
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed chemoselective synthesis of 4-trifluoromethyl pyrazoles†
Jiaqing
Lu
 a,
Yuning
Man
a,
Yabin
Zhang
a,
Bo
Lin
a,
Qi
Lin
*b and
Zhiqiang
Weng
*a
a,
Yuning
Man
a,
Yabin
Zhang
a,
Bo
Lin
a,
Qi
Lin
*b and
Zhiqiang
Weng
*a
aFujian Provincial Key Laboratory of Electrochemical Energy Storage Materials, College of Chemistry, Fuzhou University, Fuzhou, 350108, China. E-mail: zweng@fzu.edu.cn; Fax: +86 591 22866247; Tel: +86 591 22866247
bOcean College, Minjiang University, Fuzhou, 350108, China. E-mail: qlin1990@163.com
First published on 30th September 2019
Abstract
A series of 4-trifluoromethyl pyrazoles have been prepared via the copper-catalyzed cycloaddition of 2-bromo-3,3,3-trifluoropropene with a variety of N-arylsydnone derivatives under mild conditions. This new protocol under optimized reaction conditions [Cu(OTf)2/phen, DBU, CH3CN, 35 °C] afforded 4-trifluoromethyl pyrazoles in moderate to excellent yields with excellent regioselectivity.
Introduction
Methods for introduction of a fluorine atom or fluorinated group into organic molecules have emerged over the last decade as powerful synthetic tools that allow the construction of organofluorine compounds, which are of immense interest in materials sciences, agrochemical development, and medicinal chemistry.1–4 Indeed, derivatization of organic compounds with fluorinated units often alters their chemical, physical, and biological properties, by comparison with the corresponding non-fluorinated derivative and often improves their pharmacological properties.5–10Among organofluorinated molecules, trifluoromethylated pyrazoles11 are particularly important compounds because many of them exhibit diverse biological activities and have found applications as drugs and agrochemicals (Fig. 1). Key examples include Celecoxib, a nonsteroidal anti-inflammatory drug used to treat pain or inflammation,12 and Razaxaban, a factor Xa inhibitor potentially for the treatment of thrombosis.13 In addition, trifluoromethylated pyrazoles form the core of several agrochemicals currently in development. Specifically, penthiopyrad has shown fungicidal activity against foliar pathogens and soilborne pathogens,14 compounds A have shown insecticidal activity for control of Spodoptera littoralis,15 and compounds B has potential application as an acaricide for control of Tetranychus urticae.16
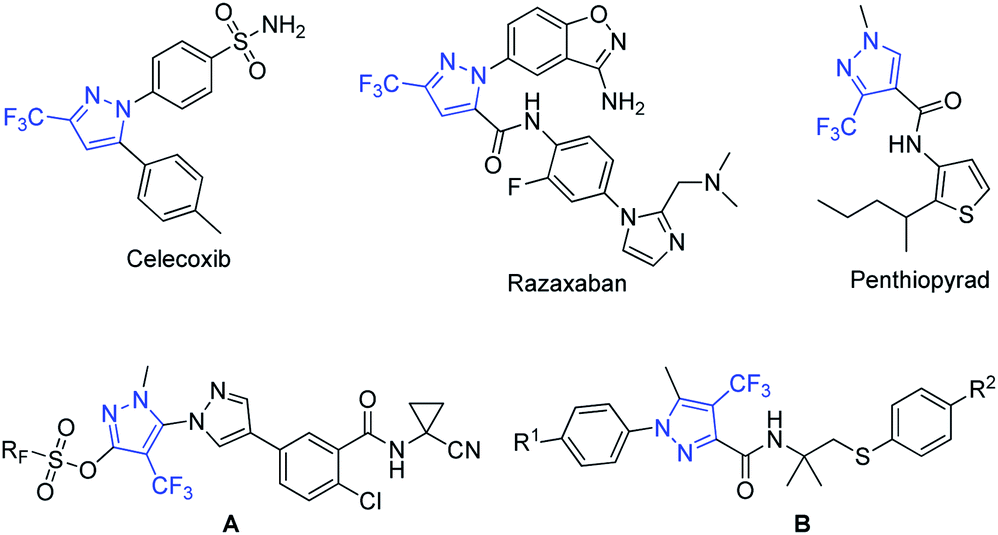 | ||
| Fig. 1 Some biologically active trifluoromethylated pyrazoles. | ||
