
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dinuclear cobalt cluster as electrocatalyst for oxygen reduction reaction†
Yun-Wu Li ,
Wen-Jie Zhang,
Chun-Xia Li,
Lin Gu,
Hong-Mei Du,
Hui-Yan Ma*,
Su-Na Wang* and
Jin-Sheng Zhao*
,
Wen-Jie Zhang,
Chun-Xia Li,
Lin Gu,
Hong-Mei Du,
Hui-Yan Ma*,
Su-Na Wang* and
Jin-Sheng Zhao*
Shandong Provincial Key Laboratory/Collaborative Innovation Center of Chemical Energy Storage and Novel Cell Technology, School of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng 252000, P. R. China. E-mail: mahuiyanyan@163.com; wangsuna@lcu.edu.cn; j.s.zhao@163.com
First published on 23rd December 2019
Abstract
Dinuclear metal clusters as metalloenzymes execute efficient catalytic activities in biological systems. Enlightened by this, a dinuclear {CoII2} cluster was selected to survey its ORR (Oxygen Reduction Reaction) catalytic activities. The crystalline {CoII2} possesses defined structure and potential catalytic active centers of {CoN4O2} sites, which was identified by X-ray single crystal diffraction, Raman and XPS. The appropriate supramolecular porosity combining abundant pyridinic-N and triazole-N sites of {CoII2} catalyst synergistically benefit the ORR performance. As a result, this non-noble metal catalyst presents a nice ORR electrocatalytic activity and abides by a nearly 4-electron reduction pathway. Thus, this unpyrolyzed crystalline catalyst clearly provide precise active sites and the whole defined structural information, which can help researcher to design and fabricate efficient ORR catalysts to improve their activities. Considering the visible crystal structure, a single cobalt center-mediated catalytic mechanism was also proposed to elucidate the ORR process.
1. Introduction
Oxygen reduction reaction (ORR) is a critical central reaction in many new energy batteries, such as fuel cells and metal–air batteries.1,2 The best and widely accepted benchmark catalyst for ORR is noble metal Pt,3 but cost effectiveness blocks its large-scale commercial applications. So lots of Pt-alloy and Pt-free catalysts, such as PtPb/Pt alloy,4a Pt/Co alloy,4b Pt/Ni alloy,4c Pt/Fe alloy,4d transition metal derived nanomaterials,5 carbon species and its composite materials,6 have been emerging as the times require. Among them, {FeNx} and {CoNx} analogues are two types of high-profile catalysts, due to their competitive ORR catalytic activities and lower cost.7,8 However, {MNx} (M = Metals) catalysts are usually prepared by pyrolysis of specific precursors, such as coordination multinuclear clusters (CMCs) and metal–organic frameworks (MOFs), which leads to presenting amorphous state and lacking defined structure. As a result, it brings difficulty to manage actual and definite activity sites in ORR progress. Most recently, several groups began to research crystalline metal coordinated complexes as ORR catalysts and have made some progress. For example, a crystal {NiII3(HITP)} catalyst with {NiN4} sites showed a good ORR activity and stability but just followed a 2e− process.9 After then, a mononuclear {CoII} coordinated complex and graphene composite with {CoNO4} activity sites for ORR,10 a family of {CoII2} clusters with {CoN5O} activity sites for ORR,11 a CoII cluster-based MOF with multiple {CoII} centers for ORR,12 have been successively reported and all displayed promising ORR activities. These meaningful works encourage us to start investigating the coordinated crystalline material for ORR.Our present interest focus on crystalline dinuclear {M2} clusters as ORR catalysts inspired by their efficient catalytic activities in biological systems, such as many dinuclear metalloenzymes including {FeII2} hydrogenase and {CuII2} tyrosinase. So we selected a crystalline {CoII2} cluster reported by our group13 to perform the ORR catalysis in view of following features: (a) the {CoII2} cluster bearing defined crystal structure; (b) the {CoII2} cluster containing precised potential catalytic active center {CoN4O2} cores; (c) the N-rich ligand in {CoII2} cluster simultaneous involving pyridinic-N and triazole-N sites; (d) the {CoII2} cluster possessing thermal stability.13 Theses characteristics could theoretically endow the {CoII2} cluster with targeted ORR activity, and as expected, it can boost the ORR and abides by a nearly 4-electrons reduction pathway. This unpyrolyzed non-noble metal crystalline catalyst here clearly provide precise active sites and the whole defined structural information, which can help researcher to design and fabricate efficient ORR catalysts to improve their activities.
2. Experimental section
2.1 Chemicals and syntheses
All chemicals were purchased without purification. Dinuclear {CoII2} (molecular formula: [Co(pmmat)ClO4]2) cluster catalyst was synthesized according our reported experiment and the detailed see ESI.† 132.2 Material characterization
N2 adsorption experiment was tested using a Micrometrics ASAP 2020M. Electron microscopy measurements were measured using field-emission scanning electron microscopy (SEM, JSM-6360). Transmission electron microscopy (TEM) was measured by JEM-2100 at 200 kV. Raman spectrum was tested using Monovista CRS500. X-ray photoelectron spectrum (XPS) were carried out using ESCALAB Xi+. Elemental analyses (C, H and N) were performed on an Elementar Vario EL analyzer. The X-ray powder diffraction (XRPD) was obtained on a D/MAX-rA (Rigaku) diffractometer with Cu Kα radiation (λ = 1.542 Å) with a scan rate of 4° min−1. FT-IR spectra were recorded on a FT6700 spectrometer (USA) using KBr disc method in the range of 400–4000 cm−1.2.3 Electrochemical measurements
The electrocatalytic properties including EIS (electrochemical impedance spectroscopy) were conducted using an Autolab PGSTAT 302 N potentiostat, equipped with an electrode rotator (AFMSRCE, Pine). A typical experiment: 1.6 mg {CoII2} catalyst and 1.6 mg high-purity carbon powder (only 3.2 mg 20% Pt/C) were dispersed in a mixture of 570 μL ultra-pure H2O and 177 μL iso-propanol under supersound, and 3 μL 5 wt% Nafion dispersed ethanol solution was added. Then, 8.5 μL catalyst ink was dropped on the glassy carbon rotating disk electrode (RDE) and air dried. So the mass loading of the {CoII2} and Pt/C catalysts on the electrodes are 18.13 μg and 36.26 μg, respectively. The corresponding metal Co and Pt loading is 2.14 μg and 7.25 μg, respectively. A three-electrode cell system was worked in 70 mL 0.1 M KOH electrolyte. The working electrode is RDE glassy carbon, the reference electrode is saturated Ag/AgCl, and the counter electrode is platinum wire. Cyclic voltammogram (CV) and linear sweep voltammogram (LSV) tests were performed with a scan rate of 100 and 5 mV s−1, respectively. For comparison, commercial Pt/C was also loaded onto the RDE by the same method. All the potentials were referred to a reversible hydrogen electrode (RHE) using equation ERHE = EAg/AgCl + 0.197 V + 0.0591 pH.The ORR kinetics was analyzed with the Koutecky–Levich (K–L) plots based on K–L equations:
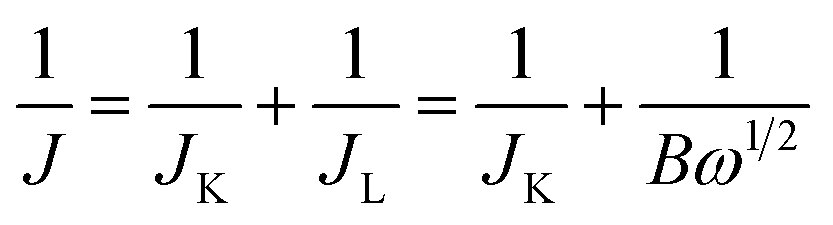 | (1a) |
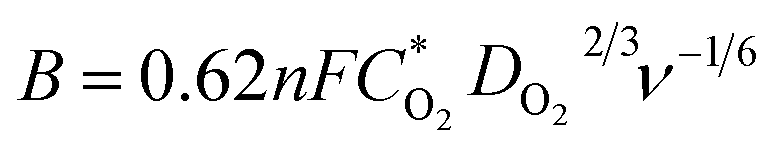 | (1b) |
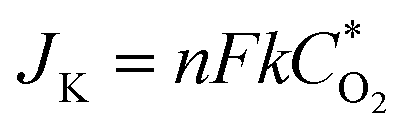 | (1c) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1); D is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1); ν is the kinetic viscosity (0.01 cm2 s−1), k is the electron transfer rate constant, and C* is the concentration of O2 (1.2 × 10−6 mol cm−3) in solution.
