
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of MeBmt and related derivatives via syn-selective ATH-DKR†
Adam Rolta,
Paul M. O'Neillb,
T. Jake Liangc and
Andrew V. Stachulski *b
*b
aDepartment of Biochemistry, University of Oxford, OX1 3QU, UK
bThe Robert Robinson Laboratories, Department of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK. E-mail: stachuls@liv.ac.uk
cLiver Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, 10 Center Drive, Bethesda, Maryland 20892, USA
First published on 5th December 2019
Abstract
The unusual α-amino, β-hydroxy acid MeBmt is a key structural feature of cyclosporin A, an important naturally occurring immunosuppressant and antiviral agent. We present a convergent synthesis of MeBmt which relies on new aspects of dynamic kinetic resolution (DKR) to establish simultaneously the chirality at C(2) and C(3). We also show that this route is applicable to the synthesis of other derivatives.
The α-amino-β-hydroxy unit is a familiar structural motif in many important natural products, notably the proteinogenic amino-acids serine and threonine, glycosphingolipids1 and more complex structures such as the polyoxins.2 We were particularly interested in the cyclic undecapeptide cyclosporin A (CsA) 1, which contains the unusual amino acid [(2S,3R,4R,6E)-3-hydroxy-4-methyl-2-(methylamino)-6-octenoic acid] (MeBmt 2) at position 1, shown in Fig. 1.
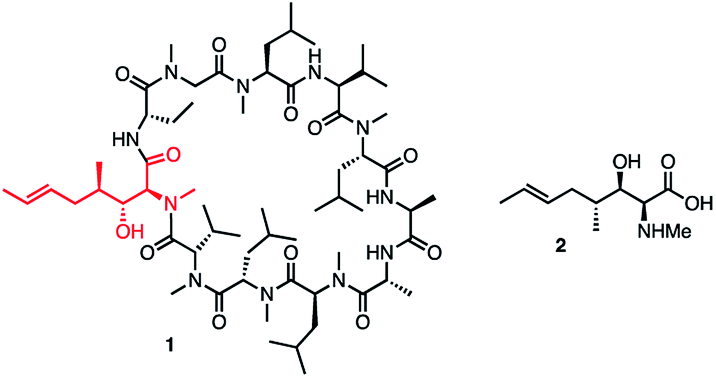 | ||
| Fig. 1 Structures of CsA 1 and MeBmt 2. | ||
While CsA 1, first attracted attention as a valuable immuno-suppressant3 used especially following transplant surgery, it was later found to have potent antiviral activity, notably against hepatitis C virus (HCV) via its inhibition of the proline cis–trans isomerase, cyclophilin A.4–6 Synthetic modification can uncouple the antiviral effects from the immunosuppressive effects,7,8 thus Debio-025 (Alisporivir), a CsA analogue modified at positions 3 and 4, retains excellent activity vs. HCV (IC50 = 30 nM) and is essentially non-immunosuppressive.9 Additionally, reported analogues of 1 modified at the MeBmt residue also show antiviral activity while being essentially inactive as immunosuppressants.7 Previous syntheses of 2 were too lengthy and linear to facilitate a medicinal chemistry campaign of CsA around 2.10–13 We therefore sought to develop a short effective synthesis of 2, which would readily permit the synthesis of position 1 analogues of CsA without relying on partial synthesis. Thus, our retrosynthesis of MeBmt 2 is shown in Scheme 1, it relies on the syn-selective dynamic kinetic resolution (DKR) of a β-ketoester precursor 3, which is in turn accessible via a crossed Claisen condensation14 of an activated form of carboxylic acid 4 with protected sarcosine ester 5.
 | ||
| Scheme 1 Proposed disconnections in the synthesis of MeBmt. R1/2 = protecting groups. | ||
Several groups have effected DKR under catalytic asymmetric transfer hydrogenation (ATH)15 conditions, the stereochemical outcome is dependent on substrate structure. The synthesis of β-hydroxy amino acid derivatives via ATH DKR is usually undertaken via reduction from the prerequisite β-keto methyl ester and the stereochemical outcome tends to the 2,3-anti-product.16,17 This anti diastereoselectivity is proposed to be the product of intramolecular hydrogen bonding.18 In contrast, γ-aryl-N-Me substrates underwent ATH DKR to give the desired syn-products,19 though this had not been demonstrated with γ-alkyl substrates. We therefore set out to study these ATH-DKR conditions, initially on a model isobutyryl substrate 6a, and we now report our findings, leading eventually to a concise synthesis of MeBmt.