While the vast array of procedures to synthesize 3-trifluoromethyl pyrazoles have been established,17–19 there were considerably less efforts devoted to the development of synthetic methods that allow effectively access to 4-trifluoromethyl pyrazoles. Conventionally, 4-trifluoromethyl pyrazoles were synthesized by the cycloaddition/cyclization reaction with CF3-containing building blocks.20–23 For example, the synthesis of 4-(trifluoromethyl)pyrazole derivatives from cycloaddition of hydrazines to β-trifluoromethylated vinamidinium salt or 2-(trifluoromethyl)-1,3-diketones was independently reported by the research groups of Yamanaka and Yamakawa (Scheme 1a).20,21 Recently, Tsui and co-workers developed a copper-mediated synthesis of 4-(trifluoromethyl)pyrazoles from reaction of α,β-alkynic tosylhydrazones with trifluoromethyltrimethylsilane through domino cyclization, trifluoromethylation, and deprotection sequence (Scheme 1b).24 Meazza and co-workers reported the preparation of trifluoromethyl-substituted pyrazoles via 1,3-dipolar cycloaddition from sydnones and 1-aryl-3,3,3-trifluoro-propynes (Scheme 1c).22 The methods described above and some other reported methods are useful,25–30 and have found extensive applications in synthesis; however, some of reactions suffered from tedious procedures for the synthesis of trifluoromethylated sources, the requirement of a stoichiometric amount of metal catalyst, and harsh reaction conditions, as well as poor regioselectivities. Therefore, the development of more general and practical approaches employing easily accessible starting materials under mild conditions is highly desirable.
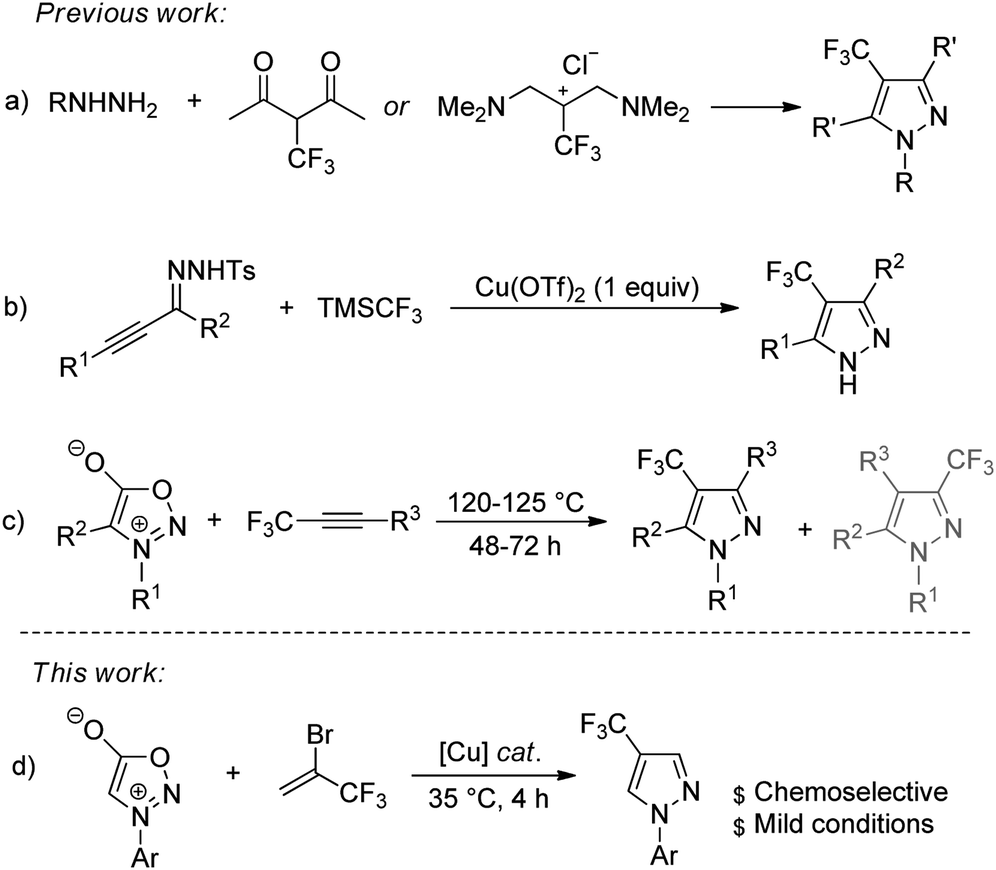 | ||
| Scheme 1 Recent examples for the preparation of 4-trifluoromethyl pyrazoles. | ||
As part of our ongoing research into the development of copper-catalyzed reactions for the preparation of organofluorine compounds,31,32 we aimed to investigate an alternative protocol for the synthesis of these scaffolds using easy-to-operate synthetic tools. Based on the fact that 3,3,3-trifluoropropyne could be generated in situ from a dehydrobromination reaction of readily available 2-bromo-3,3,3-trifluoropropene (1) under base conditions,33 and our recent work on 1,3-dipolar cycloaddition reaction,34–36 we wondered if 2-bromo-3,3,3-trifluoropropene could be used for the selective synthesis of 4-trifluoromethyl pyrazoles (Scheme 1d). We report here a new method for the synthesis of 4-trifluoromethyl pyrazoles through copper-catalyzed reaction of sydnones with 2-bromo-3,3,3-trifluoropropene (1).