485 C mol−1); D is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1); ν is the kinetic viscosity (0.01 cm2 s−1), k is the electron transfer rate constant, and C* is the concentration of O2 (1.2 × 10−6 mol cm−3) in solution.
3. Results and discussions
3.1 The structure and component of the {CoII2} catalyst
The precise structure of catalyst is a dinuclear {CoII2} cluster with two {CoN4O2} coordinated cores wrapped by two N-rich organic ligands pmmat− (Fig. 1a).13 The detailed crystal data are shown in Table S1 (ESI†). It displays a 3D supramolecular microporous structure with a 21.3% porosity using the PLATON analysis (Fig. 1b), which can favor O2 transport during ORR process. The porous fact also proved by the N2 adsorption at 77 K and 1 atm (Fig. 2a). The adsorb amount of N2 increases slowly before 0.5 atm, and then rises rapidly to achieve a maximum at 63.3 cm3 g−1. The relevant BET (Brunner–Emmett–Teller) surface areas is 134.2 m2 g−1 by calculated using above N2 adsorption data. The pore size diameter is among 1.5–15 Å, and the medium diameter is 3.9 Å, which can accommodate N2 (kinetic diameter: 3.6 Å) and O2 (kinetic diameter: 3.5 Å) appropriately.14 The other basic characterization containing IR, PXRD, magnetism, and TG, can confirm its single crystalline pure-phase and thermal stability.13 The SEM and TEM images show the {CoII2} catalyst displays a flake-like morphology with crystalline smooth surface (Fig. 1c and d). The width is ca. 3–5 μm and length is ca. 30–50 μm.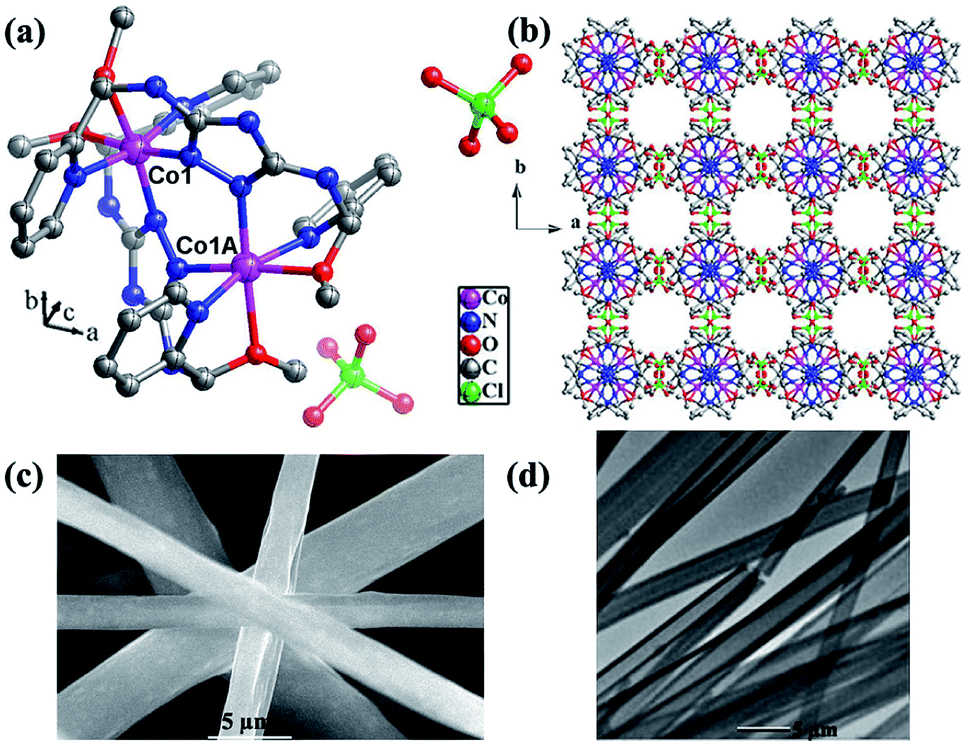 | ||
| Fig. 1 (a) The structure of {CoII2} catalyst. (b) The 3D supramolecular packing mode along c axis showing supramolecular porosity. (c) The SEM and (d) TEM images of {CoII2} catalyst. | ||
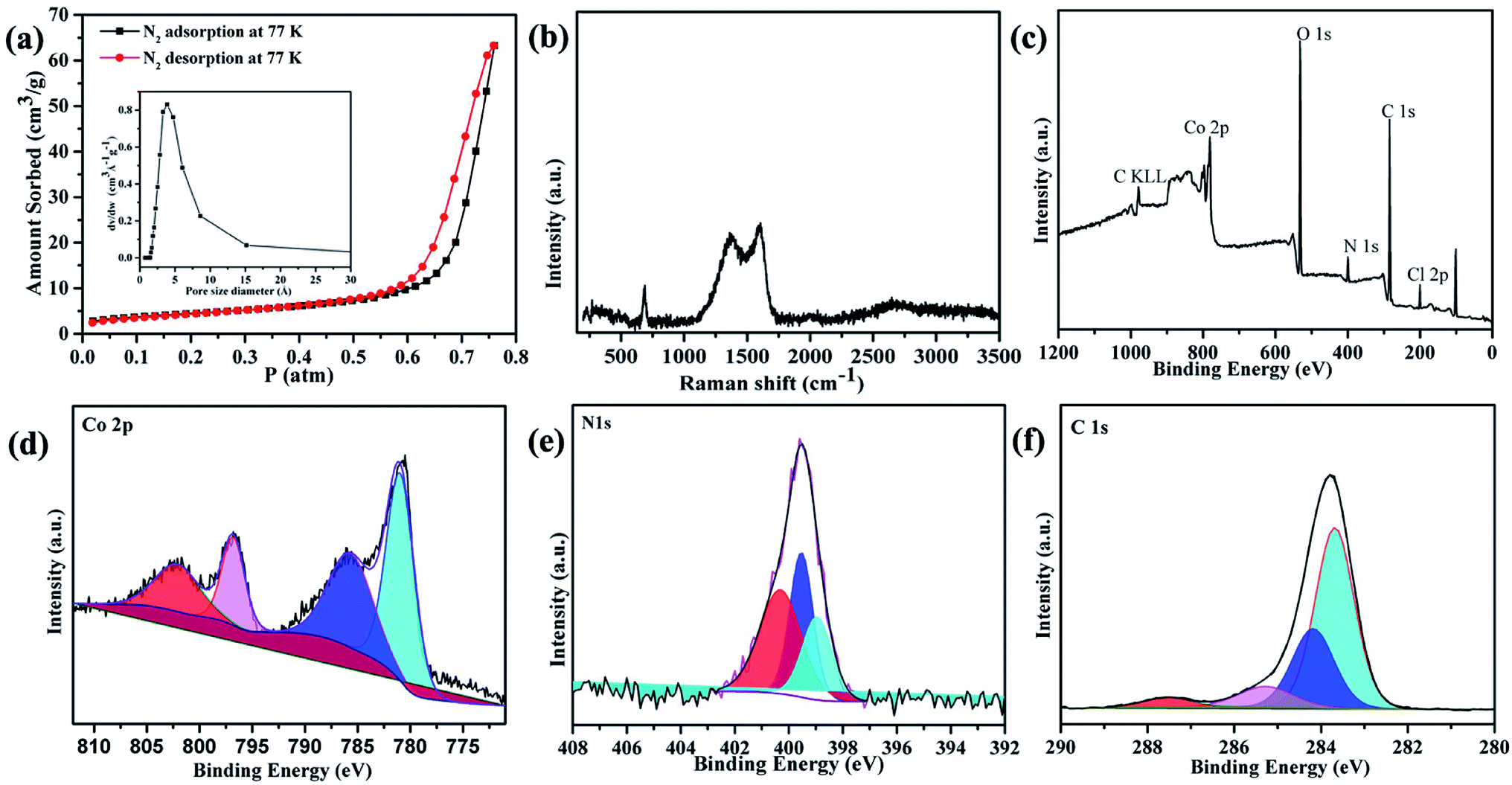 | ||
| Fig. 2 (a) N2 adsorption at 77 K and 1 atm (inset: the pore size diameter). (b) Raman spectrum. (c–f) XPS spectrum of {CoII2} catalyst. | ||
The Raman spectrum further confirms the catalyst is in crystalline state and composed of various functional modules (Fig. 2b).15 It displays five main peaks locating at 268, 690, 1365, 1603, and 2656 cm−1, respectively. The low-frequency peak at 268 cm−1 is mainly attributed to the lattice vibrations of the {CoII2} catalyst.15 In the fingerprint region, the peaks at 690 and 1365 cm−1 are mainly due to the {CoII2} cluster breathing, pyridine and triazole rings contracting, and C–H stretching; the peak at 1603 cm−1 may result from pyridine and triazole rings breathing and C–H bending.15 The high-frequency peak at 2656 cm−1 can be attributed to the C–O stretching of the organic groups. The C–N–C stretching is overlapped by strong intensities at 1365 and 1603 cm−1.
To further prove the chemical composition and oxidation states of the catalyst, the XPS were measured (Fig. 2c–f). The overall spectrum shows there exist Co, C, N, O and Cl elements (Fig. 2c). The high resolution of Co2p spectrum reveals it contains four typical characteristic peaks which are assigned to Co(II)2p3/2 (780.2 eV), Co(II)2p1/2 (796 eV), and two satellite peaks (786 and 802 eV) (Fig. 2d), respectively.16,17 The high resolution of N1s spectra are divided into three peaks (Fig. 2e). The peak at 398.96 eV is the pyridinic N species (24.08%), 399.53 eV is the amino group N species (33.15%), and 400.31 eV is the triazole N species (42.77%), respectively.16,17 As references reported, the pyridinic-N and triazole-N can synergistically offer ORR active sites.16,17 The high resolution of C1s spectrum can be deconvoluted into four peaks at 283.68, 284.18, 287.52, and 285.29 eV (Fig. 2f), regarding as C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN/C–O, and C–N/CO, respectively.16,17 The XPS results are aligned with the X-ray single crystal structure analysis.13
C, CN/C–O, and C–N/CO, respectively.16,17 The XPS results are aligned with the X-ray single crystal structure analysis.13
3.2 ORR electrocatalytic activities
The stability of the catalyst in the ink is important. We soaked the crystals of as-synthesised catalyst in a similar mixture solution including the same ratio of H2O/iso-propanol/ethanol under supersound. After 2 days, we filtered the catalyst and carried out the PXRD test. The PXRD patterns indicate that the soaked catalyst also has crystalline forms (Fig. S3, ESI†), which can proved the stability and insolubility of the catalyst in the ink.Then, the ORR activities of the crystal {CoII2} cluster and 20% Pt/C were both tested under the same condition. The CV curves exhibit an obvious oxygen redox peak in O2-saturated electrolyte compared with N2 environment (Fig. 3a), which reveals the potential ORR activities of the {CoII2} catalyst. From LSV curves of the {CoII2} catalyst (Fig. 3b), the limiting current densities (JL) increase evidently along with the raising rotation rates from 400 to 1600 rpm. JL reaches maximum value of 4.43 mA cm−2 at highest rotation rate 1600 rpm, implying the fastest O2 diffuse around the electrode surface. As seen, the onset potential (Eonset) and half-wave potential (E1/2) is 1.05 and 0.79 mV for the {CoII2} catalyst, respectively. For comparison, the LSV curves of commercial Pt/C in same condition show similar trend (Fig. 3c). From the curves, the activities of {CoII2} catalyst are a little weaker than Pt/C catalyst, whereas the maximum JL, Eonset, and E1/2 of Pt/C is 4.76 mA cm−2, 1.15 mV, and 0.93 mV for Pt/C, respectively. From references, crystalline coordinated clusters possessing high ORR activities are rare.9–11 Furthermore, Koutecky–Levich (K–L) equation was used to evaluate kinetic progress of the {CoII2} catalyst (Fig. 3d).18 From fitted data at various potentials from 0 to 0.20 V, the K–L curves present fine linearity with almost the same slopes. By calculated, the consistent electron transfer number (n) is 3.8, theoretically corresponding to a four-electrons reduction pathway. The good linearity also illustrates the ORR of the {CoII2} catalyst following the first-order reaction kinetics. The kinetic current density (JK) of the {CoII2} catalyst is ca. 20.72 mA cm−2 according to K–L curves at 0.20 V. The n and JK is near Pt/C (where n = 4.0, JK = 22.33 mA cm−2, Fig. 3e and f) at the same condition, which further confirms the good ORR activities of the {CoII2} catalyst. The various electrochemical parameters of the {CoII2} catalyst for evaluating the ORR performance containing Eonset, E1/2, n, and JK, are listed in column in order to help comparing with Pt/C (Fig. 3f).