Compound 6a (Scheme 2) was prepared by a crossed Claisen condensation between isobutyryl chloride and sarcosine derivative 7a (see ESI†); alternatively, using the corresponding glycine ester 7b, 6b was readily obtained.14
 | ||
| Scheme 2 Synthesis of α-acylamino-β-keto-esters and ATH DKR discrepancy. (i) TiCl4, Bu3N, N-Me imidazole, CH2Cl2, −40 °C; (ii) HCO2Na, TBAI, CH2Cl2/H2O; 3% Ru(p-cymene)[(R,R)-TsDPEN], 20 °C, 20 h. Isolated yields are shown. | ||
Branching of the terminal alkyl substituent in 6a/6b has been shown to improve stereoselectivity in ATH DKR of similar substrates18 thus 6a/6b are good models for MeBmt. Substrate 6b was reduced efficiently via ATH DKR to the (2R,3R)-anti product 8b (Table 1), however under these conditions the N-Me substrate 6a, which was isolated almost entirely as the enol tautomer (>95%, NMR), was unreactive towards ATH DKR. Attempted optimisation of the ATH DKR step with increased catalyst loading, elevated temperatures, and replacement of N-benzoyl with other N-protecting groups (viz. CBz (6c)) and alternative ATH DKR conditions16,20,21 led to the same result. In contrast, related γ-aryl-β-keto esters bearing an N-Me group are reported to be efficiently reduced.19
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Cmpd | R1 | R2 | R3 | Keto![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enol :enol |
Product | Isolated yield (%) | dra | erb |
| a Syn:anti ratio (1H NMR).b Ratio of (2S,3R):(2R,3S) enantiomers.c 10% [(R,R)-Teth-TsDpen], 40 h, 20 °C. |
|||||||||
| 1 | 6b | H | Bz | OMe | >95:5 |
8b | 82 | 15:85 |
ND |
| 2 | 6a | Me | Bz | OMe | >5:95 |
— | NR | ND | ND |
| 3 | 6c | Me | CBz | OMe | >5:95 |
— | NR | ND | ND |
| 4 | 13 | Me | Bz | NHPh | 28:72 |
15 | 4 | >95:5 |
ND |
| 5 | 14 | Me | FMOC | NHPh | 25:75 |
16 | 18 | >95:5 |
>99:1 |
| 6 | 9 | Me | CBz | NHPh | 30:70 |
17 | 13 | >95:5 |
>99:1 |
| 7c | 9 | Me | CBz | NHPh | 30:70 |
17 | 64 | >95:5 |
>99:1 |
 |
|||||||||

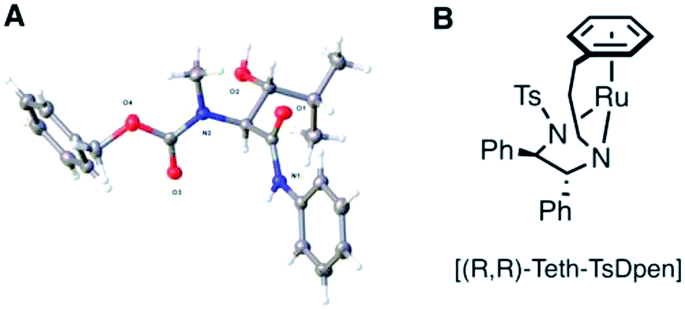
We considered that the previously proposed intramolecular hydrogen bonding18 (Fig. 2A and B) of 6b could also be responsible for the reactivity of the compounds in general through stabilisation or activation of the keto tautomer. The successful syn-reduction of a γ-aryl-N-Me, examples is, we believe, largely due to the great preference for the keto tautomer in such examples.
 | ||
| Fig. 2 (A) Proposed intramolecular H-bonding accounting for the anti-reduction of 6b18 (B) removing the H-bond leads to a completely unreactive enol tautomer; (C) restoring an intra-H bond via an anilide. | ||
In contrast, our earlier γ-alkyl-N-Me, substrate 6a existed almost entirely as the enol tautomer (>95:5 enol) by 1H NMR. We therefore proposed that a favourable intramolecular H-bond could be reintroduced by employing an anilide rather than an ester, in a substrate such as 9, Fig. 2C, ensuring a significant percentage of the necessary keto tautomer shown, indeed β-keto anilides have been utilized in ATH DKR previously though not in this context.22 Thus, the anilides were prepared according to Scheme 3.