Results and discussion
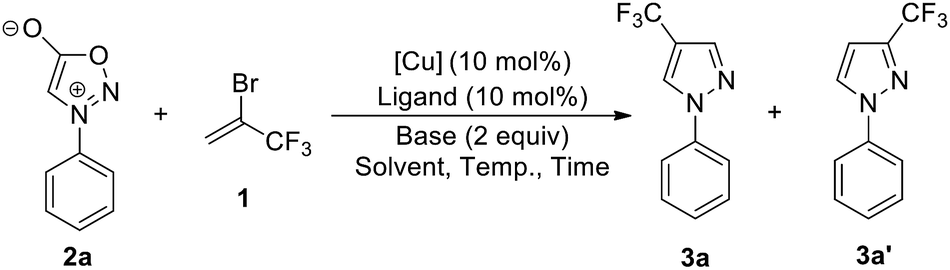
We commenced our investigation by using N-phenylsydnone (2a) as a model substrate in the reaction with 3.0 equiv. of 1 in the presence of CuI (10 mol%)/phen (10 mol%) as a catalyst and 2.0 equiv. of DBU as a base (Table 1). Gratifyingly, after 4 h at 35 °C in CH3CN under a nitrogen atmosphere, 4-trifluoromethylated pyrazole 3a was obtained in 47% yield with excellent regioselectivity (Entry 1). The structure of 3a was unambiguously determined by NMR spectroscopy and X-ray crystallography (Fig. 2). Encouraged by this initial result, we carried out the reaction under different conditions to optimize the product yield. First, copper catalysts and ligands were screened (Entries 2–10), and the results showed that the use of Cu(OTf)2/phen presented the highest catalytic activity for this reaction, providing product 3a in 99% yield (Entry 6). It is worth mentioning that no product formation was observed in the absence of copper salts and ligands (Entry 11), thus implying the importance of the copper catalyst in this transformation. The effects of the base were also examined. When DBU was replaced with NEt3, NaOt-Bu, KOt-Bu, NaOH, KOH, or K3PO4 the reactions only produced trace amounts or no desired product (Entries 12−17). Commonly used organic solvents were also screened. It was found that CH3CN gave the best yield (Entry 6); solvents such as DMSO gave moderate yields of the product (Entry 18), and THF, toluene, dioxane, and DMF retarded the reaction (Entries 19–22). Reactions performed either at lower temperatures (25 °C) or reducing the reaction time to 2 h resulted in substantially lower product yields (44% and 60% yields, respectively; Entry 23 and 24).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | [Cu] | Ligand | Base | Solvent | Temp. (°C) | Time (h) | Yieldb (%) | 3a:3a′ |
| a Reaction conditions: 1 (0.30 mmol, 3.0 equiv.), 2a (0.10 mmol), solvent (1.0 mL), N2; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; phen = 1,10-phenanthroline; bpy = 2,2′-bipyridine; dmbpy = 4,4′-dimethyl-2,2′-bipyridine; TMEDA = tetramethylethylenediamine. b The yield was determined by 19F NMR spectroscopy with PhOCF3 as internal standard. | ||||||||