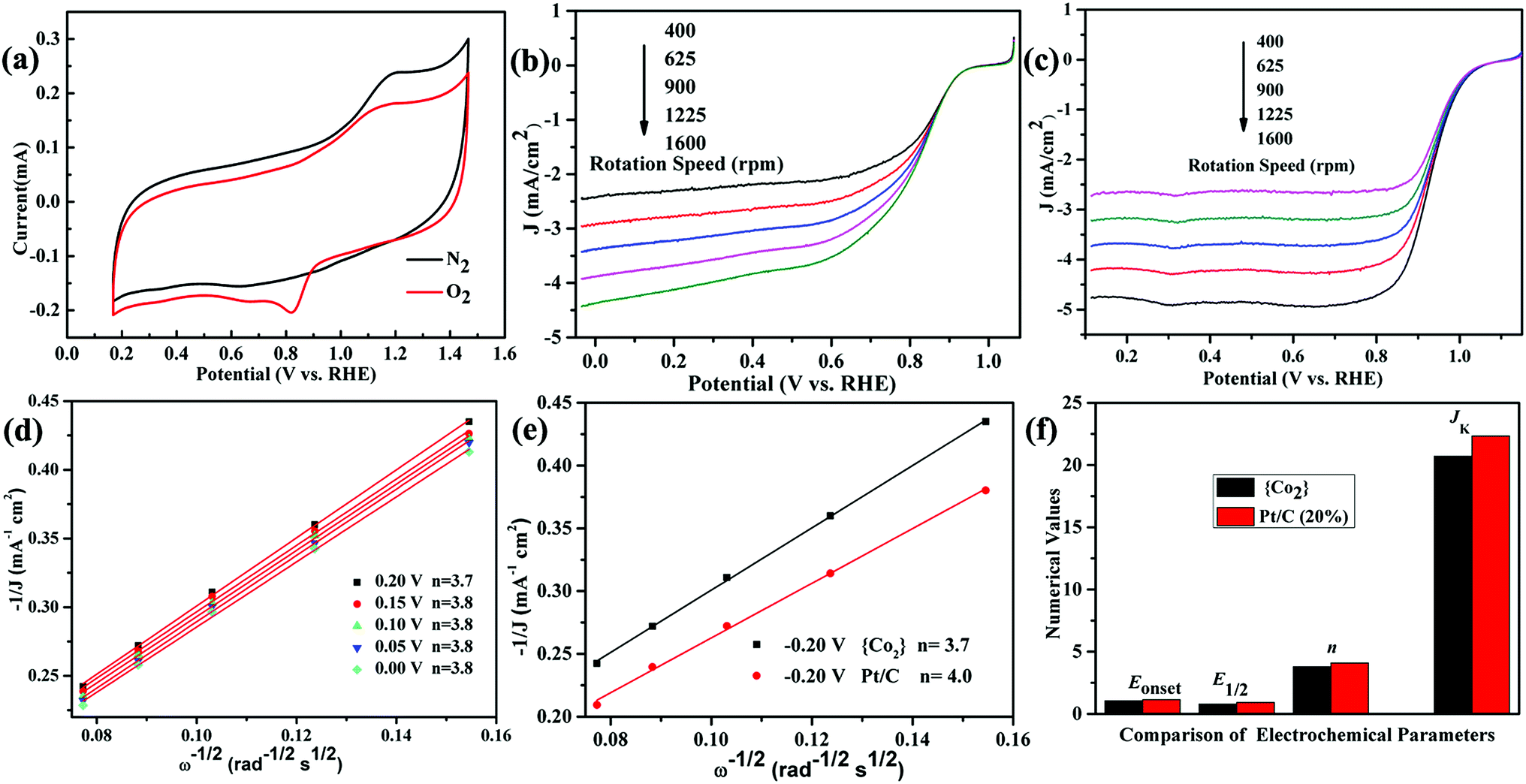 | ||
| Fig. 3 (a) CV curves of the {CoII2} catalyst. (b) and (c) LSV curves of the {CoII2} catalyst and commercial Pt/C. (d) K–L plots of the {CoII2} catalyst at different potentials from 0 to 0.20 V. (e) Comparison K–L plots of the {CoII2} catalyst and commercial Pt/C at 0.2 V. (f) The comparison various electrochemical parameters including Eonset, E1/2, n and JK of the {CoII2} catalyst and commercial Pt/C. | ||
The long-term durability of {CoII2} catalyst and the 20% commercial Pt/C was also evaluated by chronoamperometric measurements in O2 saturated 0.1 M KOH solutions. As revealed in the current–time (i–t) curves (Fig. 4a), {CoII2} catalyst retains 83% of the initial current after 36000 seconds, whereas the value for the commercial Pt/C is only 73%, indicating that {CoII2} catalyst exhibits better stability than the Pt/C in alkaline solution. Moreover, the EIS was further performed to investigate the catalytic kinetics of {CoII2} catalyst and commercial Pt/C. As shown in Fig. 4b, the Nyquist plots and the simulated equivalent circuits were presented for the two catalysts. As known, the radius of Nyquist plot can reflect the charge transfer resistance (Rp) and further to evaluate the ORR electrocatalytic activity. The Rp of {CoII2} catalyst (432 Ω) is higher than commercial Pt/C (228 Ω), which is consistent with the slightly worse ORR performance of {CoII2} catalyst compared to commercial Pt/C. Hence, the {CoII2} catalyst is one efficient ORR catalyst with good stability and electrocatalytic activity.
