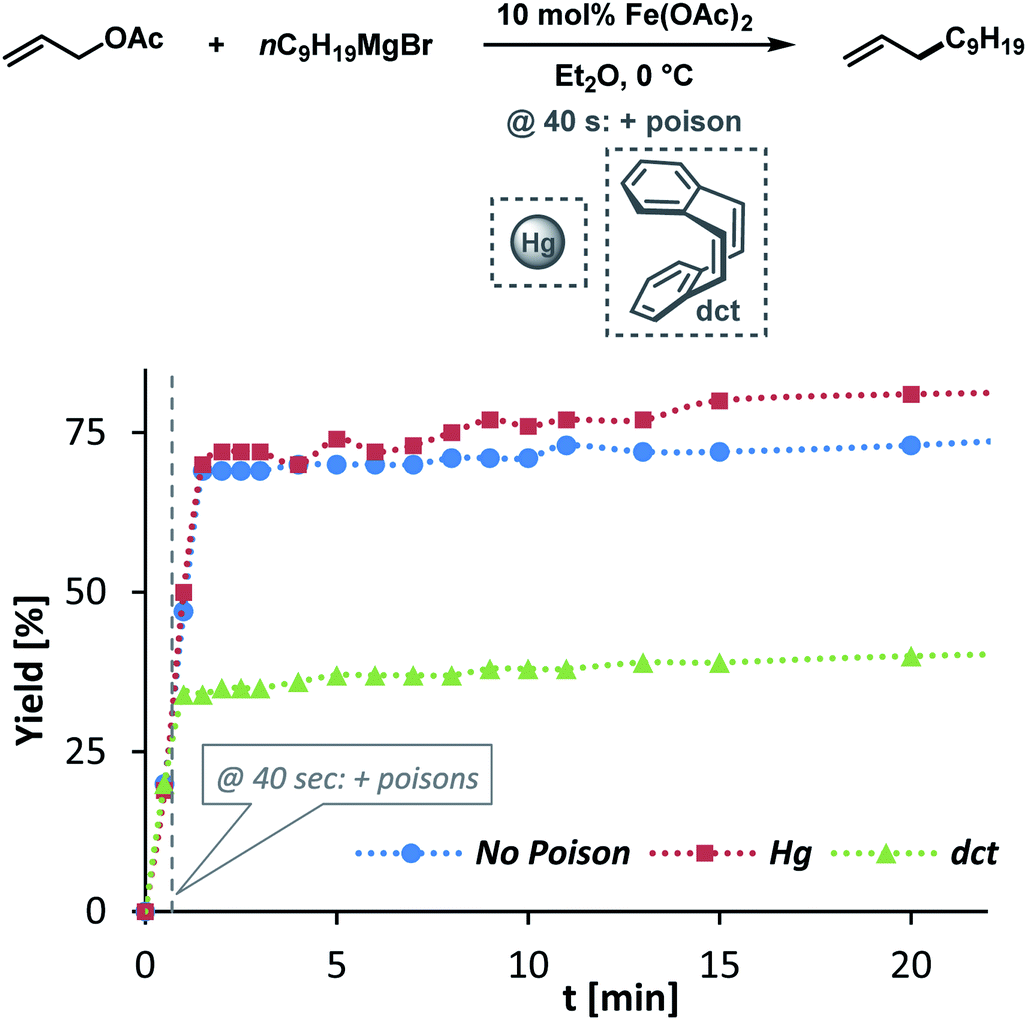
The clear distinction whether homogeneous or heterogeneous catalysis is operating can be intricate, as the catalysts can be part of an equilibrium between several species and the spectroscopic tools often perturb the system under investigation. We performed Maitlis' hot filtration test which showed comparable catalytic activity in both the filtrate and filter phases.43 The most reliable insight can be derived from kinetic experiments that are conducted in operando under the catalytic reaction conditions.43,44 Reaction progress analyses showed no sigmoidal curvature that would have been indicative of an initial catalyst nucleation and particle growth. On the contrary, the highest catalyst activity was recorded at the onset of conversion within the first 30 s of the reaction. Furthermore, kinetic poisoning experiments were conducted (Scheme 4). Amalgamation of a potential Fe(0) catalyst was not observed upon addition of 100 equiv. Hg per Fe.45,46 The addition of dibenzo[a,e]cyclooctatetraene (dct, 2 equiv. per Fe),16,47,48 a selective homotopic poison of low-valent late transition metals, resulted in immediate and complete inhibition of catalytic activity. This observation constitutes a strong indication of a homogeneous catalysis mechanism. We have collected further mechanistic insight from a set of key experiments: the strongly reducing conditions in the presence of a large excess of Grignard reagent led to rapid deactivation of the catalyst,41 an effect that already became apparent from the adaptation of slow-addition protocols (vide supra). Stoichiometric reactions between all components of this allylation protocol provided further insight (Scheme 5A). No direct reaction between allyl acetate and Fe(OAc)2 occured so that an allyliron or reduced iron species (both form in the presence of the Grignard reagent) may act as active catalyst in the formal oxidative addition of allyl acetate. Reaction with equimolar substrates and pre-catalyst gave 29% product yield, whereas the presence of 2 equiv. nonylmagnesium bromide effectively suppressed product formation (Scheme 5A). It is well known that reactions of ferrous salts with 2 equiv. alkyl-Grignard reagents form iron(0) species which rapidly nucleate and grow to particles if no suitable ligands are available. The formation of catalytically inactive Fe(0) particles in the presence of excess Grignard reagent may indicate that higher oxidation state catalysts are operating, presumably Fe(I) which is in accord with recent literature reports under very similar conditions.29,49,50 However, we cannot exclude the