| 1 | CuI | phen | DBU | CH3CN | 35 | 4 | 47 | 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 :0 |
| 2 | CuBr | phen | DBU | CH3CN | 35 | 4 | 36 | 100:0 |
| 3 | CuSCN | phen | DBU | CH3CN | 35 | 4 | 71 | 100:0 |
| 4 | CuTc | phen | DBU | CH3CN | 35 | 4 | 74 | 100:0 |
| 5 | CuSO4 | phen | DBU | CH3CN | 35 | 4 | 9 | 100:0 |
| 6 | Cu(OTf)2 | phen | DBU | CH3CN | 35 | 4 | 99 | 100:0 |
| 7 | Cu(TFA)2 | phen | DBU | CH3CN | 35 | 4 | 85 | 100:0 |
| 8 | Cu(OTf)2 | bpy | DBU | CH3CN | 35 | 4 | 22 | 97:3 |
| 9 | Cu(OTf)2 | dmbpy | DBU | CH3CN | 35 | 4 | 27 | 99:1 |
| 10 | Cu(OTf)2 | tmeda | DBU | CH3CN | 35 | 4 | 23 | 99:1 |
| 11 | — | — | DBU | CH3CN | 35 | 4 | 0 | — |
| 12 | Cu(OTf)2 | phen | NEt3 | CH3CN | 35 | 4 | 0 | — |
| 13 | Cu(OTf)2 | phen | NaOt-Bu | CH3CN | 35 | 4 | <1 | — |
| 14 | Cu(OTf)2 | phen | KOt-Bu | CH3CN | 35 | 4 | <1 | — |
| 15 | Cu(OTf)2 | phen | NaOH | CH3CN | 35 | 4 | <1 | — |
| 16 | Cu(OTf)2 | phen | KOH | CH3CN | 35 | 4 | <1 | — |
| 17 | Cu(OTf)2 | phen | K3PO4 | CH3CN | 35 | 4 | 0 | — |
| 18 | Cu(OTf)2 | phen | DBU | DMSO | 35 | 4 | 50 | 100:0 |
| 19 | Cu(OTf)2 | phen | DBU | THF | 35 | 4 | <1 | — |
| 20 | Cu(OTf)2 | phen | DBU | Toluene | 35 | 4 | 0 | — |
| 21 | Cu(OTf)2 | phen | DBU | Dioxane | 35 | 4 | <1 | — |
| 22 | Cu(OTf)2 | phen | DBU | DMF | 35 | 4 | <1 | — |
| 23 | Cu(OTf)2 | phen | DBU | CH3CN | 25 | 4 | 44 | 100:0 |
| 24 | Cu(OTf)2 | phen | DBU | CH3CN | 35 | 2 | 60 | 100:0 |
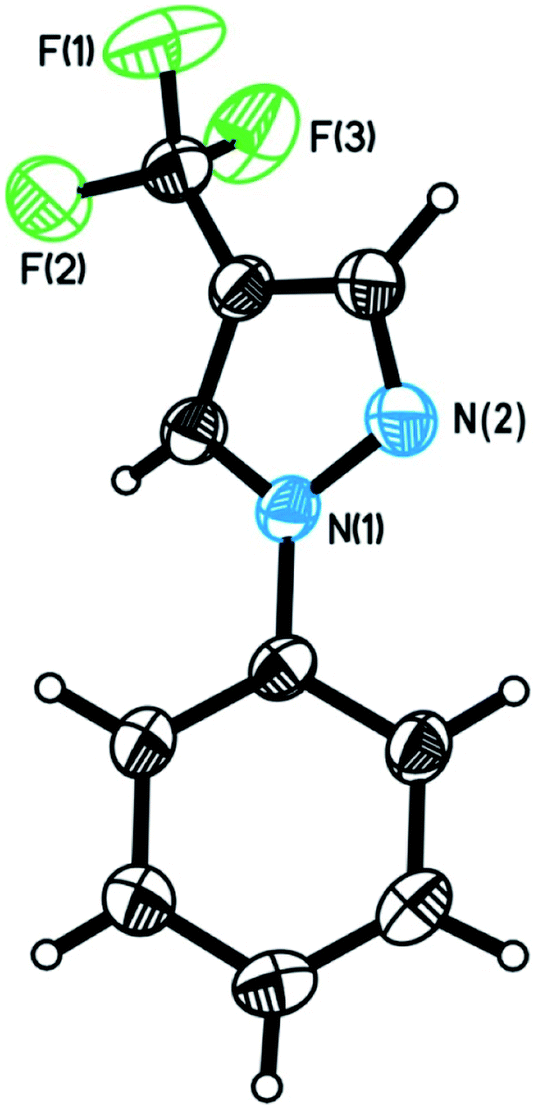 | ||
| Fig. 2 ORTEP drawing of 3a. Thermal ellipsoids are drawn at 40% probability. | ||
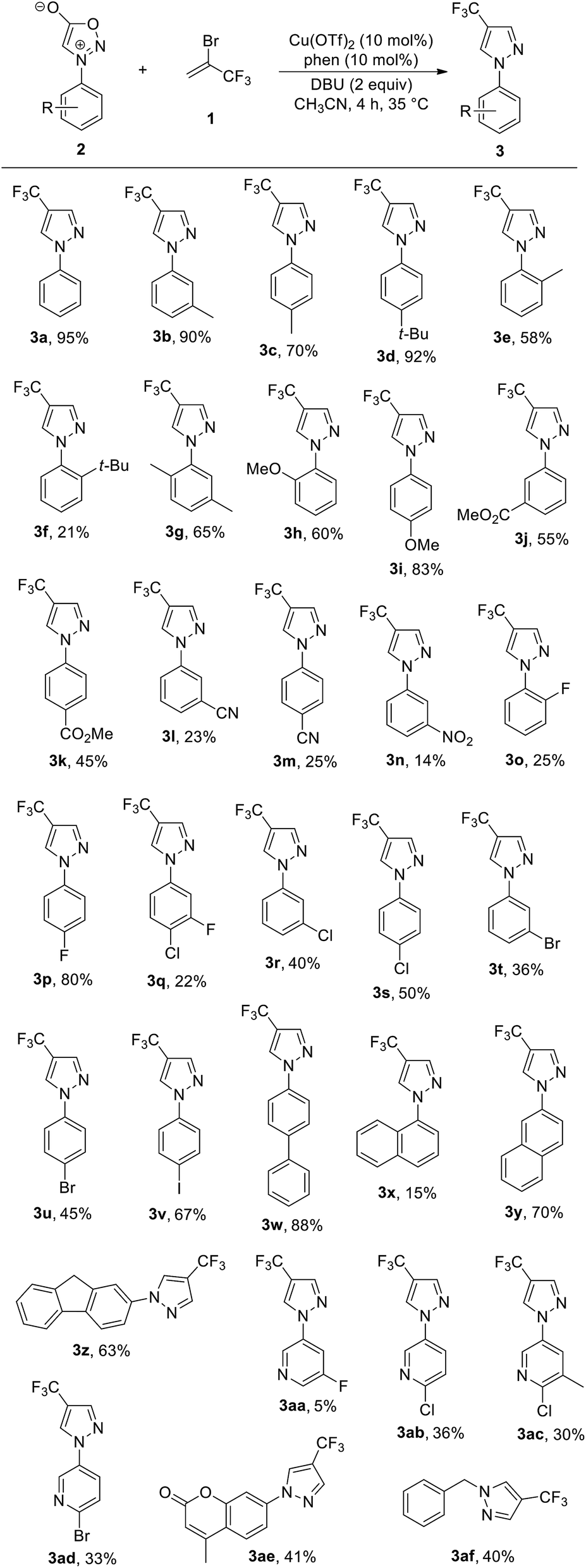