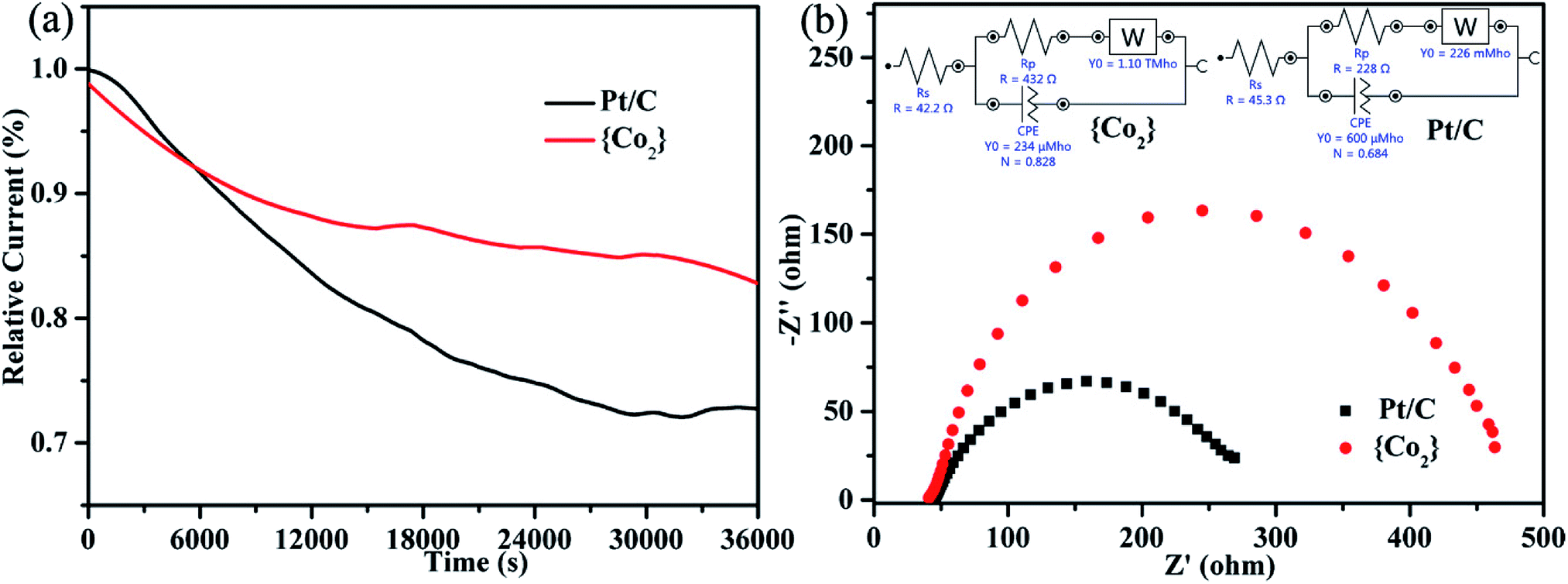 | ||
| Fig. 4 (a) The current–time (i–t) chronoamperometry curves of {CoII2} catalyst and commercial Pt/C in O2-saturated 0.1 M KOH solution.(b) Nyquist plots of EIS for {CoII2} catalyst and commercial Pt/C (inset: simulated equivalent circuits). | ||
For comparison, we also tested the ORR activities of the free ligand Hpmat in the same condition. As shown from the Fig. S4 (ESI†), the corresponding parameters Eonset, E1/2, n, and JK are worse than the {CoII2} catalyst. So it can be speculated that the CoII-coordinated centres of the {CoII2} cluster will be the catalytic active sites. Combination of X-ray single crystal structure analysis, XPS results, the worse ORR activities of the free ligand, and the references including experimental and theoretical insights,7,8,19,20 it can be speculation that the ORR active sites of the {CoII2} catalyst are probable the {CoN4O2} coordinated cores.7,8,10,12,19,20
Stated thus, the good ORR activities of {CoII2} catalyst may be attributed to the following two factors: on one hand, the appropriate porosity and the BET surface area provide effective mass transport pathway for the defined CoII-coordinated {CoN4O2} active sites.7,8,10,12,16,17,19,20 On the other hand, the abundant pyridinic-N and triazole-N synergistically benefit the ORR activities.7,8,16,17,19,20 These two types of N not only can enhance the efficient electron transfer, but also lead to more defects and positive {CoN4O2} active sites, which can both improve the ORR performance.7,8,10,12,16,17,19,20 To date, most ORR researches are focus on pyrolysis various precursors (such as CMCs and MOFs) to obtain {MNx} catalysts, their precise active sites and the catalytic mechanism are still not very clear, although they show higher ORR performance. In comparison, unpyrolyzed coordinated polymers (including CMCs and MOFs) often display slightly worse ORR activities, but they can provide precise active sites and the whole structural information, which can help researcher to design and fabricate efficient ORR catalysts to improve their activities.