formation of soluble ferrate(0) complexes.51 Radical clock experiments did not fully support the potential intermediacy of organic radicals.52 No ring-opening was observed with the 1-cyclopropyl-bearing allyl acetate under the standard conditions. The operation of radical addition of an open-shell Fe catalyst to the alkene group was excluded by the absence of any reactivity with α-cyclopropyl styrene. 5-Hexenylmagnesium bromide underwent only minor ring-closure. Based on the collected data from these key experiments, we postulate a mechanism that commences with reduction of the Fe(II) pre-catalyst to an active Fe(I) species. Over-reduction to inactive Fe(0) particles in the presence of high concentrations of the Grignard reagent can be healed by addition of suitable (buffering) electrophiles such as chloroform, esters, or allyl acetate itself. Addition of allyl acetate leads to an η3-allyliron complex that undergoes reductive elimination of the C–C coupling product.
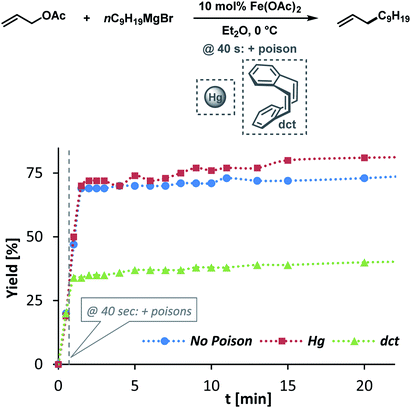 |
| | Scheme 4 Poisoning experiments with Hg (100 equiv. per Fe) and dct (2 equiv. per Fe). | |
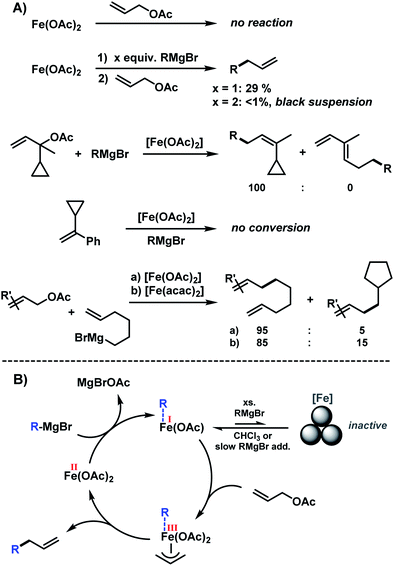 |
| | Scheme 5 (A) Key mechanistic experiments; (B) postulated mechanism. | |
Sequential allylation–hydrogenation
Having established reaction conditions for the allylation of alkylmagnesium halides, the development of a conceptually distinct alternative15 to the rather challenging alkyl–alkyl cross-coupling reactions still requires an effective Fe-catalysed alkene hydrogenation.31–37 Several hydrogenation protocols with a variety of homogeneous and heterogeneous iron catalysts have been reported in the past decade.31,34,53–58 Interestingly, hydrogenation catalysts have also been reported to form from ferrous salts and alkylmagnesium halides, which may enable sequential allylation–hydrogenation reactions with the same catalyst mixture. However, there is very little knowledge on the conversion of in situ prepared product mixtures (without workup) and the potential cross-contamination of iron catalysts by residual byproducts from the preceding steps. We therefore performed hydrogenation reactions in the presence of all possible byproducts from the allylation step under conditions A and B (Table 6). While residual allyl acetate showed no detrimental effect on the activity of the Fe catalyst, the presence of inorganic bromide salts (MgBr2 or LiBr) inhibited the hydrogenation step (entries 2 and 4). An alternative method of hydrogenation can be applied if the crude mixture of the allylation reaction is worked up (by aqueous extraction of MgBr2) prior to the hydrogenation.