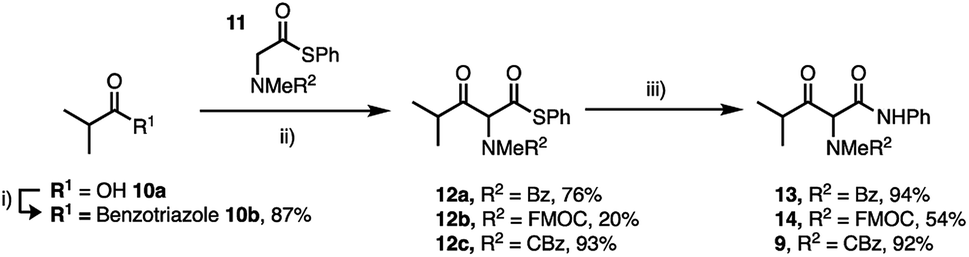 | ||
| Scheme 3 Synthesis of γ-alkyl-β-keto-anilides (i) 1H-benzotriazole, SOCl2, DCM, 20 °C (ii) MgBr2·OEt2, iPrNEt2, DCM, 0–20 °C, 16 h (iii) PhNH2, AgCO2CF3, THF, 20 °C, 16 h. Isolated yields are shown. | ||
Thioesters such as 11 are excellent nucleophiles in the crossed Claisen condensation with N-acyl benzotriazoles (10b) and deliver thioesters in very good yield other than FMOC derivative 12b which underwent base catalysed degradation.23 Finally, Ag+-catalysed aminolysis of the thioesters 12a–c delivered the desired anilides 9, 13 and 14 in high yield. The anilides were isolated with a more favourable keto:enol ratio of ∼30:70 depending on R2 (determined by NMR in CDCl3).
Accordingly, incorporation of the anilide in the isobutyryl series both re-establishes reactivity and reverses the original anti diastereoselectivity, delivering the syn product 15, although in low isolated yield (entries 4–6, Table 1). Other N-substituents, viz. Z, entry 5, and Fmoc, entry 6, were also compatible with these conditions, though Fmoc proceeds with much lower yields in the preceding crossed-Claisen step, presumably due to decomposition under basic conditions. To permit a practical transformation, we screened reaction conditions and a number of commercially available catalysts and ligands to increase yield and stereoselectivity17,24–28 (see ESI†). The most efficient proved to be the tethered catalyst described by Wills et al.; entry 7 and illustrated in Table 1 B, this delivered a 64% isolated yield of syn-17 after 40 h, in >95:5 dr and >99:1 er with full consumption of starting material. The full restoration of DKR reaction with just 25–30% keto tautomer in the precursor is emphasized.
γ-Alkyl-β-hydroxy anilide 17 was crystallised in a form suitable for single crystal X-ray structure determination, confirming the absolute configuration; shown in Table 1. Replacement of the anilide with benzylamide did not permit ATH DKR, presumably because of the lower acidity of the resultant NH. With the DKR step optimized for the model substrate, we proceeded to complete the synthesis of MeBmt 2, Scheme 4. Based on a method originally reported by Rich et al.29 starting from (±)-3-buten-2-ol 18, a Johnson–Claisen rearrangement followed by transformation of the crude ethyl ester gave (S,R)-phenylglycinol amide 19, which could be separated from its (S,S) diastereomer by gradient column chromatography in multigram amounts. The amide was quantitatively hydrolysed under acid catalysis aided by neighbouring OH-group participation to afford carboxylic acid (2R)-20. Conversion to the activated N-acyl-benzotriazolyl (Bt) electrophile30 (2R)-21 (stable at room temperature in air), followed by a crossed Claisen condensation with sarcosine derivative 22 (see ESI†), afforded enol thioester 23, which was converted to β-keto-anilide 24 in excellent yield under mild conditions. Syn selective ATH DKR afforded β-hydroxy-amide 25, without observable epimerization at C4 (judged by 1H NMR). Base hydrolysis of 25 led to universal deprotection,31 yielding MeBmt 2 without any detectable racemization (1H NMR).