3.3 The proposed ORR electrocatalytic mechanism
Considering the nearly symmetric structure and coordination environment of the two CoII ions, a single cobalt center-mediated reduction mechanism was proposed, which abides by a four-electron reduction pathway (see Fig. 5).10–12,21–25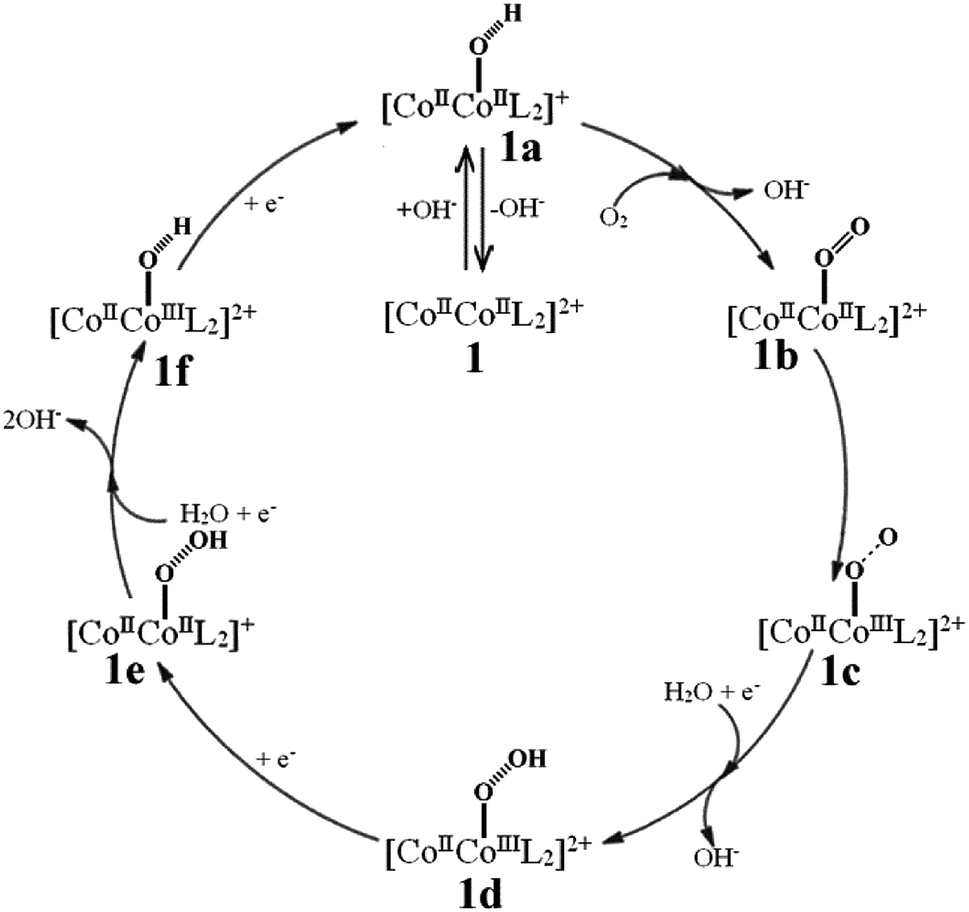 | ||
| Fig. 5 The proposed ORR mechanism of the {CoII2} catalyst in alkaline medium. | ||
(1): [CoIICoIIL2]2+ (abbr. 1) catalyst captures one OH− in a alkaline solution to form a [CoIICoII(OH)L2]+ intermediate abbreviated as 1a, the reaction equation is:
| [CoIICoIIL2]2+ + OH− → [CoIICoII(OH)L2]+ | (2a) |
(2): 1a containing [CoIICoII(OH)L2]+ adsorbs one O2 molecule on active sites and releases one OH−, generating a [CoIICoII(OO)L2]2+ intermediate abbreviated as 1b, the reaction equation is:
|
[CoIICoII(OH)L2]+ + O2 → [CoIICoII(OO)L2]2+ + OH−
| (2b) |
(3): O2 is feasible to oxidize one CoII ion to CoIII ion, and transfers to a [CoIICoIII(O–O)L2]2+ intermediate abbreviated as 1c, the reaction equation is:
|
[CoIICoII(OO)L2]2+ → [CoIICoIII(O–O)L2]2+
| (2c) |
(4): [CoIICoIII(O–O)L2]2+ fixes one H2O molecule and obtains one electron at the same time releases one OH−, generating a [CoIICoIII(O–OH)L2]2+ intermediate abbreviated as 1d, the reaction equation is:
| [CoIICoIII(O–O)L2]2+ + H2O + e− → [CoIICoIII(O–OH)L2]2+ + OH− | (2d) |
(5): [CoIICoIII(O–OH)L2]2+ continuously obtains one electron to reduce the CoIII to CoII and generates a [CoIICoII(O–OH)L2]+ intermediate abbreviated as 1e, the reaction equation is:
| [CoIICoIII(O–OH)L2]2+ + e− → [CoIICoII(O–OH)L2]+ | (2e) |
(6): [CoIICoII(O–OH)L2]+ further fixes one H2O molecule and obtains one electron at the same time releases two OH−, generating a [CoIICoIII(OH)L2]2+ intermediate abbreviated as 1f, the reaction equation is:
| [CoIICoII(O–OH)L2]+ + H2O + e− → [CoIICoIII(OH)L2]2+ + 2OH− | (2f) |
(7): [CoIICoIII(OH)L2]2+ further obtains one electron to reduce CoIII ion into CoII ion, returning to [CoIICoII(OH)L2]+ 1a intermediate, the reaction equation is:
| [CoIICoIII(OH)L2]2+ + e− → [CoIICoII(OH)L2]+ | (2g) |
(8): the [CoIICoII(OH)L2]+ releases the OH− and goes back to [CoIICoIIL2]2+ 1, completing one catalytic cycle progress:
| [CoIICoII(OH)L2]+ → [CoIICoIIL2]2+ + OH− | (2h) |
4. Conclusion
In conclusion, a Pt-free dinuclear {CoII2} cluster was elaborately selected to research its ORR catalytic activities inspired by dinuclear metalloenzymtic catalysis in biological systems. The {CoII2} catalyst possesses defined crystal structure, precised {CoN4O2} potential catalytic active centers, N-rich ligand involving pyridinic-N and triazole-N, and thermal stability. These features endow the {CoII2} catalyst with a nice ORR electrocatalytic performance by a nearly 4-electrons reduction pathway. Considering the visible crystal structure, a single cobalt center-mediated catalytic mechanism was also proposed to elucidate the ORR process, which can help researcher to design and fabricate efficient ORR catalysts to improve their activities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NNSF of China (21771095, 21571092 and 21601079), the NSF of Shandong Province (ZR2017JL013), and the Youth Innovation Team of Shandong Colleges and Universities (2019KJC027).References
- (a) R. Devanathan, Energy Environ. Sci., 2008, 1, 101–119 RSC; (b) X. X. Wang, D. A. Cullen, Y. T. Pan, S. Hwang, M. Y. Wang, Z. X. Feng, J. Y. Wang, M. H. Engelhard, H. G. Zhang, Y. H. He, Y. Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed.
- (a) T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC; (b) P. Tan, B. Chen, H. R. Xu, H. C. Zhang, W. Z. Cai, M. Ni, M. L. Liu and Z. P. Shao, Energy Environ. Sci., 2017, 10, 2056–2080 RSC; (c) M. H. Yu, Z. K. Wang, C. Hou, Z. L. Wang, C. L. Liang, C. Y. Zhao, Y. X. Tong, X. H. Lu and S. H. Yang, Adv. Mater., 2017, 29, 1602868 CrossRef PubMed.