Table 6 Halide-containing additives as poisons of iron-catalysed alkene hydrogenations
|
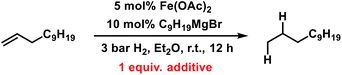
|
| Entry |
Additive |
Conversiona [%] |
Yielda [%] |
|
Yields determined by quantitative GC-FID vs. internal n-pentadecane.
|
| 1 |
— |
100 |
77 |
| 2 |
MgBr2 |
19 |
<1 |
| 3 |
MgCl2 |
100 |
72 |
| 4 |
LiBr |
3 |
<1 |
| 5 |
CHCl3 |
0 |
0 |
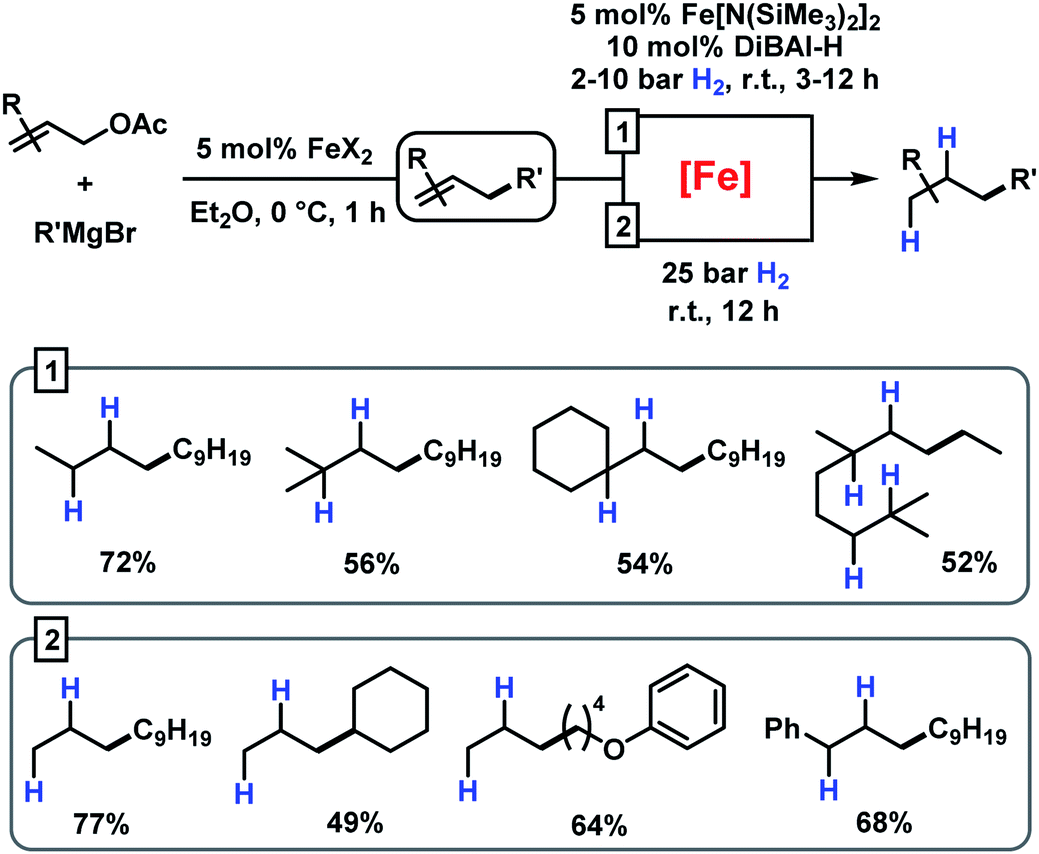
This strategy has enabled the hydrogenation of allylation products in good yields with a Fe[N(SiMe3)2]2/DiBAl-H (diisobutylaluminium hydride) catalyst under mild conditions (method 1 in Scheme 6).31 When not making the effort to work up the crude allylation reactions, sequential hydrogenations can be performed at elevated pressure (25 bar H2) to afford the alkanes with very similar yields (method 2 in Scheme 6).