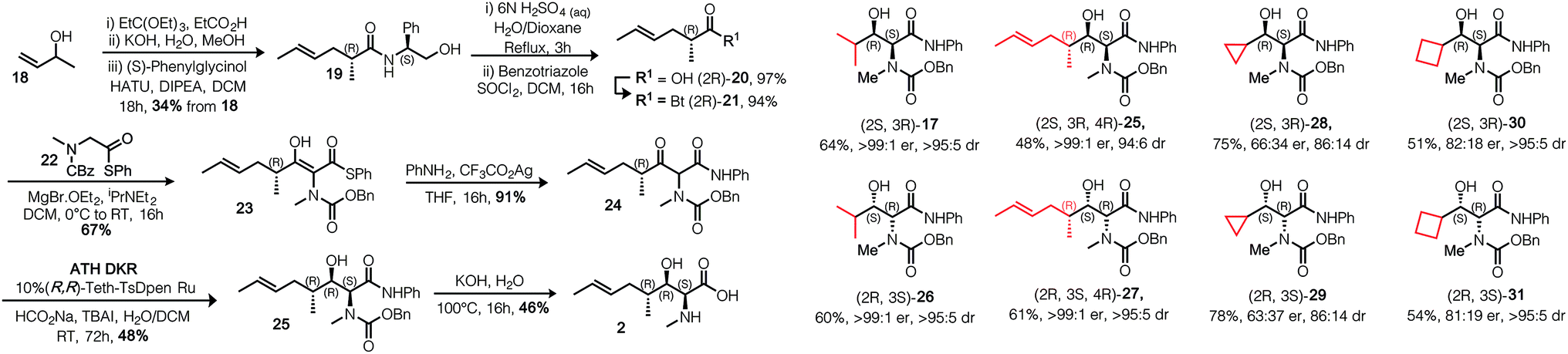 | ||
| Scheme 4 Synthesis of MeBmt via ATH DKR and demonstration of substrate scope. Analogues were synthesized according to the optimised procedures. Isolated yield following aqueous workup and column chromatography are shown, dr is expressed as syn:anti in crude product, er is expressed as major:minor in purified major diastereomer. Absolute stereochemistry in products is dependent on catalyst configuration. | ||
Finally, in Scheme 4 we demonstrate that the route is applicable to the synthesis of several γ-alkyl-α-N-Me derivatives including model isopropyl examples 17 and 26, MeBmt precursor 25 and isomer 27. Structurally novel cycloalkyl examples 28–31 were also synthesized starting from their constituent carboxylic acids (see ESI†). Absolute stereochemistry is controlled by using the opposite enantiomer of catalyst, in all cases the major and minor diastereomers could be separated by column chromatography. From this small set, it can be seen that stereoselectivity increases as the steric bulk of the side chain increases across the series. Interestingly, a match/mismatch effect in the reduction of 24 with (S,S)-Teth-TsDPEN was observed: reduction from the re-face of the molecule was slower than reduction from the si-face (cf. 25 and 27), judged by TLC, which was also reflected in the isolated yields and the diastereoselectivity. This is expected to be a consequence of the increased steric bulk of the (4R)-butenyl group in substrate 24 producing facial discrepancies in catalyst approach.
In conclusion, through analysis of intramolecular hydrogen bonding, we have expanded the scope of the ATH DKR reaction, giving access to biologically relevant γ-alkyl-β-hydroxy-α-Me-amino acids from readily accessible β-keto precursors. This was accomplished by switching from β-keto-esters to β-keto-anilides, which was sufficient to restore reactivity and reverse the original anti diastereoselectivity, delivering the syn products. Accordingly, we demonstrated a synthesis of MeBmt, 2 in five linear steps from precursor carboxylic acid (2R)-20 and have further shown that this route is also applicable for the synthesis of alternative γ-alkyl derivatives. The route is reasonably short, while also being modular and flexible in terms of side chain choice and absolute stereochemical outcome.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Wellcome Trust for an NIH-Wellcome PhD studentship grant (098256/Z/12/Z) to ACR.Notes and references
- D. Leichnitz, S. Pflanze and C. Beemelmanns, Org. Biomol. Chem., 2019, 17, 6964–6969 RSC
.