- (a) J. B. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed; (b) L. Zhang, L. T. Roling, X. Wang, M. Vara, M. F. Chi, J. Y. Liu, S. II Choi, J. Park, J. A. Herron, Z. X. Xie, M. Mavrikakis and Y. N. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- (a) L. Z. Bu, N. Zhang, S. J. Guo, X. Zhang, J. Li, J. L. Yao, T. Wu, G. Lu, J. Y. Ma, D. Su and X. Q. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed; (b) W. H. Lai, B. W. Zhang, Z. P. Hu, X. M. Qu, Y. X. Jiang, Y. X. Wang, J. Z. Wang, H. K. Liu and S. L. Chou, Adv. Funct. Mater., 2019, 29, 1807340 CrossRef; (c) C. Chen, Y. J. Kang, Z. Y. Huo, Z. W. Zhu, W. Y. Huang, H. L. L. Xin, J. D. nyder, D. G. Li, J. A. Herron, M. Mavrikakis, M. F. Chi, K. L. More, Y. D. Li, N. M. Markovic, G. A. Somorjai, P. D. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed; (d) C. Wang, Y. Hou, J. Kim and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 6333–6335 CrossRef CAS PubMed.
- (a) W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC; (b) D. F. Yan, Y. X. Li, J. Huo, R. Chen, L. M. Dai and S. Y. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed; (c) H. J. Qiu, P. Du, K. L. Hu, J. J. Gao, H. L. Li, P. Liu, T. Ina, K. Ohara, Y. Ito and M. W. Chen, Adv. Mater., 2019, 31, 1900843 CrossRef PubMed; (d) W. Xia, A. Mahmood, Z. B. Liang, R. Q. Zou and S. J. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650–2676 CrossRef CAS PubMed.
- (a) C. G. Hu and L. M. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736–11758 CrossRef CAS PubMed; (b) N. Y. Cheng, L. Ren, X. Xu, Y. Du and S. X. Dou, Adv. Energy Mater., 2018, 8, 1801257 CrossRef; (c) Y. Y. Huang, Y. Q. Wang, C. Tang, J. Wang, Q. Zhang, Y. B. Wang and J. T. Zhang, Adv. Mater., 2019, 31, 1803800 CrossRef PubMed; (d) H. T. Sun, L. Mei, J. F. Liang, Z. P. Zhao, C. Lee, H. L. Fei, M. N. Ding, J. Lau, M. F. Li, C. Wang, X. Xu, G. L. Hao, B. Papandrea, I. Shakir, B. Dunn, Y. Huang and X. F. Duan, Science, 2017, 356, 599–604 CrossRef CAS PubMed.
- (a) Q. T. Liu, X. F. Liu, L. R. Zheng and J. L. Shui, Angew. Chem., Int. Ed., 2018, 57, 1204–1208 CrossRef CAS PubMed; (b) J. Y. Liu, Y. Liu, P. Li, L. H. Wang, H. R. Zhang, H. Liu, J. L. Liu, Y. X. Wang, W. Tian, X. B. Wang, Z. T. Li and M. B. Wu, Carbon, 2018, 126, 1–8 CrossRef CAS; (c) P. Peng, L. Shi, F. Huo, C. X. Mi, X. H. Wu, S. J. Zhang and Z. H. Xiang, Sci. Adv., 2019, 5, eaaw2322 CrossRef PubMed; (d) F. L. Li, P. T. Wang, X. Q. Huang, D. J. Young, H. F. Wang, P. Braunstein and J. P. Lang, Angew. Chem., Int. Ed., 2019, 58, 7051–7056 CrossRef CAS PubMed.
- (a) M. D. Zhang, Q. B. Dai, H. G. Zheng, M. D. Chen and L. M. Dai, Adv. Mater., 2018, 30, 1705431 CrossRef PubMed; (b) J. Zhou, Y. B. Dou, A. W. Zhou, R. M. Guo, M. J. Zhao and J. R. Li, Adv. Energy Mater., 2017, 7, 1602643 CrossRef; (c) Y. P. Wu, J. W. Tian, S. Liu, B. Li, J. Zhao, L. F. Ma, D. S. Li, Y. Q. Lan and X. H. Bu, Angew. Chem., Int. Ed., 2019, 58, 12185–12189 CrossRef CAS PubMed; (d) C. H. Liu, Y. J. Tang, X. L. Wang, W. Huang, S. L. Li, L. Z. Dong and Y. Q. Lan, J. Mater. Chem. A, 2016, 4, 18100–18106 RSC.
- E. M. Miner, T. Fukushima, D. Sheberla, L. Sun, Y. Surendranath and M. Dincă, Nat. Commun., 2016, 7, 10942 CrossRef CAS PubMed.
- J. Han, Y. J. Sa, Y. Shim, M. Choi, N. Park, S. H. Joo and S. Park, Angew. Chem., Int. Ed., 2015, 54, 12622–12626 CrossRef CAS PubMed.
- C. D. Giovanni, C. Gimbert-Suriñach, M. Nippe, J. Benet-Buchholz, J. R. Long, X. Sala and A. Llobet, Chem.–Eur. J., 2016, 22, 361–369 CrossRef PubMed.
- P. Mani, A. Sheelam, S. Das, G. X. Wang, V. K. Ramani, K. Ramanujam, S. K. Pati and S. Mandal, ACS Omega, 2018, 3, 3830–3834 CrossRef CAS PubMed.
- H. Y. Ma, Y. W. Li, S. Y. Zeng, S. N. Wang, N. Yang and X. Shao, J. Cluster Sci., 2016, 27, 1945–1952 CrossRef CAS.
- (a) Y. W. Li, J. Xu, D. C. Li, J. M. Dou, H. Yan, T. L. Hu and X. H. Bu, Chem. Commun., 2015, 51, 14211–14214 RSC; (b) Y. W. Li, H. Yan, T. L. Hu, H. Y. Ma, D. C. Li, S. N. Wang, Q. X. Yao, J. M. Dou, J. Xu and X. H. Bu, Chem. Commun., 2017, 53, 2394–2397 RSC; (c) D. S. Zhang, Y. Z. Zhang, X. L. Zhang, F. Wang, J. Zhang, H. Hu, J. Gao, H. Yan, H. L. Liu, H. Y. Ma, L. L. Geng and Y. W. Li, ACS Appl. Mater. Interfaces, 2019, 11, 20104–20109 CrossRef CAS PubMed; (d) H. Y. Ma, Y. Z. Zhang, H. Yan, W. J. Zhang, Y. W. Li, S. N. Wang, D. C. Li, J. M. Dou and J. R. Li, Dalton Trans., 2019, 48, 13541–13545 RSC.
- (a) Y. X. Shen, B. Shan, H. Cai, Y. Qin, A. Agarwal, D. B. Trivedi, B. Chen, L. Liu, H. L. Zhuang, B. Mu and S. Tongay, Adv. Mater., 2018, 30, 1802497 CrossRef PubMed; (b) C. Shi, M. W. Chen, X. Han, Y. F. Bi, L. L. Huang, K. Zhou and Z. P. Zheng, Inorg. Chem. Front., 2018, 5, 1329–1335 RSC.