With the optimised conditions in hand, the substrate scope of the reaction was evaluated using a range of electronically and sterically differentiated N-arylsydnone derivatives (Table 2). Overall, it was observed that the reaction could tolerate a variety of functional groups on the phenyl rings of sydnones. For example, N-arylsydnones bearing para- or meta-alkyl-substituted aromatic rings all reacted well with 1 to give the corresponding products 3b–3d in 70–92% yields. As an exception, ortho-alkyl-substituted on phenyl rings of N-arylsydnones afforded the products 3e and 3f in lower yields (21% and 58%, respectively), probably due to steric hindrance. The dimethyl-substituted N-arylsydnone also took part in the reaction to afford the desired product 3g in 65% yield. N-arylsydnone substrates bearing electron-donating substituents, such as methoxy at the para-position on the aryl ring was smoothly converted to the corresponding product 3i in 83% yield, with the exception of substrate bearing a OMe group at the ortho position (3h, 60% yield). However, reactions of N-arylsydnones containing electron-withdrawing substituents, such as ester, cyano, and nitro groups on the aryl ring furnished the desired products 3j–3n in relatively low yields (14–55%). Likewise, N-arylsydnone derivatives bearing electron-withdrawing halogen groups, such as fluoro, chloro, bromo, and iodo on the aryl rings were accommodated and furnished the desired products 3o–3v in 22–80% yields, thereby providing possibilities for following chemical transformations. Analogously, the reaction with polyaromatic sydnones provided the corresponding trifluoromethylated product 3w, 3y, and 3z in 63–88% yields. In the case of the N-1-naphthylsydnone substrate, the desired product 3x could only be obtained in 15% isolated yield, with the majority of the remaining material attributed to unreacted starting material. This may be due to the steric bulk of the 1-naphthyl group hindering access to the cycloaddition site. The process was applied also to heteroaromatic sydnones, such as N-pyridyl- and cumarin sydnone derivatives, the corresponding products 3aa-3ae were obtained in low to moderate yields (5–41%). Notably, a N-benzylsydnone derivative was also proved to be suitable substrate, producing the corresponding trifluoromethylated pyrazole 3af in promising yield (40%).