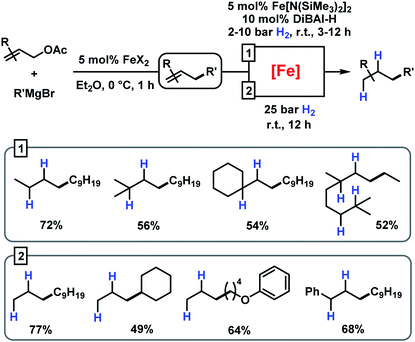 |
| | Scheme 6 Sequential iron-catalysed allylation and hydrogenation. | |
Conclusions
The iron-catalysed allylation of alkyl and aryl Grignard reagents with allyl acetate derivatives operates under very mild conditions with a commercial catalyst. Kinetic experiments suggest a highly active homotopic catalyst. Slow Grignard addition protocols or the presence of electrophilic additives (e.g. chloroform) enhanced catalyst activity and lifetime. The sequential combination of the iron-catalysed allylation of alkyl-magnesium halides with an iron-catalysed hydrogenation of the resultant alkenes results in a formal sp3–sp3-cross-coupling that proved otherwise challenging. The presence of high bromide concentrations inhibited the hydrogenation step.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was generously supported by the European Research Council (CoG 683150) and the University of Hamburg.
Notes and references
-
A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2004 Search PubMed.
- E.-i. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738 CrossRef CAS.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778 CrossRef.
- W. M. Czaplik, M. Mayer, J. Cvengros and A. Jacobi von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS.
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS.
- M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190 CrossRef CAS.
- A. S. Jones, J. F. Paliga, M. D. Greenhalgh, J. M. Quibell, A. Steven and S. P. Thomas, Org. Lett., 2014, 16, 5964 CrossRef CAS.
-
B. Plietker and M. Beller, Iron Catalysis: Fundamentals and Applications, Springer, Berlin, 2011 Search PubMed.
- D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486 CrossRef CAS.
- A. L. Silberstein, S. D. Ramgren and N. K. Garg, Org. Lett., 2012, 14, 3796 CrossRef CAS.
- T. Mesganaw and N. K. Garg, Org. Process Res. Dev., 2013, 17, 29 CrossRef CAS.
- T. Agrawal and S. P. Cook, Org. Lett., 2013, 15, 96 CrossRef CAS.
-
(a) M. Mayer, W. M. Czaplik and A. Jacobi von Wangelin, Adv. Synth. Catal., 2010, 352, 2147 CrossRef CAS;
(b) D. J. Frank, L. Guiet, A. Käslin, E. Murphy and S. P. Thomas, RSC Adv., 2013, 3, 25698 RSC.
- D. Gärtner, A. L. Stein, S. Grupe, J. Arp and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2015, 54, 10545 CrossRef.
- S. N. Kessler and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2016, 55, 3734 CrossRef CAS.
- B. Plietker, Angew. Chem., Int. Ed., 2006, 45, 1469 CrossRef CAS.
- G. K. Jarugumilli and S. P. Cook, Org. Lett., 2011, 13, 1904 CrossRef CAS.
- S. J. Ladoulis and K. M. Nicholas, J. Organomet. Chem., 1985, 285, C13–C16 CrossRef CAS.
- J. L. Roustan, J. Y. Mérour and F. Houlihan, Tetrahedron Lett., 1979, 20, 3721 CrossRef.
- Y. Xu and B. Zhou, J. Org. Chem., 1987, 52, 974 CrossRef CAS.
- B. Plietker, Angew. Chem., Int. Ed., 2006, 45, 6053 CrossRef CAS.
- R. Trivedi and J. A. Tunge, Org. Lett., 2009, 11, 5650 CrossRef CAS.
- M. Jegelka and B. Plietker, Org. Lett., 2009, 11, 3462 CrossRef CAS.