- (a) R. Z. Zhang, S. J. He, Y. Z. Lu and W. Chen, J. Mater. Chem. A, 2015, 3, 3559–3567 RSC; (b) Y. Gartia, C. M. Parnell, F. Watanabe, P. Szwedo, A. S. Biris, N. Peddi, Z. A. Nima and A. Ghosh, ACS Sustainable Chem. Eng., 2015, 3, 97–102 CrossRef CAS; (c) X. Qing, J. J Shi, C. Y. Ma, M. Y. Fan, Z. Y. Bai, Z. W. Chen, J. L. Qiao and J. J. Zhang, J. Power Sources, 2014, 266, 88–98 CrossRef CAS.
- (a) J. Y. Liu, L. Xu, Y. L. Deng, Y. L. Deng, X. W. Zhu, J. J. Deng, J. B. Lian, J. J. Wu, J. C. Qian, H. Xu, S. Q. Yuan, H. M. Li and P. M. Ajayan, J. Mater. Chem. A, 2019, 7, 14291–14301 RSC; (b) D. R. Kumar, S. Kesavan, M. L. Baynosa and J. J. Shim, Electrochim. Acta, 2017, 246, 1131–1140 CrossRef CAS; (c) H. Koshikawa, S. Nakanishi, K. Hashimoto and K. Kamiya, Electrochim. Acta, 2015, 180, 173–177 CrossRef CAS.
- (a) P. Strasser, Acc. Chem. Res., 2016, 49, 2658–2668 CrossRef CAS PubMed; (b) R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- (a) J. Gao, N. Ma, Y. M. Zheng, J. F. Zhang, J. Z. Gui, C. K. Guo, H. Q. An, X. Y. Tan, Z. Yin and D. Ma, ChemCatChem, 2017, 9, 1601–1609 CrossRef CAS; (b) H. Yang, X. Chen, W. T. Chen, Q. Wang, N. C. Cuello, A. Nafady, A. M. Al-Enizi, G. I. N. Waterhouse, G. A. Goenaga, T. A. Zawodzinski, P. E. Kruger, J. E. Clements, J. Zhang, H. Tian, S. G. Telfer and S. Q. Ma, ACS Nano, 2019, 13, 8087–8098 CrossRef CAS PubMed.
- (a) Y. Tong, P. Z. Chen, T. P. Zhou, K. Xu, W. S. Chu, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 7121–7125 CrossRef CAS PubMed; (b) X. X. Sun, K. Li, C. Yin, Y. Wang, M. G. Jiao, F. He, X. W. Ba, H. Tang and Z. J. Wu, Carbon, 2016, 108, 541–550 CrossRef CAS.
- (a) C. C. Ren, H. B. Li, R. Li, S. L. Xu, D. H. Wei, W. J. Kang, L. Wang, L. P. Jia, B. C. Yang and J. F. Liu, RSC Adv., 2016, 6, 33302–33307 RSC; (b) X. Mao, C. Y. Ling, C. Tang, C. Yan, Z. H. Zhu and A. J. Du, J. Catal., 2018, 367, 206–211 CrossRef CAS; (c) H. C. Li, Y. J. Zhang, X. Hu, W. J. Liu, J. J. Chen and H. Q. Yu, Adv. Energy Mater., 2018, 8, 1702734 CrossRef; (d) Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS PubMed.
- (a) T. Ouyang, H. H. Huang, J. W. Wang, D. C. Zhong and T. B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed; (b) T. Ouyang, H. J. Wang, H. H. Huang, J. W. Wang, S. Guo, W. J. Liu, D. C. Zhong and T. B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16480–16485 CrossRef CAS PubMed.
- (a) S. Li, L. Zhang, J. Kima, M. Pan, Z. Shi and J. J. Zhang, Electrochim. Acta, 2010, 55, 7346–7353 CrossRef CAS; (b) M. Lions, J. B. Tommasino, R. Chattot, B. Abeykoon, N. Guillou, T. Devic, A. Demessence, L. Cardenas, F. Maillard and A. Fateeva, Chem. Commun., 2017, 53, 6496–6499 RSC; (c) Z. H. Huang, N. H. Xie, M. Zhang and B. Q. Xu, ChemSusChem, 2019, 12, 200–207 CrossRef CAS PubMed; (d) H. X. Zhong, K. H. Ly, M. C. Wang, Y. Krupskaya, X. C. Han, J. C. Zhang, J. Zhang, V. Kataev, B. Bgchner, I. M. Weidinger, S. Kaskel, P. Liu, M. W. Chen, R. H. Dong and X. L. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677–10682 CrossRef CAS PubMed; (e) F. H. Sun and X. Chen, Electrochem. Commun., 2017, 82, 89–92 CrossRef CAS; (f) Y. Zhou, Y. F. Xing, J. Wen, H. B. Ma, F. B. Wang and X. H. Xia, Sci. Bull., 2019, 64, 1158–1166 CrossRef CAS; (g) N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed.
- (a) H. Y. He, M. Wang, J. S. Zhao and Y. Zhang, Chem. Eng. J., 2017, 316, 680–691 CrossRef CAS; (b) L. Gu, Y. Chu, H. M. Du, Y. Zhang, J. S. Zhao and Y. Xie, Front. Chem., 2019, 7, 622 CrossRef PubMed; (c) Y. Y. Zhao, Y. Chu, X. P. Ju, J. S. Zhao, L. Q. Kong and Y. Zhang, Catalysts, 2018, 8, 53 CrossRef; (d) Y. Q. Lu, X. L. Wang, M. Wang, L. Q. Kong and J. S. Zhao, Electrochim. Acta, 2015, 180, 86–95 CrossRef CAS; (e) Y. Chu, L. Gu, H. H. Du, K. G. Qu, Y. Zhang, J. S. Zhao and Y. Xie, Int. J. Hydrogen Energy, 2018, 43, 21810–21823 CrossRef CAS.
- (a) R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed; (b) W. J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L. J. Zhang, J. Q. Wang, J. S. Hu, Z. D. Wei and L. J. Wan, J. Am. Chem. Soc., 2016, 138, 3570–3578 CrossRef CAS PubMed; (c) K. Iwase, T. Yoshioka, S. Nakanishi, K. Hashimoto and K. Kamiya, Angew. Chem., Int. Ed., 2015, 54, 11068–11072 CrossRef CAS PubMed; (d) M. Kato, K. Kimijima, M. Shibata, H. Notsu, K. Ogino, K. Inokuma, N. Ohta, H. Uehara, Y. Uemura, N. Oyaizu, T. Ohba, S. Takakusagi, K. Asakura and I. Yagi, Phys. Chem. Chem. Phys., 2015, 17, 8638–8641 RSC; (e) Y. Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. K. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. L. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08068f |
| This journal is © The Royal Society of Chemistry 2019 |