| a Reaction conditions: 1 (1.5 mmol, 3.0 equiv.), 2 (0.50 mmol, 1.0 equiv.), Cu(OTf)2 (0.050 mmol, 10 mol%), phen (0.050 mmol, 10 mol%), DBU (1.0 mmol, 2.0 equiv.), CH3CN (5.0 mL), 35 °C, 4 h, N2; isolated yields. |
|---|
|
To illustrate the further synthetic utility of this method, a gram-scale reaction of N-phenylsydnone (2a) with 2-bromo-3,3,3-trifluoropropene (1) was performed, and the corresponding 4-trifluoromethyl pyrazole product 3a was given in 80% isolated yield (Scheme 2). This result demonstrates the scalability of the reaction.
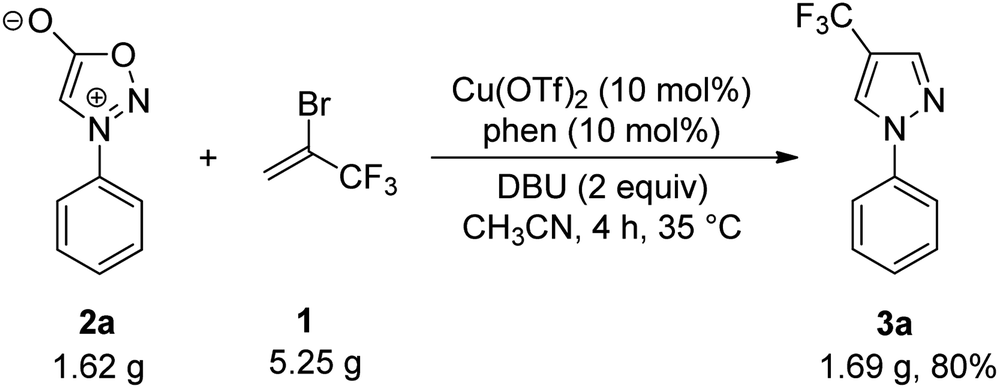 | ||
| Scheme 2 Gram scale experiment. | ||
Based on the above results and previous reports by Taran,37 Gomez-Bengoa, and Harrity,38 a plausible reaction mechanism is proposed in Scheme 3. Firstly, the copper species reacted with the in situ generated 3,3,3-trifluoropropyne to give Cu(I) acetylide species I. In the second step, the cycloaddition of I with the sydnone 2 would form the transition state II. The tight copper–nitrogen interaction in II is probably responsible for its lower the activation barrier compared with other modes of interaction between arylsydnone and Cu(I) acetylide.38,39 Subsequently, the Cu–N dissociation and C–N bond formation could generate III, which upon CO2 elimination provided 3-copper(I) pyrazolide IV. Finally, protonation of IV with new 3,3,3-trifluoropropyne produced the desired 4-trifluoromethyl pyrazole regiomer 3 along with regeneration of the starting copper species to complete the catalytic cycle.