- H. Fujino, M. Nagatomo, A. Paudel, S. Panthee, H. Hamamoto, K. Sekimizu and M. Inoue, Angew. Chem., 2017, 56, 11865–11869 CrossRef CAS PubMed
- S. Mukherjee and U. Mukherjee, J. Transplant., 2009, 701464 Search PubMed
- F. Fernandes, I.-u. H. Ansari and R. Striker, PLoS One, 2010, 5, e9815 CrossRef PubMed
- A. Badillo, V. Receveur-Brechot, S. Sarrazin, F.-X. Cantrelle, F. Delolme, M.-L. Fogeron, J. Molle, R. Montserret, A. Bockmann, R. Bartenschlager, V. Lohmann, G. Lippens, S. Ricard-Blum, X. Hanoulle and F. Penin, Biochemistry, 2017, 56, 3029–3048 CrossRef CAS PubMed
- K. Inoue, K. Sekiyama, M. Yamada, T. Watanabe, H. Yasuda and M. Yoshiba, Gastroenterology, 2003, 38, 567–572 CAS
- M. Peel and A. Scribner, Bioorg. Med. Chem. Lett, 2013, 23, 4485–4492 CrossRef CAS PubMed
- A. Scribner, D. Houck, Z. Huang, S. Mosier, M. Peel and B. Scorneaux, Bioorg. Med. Chem. Lett, 2010, 20, 6542–6546 CrossRef CAS PubMed
- L. Coelmont, S. Kaptein, J. Paeshuyse, I. Vliegen, J.-M. Dumont, G. Vuagniaux and J. Neyts, Antimicrob. Agents Chemother., 2009, 53, 967–976 CrossRef CAS PubMed
- R. M. Wenger, Helv. Chim. Acta, 1983, 66, 2308–2321 CrossRef CAS
- U. Schmidt and W. Siegel, Tetrahedron Lett., 1987, 28, 2849–2852 CrossRef CAS
- M. Kirihata, Y. Nakao, M. Fukuari and I. Ichimoto, Biosci., Biotechnol., Biochem., 1995, 59, 2228–2230 CrossRef
- D. A. Evans and A. E. Weber, J. Am. Chem. Soc., 1986, 108, 6757–6761 CrossRef CAS
- T. Misaki, R. Nagase, K. Matsumoto and Y. Tanabe, J. Am. Chem. Soc., 2005, 127, 2854–2855 CrossRef CAS PubMed
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed
- B. Seashore-Ludlow, P. Villo, C. Häcker and P. Somfai, Org. Lett., 2010, 12, 5274–5277 CrossRef CAS PubMed
- P.-G. Echeverria, J. Cornil, C. Férard, A. Guérinot, J. Cossy, P. Phansavath and V. Ratovelomanana-Vidal, RSC Adv., 2015, 5, 56815–56819 RSC
- B. Seashore-Ludlow, P. Villo and P. Somfai, Chem.–Eur. J., 2012, 18, 7219–7223 CrossRef CAS PubMed
- B. Mohar, A. Valleix, J.-R. Desmurs, M. Felemez, A. Wagner and C. Mioskowski, Chem. Commun., 2001, 2572–2573 RSC
- G. Kumaraswamy, A. Narayana Murthy, V. Narayanarao, S. P. B. Vemulapalli and J. Bharatam, Org. Biomol. Chem., 2013, 11, 6751–6765 RSC
- K. M. Steward, E. C. Gentry and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 7329–7332 CrossRef CAS PubMed
- J. Limanto, S. W. Krska, B. T. Dorner, E. Vazquez, N. Yoshikawa and L. Tan, Org. Lett., 2010, 12, 512–515 CrossRef CAS PubMed
- G. Zhou, D. Lim and D. M. Coltart, Org. Lett., 2008, 10, 3809–3812 CrossRef CAS PubMed
- Z. Fang and M. Wills, J. Org. Chem., 2013, 78, 8594–8605 CrossRef CAS PubMed
- G. Sun, Z. Zhou, Z. Luo, H. Wang, L. Chen, Y. Xu, S. Li, W. Jian, J. Zeng, B. Hu, X. Han, Y. Lin and Z. Wang, Org. Lett., 2017, 19, 4339–4342 CrossRef CAS PubMed
- Z. Luo, G. Sun, Z. Zhou, G. Liu, B. Luan, Y. Lin, L. Zhang and Z. Wang, Chem. Commun., 2018, 54, 13503–13506 RSC
- A. M. Hayes, D. J. Morris, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2005, 127, 7318–7319 CrossRef CAS PubMed
- T. Touge, T. Hakamata, H. Nara, T. Kobayashi, N. Sayo, T. Saito, Y. Kayaki and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 14960–14963 CrossRef CAS PubMed
- D. T. Deyo, J. D. Aebi and D. H. Rich, Synthesis, 1988, 608–610 CrossRef CAS
- A. R. Katritzky, Y. Zhang and S. K. Singh, Synthesis, 2003, 2795–2798 CrossRef CAS
- A. Togni, S. D. Pastor and G. Rihs, Helv. Chim. Acta, 1989, 72, 1471–1478 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data of all new molecules. CCDC 1897285. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08256e |
| This journal is © The Royal Society of Chemistry 2019 |