- B. Plietker, A. Dieskau, K. Möws and A. Jatsch, Angew. Chem., Int. Ed., 2008, 47, 198 CrossRef CAS.
- L. Qi, E. Ma, F. Jia and Z. Li, Tetrahedron Lett., 2016, 57, 2211 CrossRef CAS.
- K. G. Dongol, H. Koh, M. Sau and C. L. L. Chai, Adv. Synth. Catal., 2007, 349, 1015 CrossRef CAS.
- M. Guisán-Ceinos, F. Tato, E. Buñuel, P. Calle and D. J. Cárdenas, Chem. Sci., 2013, 4, 1098 RSC.
- T. Hatakeyama, T. Hashimoto, K. K. A. D. S. Kathriarachchi, T. Zenmyo, H. Seike and M. Nakamura, Angew. Chem., Int. Ed., 2012, 51, 8834 CrossRef CAS.
- T. N. Gieshoff, U. Chakraborty, M. Villa and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2017, 56, 3585 CrossRef CAS.
- S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794 CrossRef CAS.
- R. J. Trovitch, E. Lobkovsky, E. Bill and P. J. Chirik, Organometallics, 2008, 27, 1470 CrossRef CAS.
- P.-H. Phua, L. Lefort, J. A. F. Boogers, M. Tristany and J. G. de Vries, Chem. Commun., 2009, 3747 RSC.
- C. Rangheard, C. de Julián Fernández, P.-H. Phua, J. Hoorn, L. Lefort and J. G. de Vries, Dalton Trans., 2010, 39, 8464 RSC.
- A. Welther and A. Jacobi von Wangelin, Curr. Org. Chem., 2013, 17, 326 CrossRef CAS.
- A. J. MacNair, M.-M. Tran, J. E. Nelson, G. U. Sloan, A. Ironmonger and S. P. Thomas, Org. Biomol. Chem., 2014, 12, 5082 RSC.
- For further details, see the ESI.†.
- G. Cahiez and H. Avedissian, Synthesis, 1998, 1199 CrossRef CAS.
- W. Schlenk and W. Schlenk, Ber. Dtsch. Chem. Ges., 1929, 62, 920 CrossRef.
- J. Kleimark, P.-F. Larsson, P. Emamy, A. Hedström and P.-O. Norrby, Adv. Synth. Catal., 2012, 354, 448 CrossRef CAS.
- Y. Gartia, S. Pulla, P. Ramidi, C. C. Farris, Z. Nima, D. E. Jones, A. S. Biris and A. Ghosh, Catal. Lett., 2012, 142, 1397 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317 CrossRef CAS.
- S. Mørup, S. Linderoth, J. Jacobsen and M. Holmblad, Hyperfine Interact., 1992, 69, 489 CrossRef.
- S. Linderoth and S. Morup, J. Phys.: Condens. Matter, 1992, 4, 8627 CrossRef CAS.
- D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 855 CrossRef CAS.
-
G. Franck, M. Brill and G. Helmchen in Organic Syntheses Based on Name Reactions, ed. A. Hassner and C. Stumer, Elsevier, Burlington, 2002, p. 55 Search PubMed.
- A. Hedström, Z. Izakian, I. Vreto, C.-J. Wallentin and P.-O. Norrby, Chem.–Eur. J., 2015, 21, 5946 CrossRef.
- R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485 CrossRef CAS.
- P. Neate, M. D. Greenhalgh, W. W. Brennessel, S. P. Thomas and M. L. Neidig, J. Am. Chem. Soc., 2019, 141, 10099 CrossRef CAS.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 2002, 13, 317 CrossRef.
- R. P. Yu, J. M. Darmon, J. M. Hoyt, G. W. Margulieux, Z. R. Turner and P. J. Chirik, ACS Catal., 2012, 2, 1760 CrossRef CAS.
- A. Welther, M. Bauer, M. Mayer and A. Jacobi von Wangelin, ChemCatChem, 2012, 4, 1088 CrossRef CAS.
- T. N. Gieshoff, M. Villa, A. Welther, M. Plois, U. Chakraborty, R. Wolf and A. Jacobi von Wangelin, Green Chem., 2015, 17, 1408 RSC.
- D. Gärtner, A. Welther, B. R. Rad, R. Wolf and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2014, 53, 3722 CrossRef.
- P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687 CrossRef CAS.
- K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07604b |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*