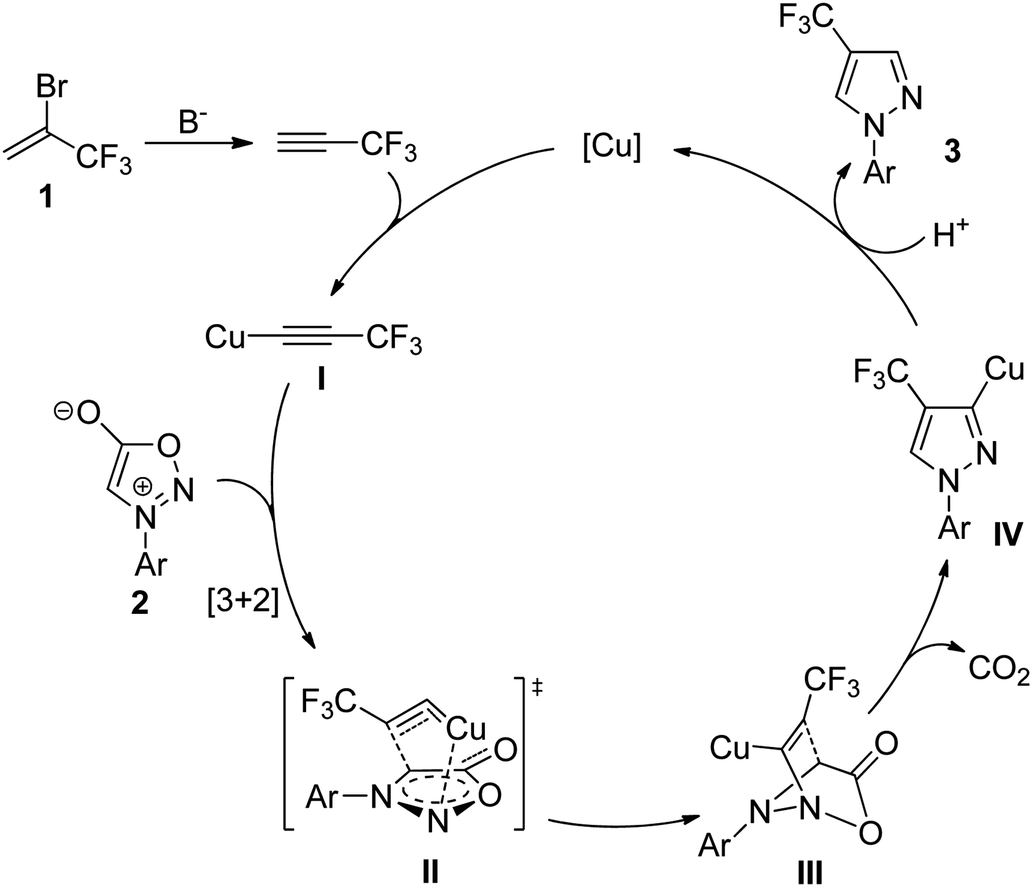 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
In summary, we have developed a simple and efficient method for the synthesis of 4-trifluoromethyl pyrazole derivatives from the copper-catalyzed cycloaddition of 2-bromo-3,3,3-trifluoropropene with a variety of N-arylsydnones under mild conditions. This approach allowed the rapid assembly of a small library of potentially medicinally relevant 4-trifluoromethylated pyrazoles in moderate to excellent yields with excellent regioselectivity. Investigations directed towards the extension of the scope of the reaction are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21772022) and Fuzhou University.Notes and references
- T. Yamazaki, T. Taguchi and I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, 2009 Search PubMed.
- J. T. Welch and S. Eswarakrishnan, Fluorine in Bioorganic Chemistry, John Wiley & Sons, New York, 1991 Search PubMed.
- K. Uneyama, Organofluorine Chemistry, Blackwell Publishing Ltd., 2006 Search PubMed.
- J. Hu and T. Umemoto, Synthetic Organofluorine Chemistry, Springer, 2018 Search PubMed.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS.
- W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS.
- L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513–1522 CrossRef CAS.
- C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef CAS.
- S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem.–Eur. J., 2014, 20, 16806–16829 CrossRef CAS.
- C. Alonso, E. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS.
- K. Kaur, V. Kumar and G. K. Gupta, J. Fluorine Chem., 2015, 178, 306–326 CrossRef CAS.
- P. L. McCormack, Drugs, 2011, 71, 2457–2489 CrossRef CAS.
- D. Zhang, N. Raghavan, S.-Y. Chen, H. Zhang, M. Quan, L. Lecureux, L. M. Patrone, P. Y. S. Lam, S. J. Bonacorsi, R. M. Knabb, G. L. Skiles and K. He, Drug Metab. Dispos., 2008, 36, 303 CrossRef CAS PubMed.
- A. K. Culbreath, T. B. Brenneman, R. C. Kemerait Jr and G. G. Hammes, Pest Manage. Sci., 2009, 65, 66–73 CrossRef CAS.
- A. Jeanguenat, M. El Qacemi, A. Stoller, A. Bigot and D. Gribkov, WO2018234240, 2018.
- K. Koyama, I. Okada, T. Fukuchi and Y. Kinoshita, JP2013216634, 2013.
- S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS.
- J. C. Sloop, C. L. Bumgardner and W. D. Loehle, J. Fluorine Chem., 2002, 118, 135–147 CrossRef CAS.
- J. C. Sloop, C. Holder and M. Henary, Eur. J. Org. Chem., 2015, 2015, 3405–3422 CrossRef CAS.
- H. Yamanaka, T.-i. Takekawa, K. Morita, T. Ishihara and J. T. Gupton, Tetrahedron Lett., 1996, 37, 1829–1832 CrossRef CAS.
- Y. Ohtsuka, D. Uraguchi, K. Yamamoto, K. Tokuhisa and T. Yamakawa, Tetrahedron, 2012, 68, 2636–2649 CrossRef CAS.
- G. Meazza, G. Zanardi and P. Piccardi, J. Heterocycl. Chem., 1993, 30, 365–371 CrossRef CAS.
- T. Hanamoto, M. Egashira, K. Ishizuka, H. Furuno and J. Inanaga, Tetrahedron, 2006, 62, 6332–6338 CrossRef CAS.
- Q. Wang, L. He, K. K. Li and G. C. Tsui, Org. Lett., 2017, 19, 658–661 CrossRef CAS.
- I. I. Gerus, R. X. Mironetz, I. S. Kondratov, A. V. Bezdudny, Y. V. Dmytriv, O. V. Shishkin, V. S. Starova, O. A. Zaporozhets, A. A. Tolmachev and P. K. Mykhailiuk, J. Org. Chem., 2012, 77, 47–56 CrossRef CAS.
- Y.-P. Xiong, M.-Y. Wu, X.-Y. Zhang, C.-L. Ma, L. Huang, L.-J. Zhao, B. Tan and X.-Y. Liu, Org. Lett., 2014, 16, 1000–1003 CrossRef CAS.
- C.-P. Zhang, Z.-L. Wang, Q.-Y. Chen, C.-T. Zhang, Y.-C. Gu and J.-C. Xiao, Angew. Chem., Int. Ed., 2011, 50, 1896–1900 CrossRef CAS.
- M. Chen and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 11628–11631 CrossRef CAS.
- M. Presset, D. Oehlrich, F. Rombouts and G. A. Molander, J. Org. Chem., 2013, 78, 12837–12843 CrossRef CAS.
- Z. Gonda, S. Kovács, C. Wéber, T. Gáti, A. Mészáros, A. Kotschy and Z. Novák, Org. Lett., 2014, 16, 4268–4271 CrossRef CAS.
- H. Zheng, Y. Huang and Z. Weng, Tetrahedron Lett., 2016, 57, 1397–1409 CrossRef CAS.
- W. Wu and Z. Weng, Synthesis, 2018, 50, 1958–1964 CrossRef CAS.
- T. Yamazaki, K. Mizutani and T. Kitazume, J. Org. Chem., 1995, 60, 6046–6056 CrossRef CAS.
- W. Wu, J. Wang, Y. Wang, Y. Huang, Y. Tan and Z. Weng, Angew. Chem., Int. Ed., 2017, 56, 10476–10480 CrossRef CAS.
- B. Lin, W. Wu and Z. Weng, Tetrahedron, 2019, 75, 2843–2847 CrossRef CAS.
- Z. Wang, X. Ding, S. Li and Z. Weng, Tetrahedron, 2018, 74, 6631–6634 CrossRef CAS.
- S. Kolodych, E. Rasolofonjatovo, M. Chaumontet, M.-C. Nevers, C. Créminon and F. Taran, Angew. Chem., Int. Ed., 2013, 52, 12056–12060 CrossRef CAS.
- J. Comas-Barceló, R. S. Foster, B. Fiser, E. Gomez-Bengoa and J. P. A. Harrity, Chem.–Eur. J., 2015, 21, 3257–3263 CrossRef.
- V. Hladikova, J. Vana and J. Hanusek, Beilstein J. Org. Chem., 2018, 14, 1317–1348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1934789. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra07694h |
| This journal is © The Royal Society of Chemistry 2019 |