
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual constituents from the medicinal mushroom Ganoderma lingzhi†
Zhen-Zhu Zhaoab,
Xu-Bo Lianga,
Wei-Sheng Feng ab,
Ya Wuab,
Yan-Le Zhiab,
Gui-Min Xueab,
He-Ping Chen*c and
Ji-Kai Liu*c
ab,
Ya Wuab,
Yan-Le Zhiab,
Gui-Min Xueab,
He-Ping Chen*c and
Ji-Kai Liu*c
aCollege of Pharmacy, Henan University of Chinese Medicine, Zhengzhou 450046, China
bCollaborative Innovation Center for Respiratory Disease Diagnosis and Treatment and Chinese Medicine Development of Henan Province, Zhengzhou 450046, China
cSchool of Pharmaceutical Sciences, South-Central University for Nationalities, Wuhan 430074, China. E-mail: chenhp@mail.scuec.edu.cn; liujikai@mail.scuec.edu.cn
First published on 13th November 2019
Abstract
Extensive studies have revealed that triterpenoids, meroterpenoids, and polysaccharides are the main constituents of the well-known traditional Chinese medicinal mushroom Ganoderma. In this study, we report seven previously undescribed sesquiterpenoids, including six gymnomitranes (1–6) and a novel type of sesquiterpenoid (8), together with a polyketide (7) and a known steroid (9) from the fruiting bodies of Ganoderma lingzhi, a fungus used as traditional medicine and food supplement in East Asia for ages. The structures of 1–8 were deduced by analysis of spectroscopic data, X-ray single crystal diffractions and TDDFT/ECD calculations. Compound 8 possessed an unusual 14(7→6)-cuparane scaffold. Compound 9 exhibited weak cytotoxicity against the five human cancer cell lines HL-60, MCF-7, SW480, A549, and SMMC-7721 with IC50 values of 18.0–32.3 μM. A simple structure-activity-relationship (SAR) investigation by acetylating the 5-OH of 9 (9a) suggested that the 5-OH is essential for its cytotoxicity. Additionally, the biosynthetic pathways for compounds 2 and 8 are discussed.
Introduction
Ganoderma have been used as traditional and folk medicines for thousands of years in East Asia. Ganoderma natural products have long been a hot topic for their health-preserving and therapeutic effects.1,2 The Ganoderma lucidum complex includes more than 23 described species.3 This genus has proved to be a prolific source of triterpenes, and to date, more than 400 examples have been reported.4 Ganoderma constituents have shown anti-tumour, anti-inflammatory, anti-microbial, acetylcholinesterase inhibitory,5 and pancreatic lipase inhibitory,6 and have emerging potency as anti-aging agents.7 Extensive studies have revealed that the polysaccharides and triterpenes are responsible for the anti-tumour and immunomodulatory functions.5 Notably, Ganoderma meroterpenoids have widely attracted attention in the past six years due to their intriguing structures and diverse biological activities.8 All these efforts have illuminated and enriched the chemical profile of Ganoderma secondary metabolites, which has made it possible for Ganoderma to be developed as a regular drug rather than as a dietary supplement.9,10Sesquiterpenoids are the most abundant type of secondary metabolites in mushrooms.11 However, in contrast to the attention that has been paid on the triterpenes, research on sesquiterpenes from Ganoderma fruiting bodies is still unfolding. The Ganoderma genome-sequencing, as part of the herb genomics plan, revealed that Ganoderma encodes terpene synthases for monoterpene, sesquiterpene, and diterpene backbones.12 However, hitherto only fourteen sesquiterpenoids have been reported from the fermentation products of three different Ganoderma species,4 while only two other sesquiterpenes were isolated from Ganoderma fruiting bodies, i.e. ganosinensine13 and gymnomitrane-3α,5α,9β,15-tetrol.14 Besides, the first sesquiterpene ganosinensine was then regarded as a metabolite of the symbiotic bacteria of G. sinense.
Ganoderma lingzhi, widely distributed in China, Japan, and Korea, is a new species firstly proposed by Y.-C. Dai in 201215 and then regarded as a later synonym of G. sichuanense, although there is still a controversy about the taxonomy between G. lingzhi and G. sichuanense.16,17 Comparing with the other Ganoderma species, the secondary metabolites of G. lingzhi have rarely been investigated. Y. M. Yan et al. reported six meroterpenoids with neural stem cells proliferation-promoting activity from this fungus.18
In order to explore diverse constituents for the identification of bioactive natural products from Ganoderma, a detailed chemical investigation on G. lingzhi led to the isolation of six rarely-encountered gymnomitrane-type sesquiterpenoids (1–6), an unusual type sesquiterpene (8), a polyketide (7) and an unusual ergosterol (9) from the low-polarity fractions of the crude extract (Fig. 1). Herein we report the isolation, structural characterization, biological activity, and feasible biosynthetic pathways of the compounds 1–9.
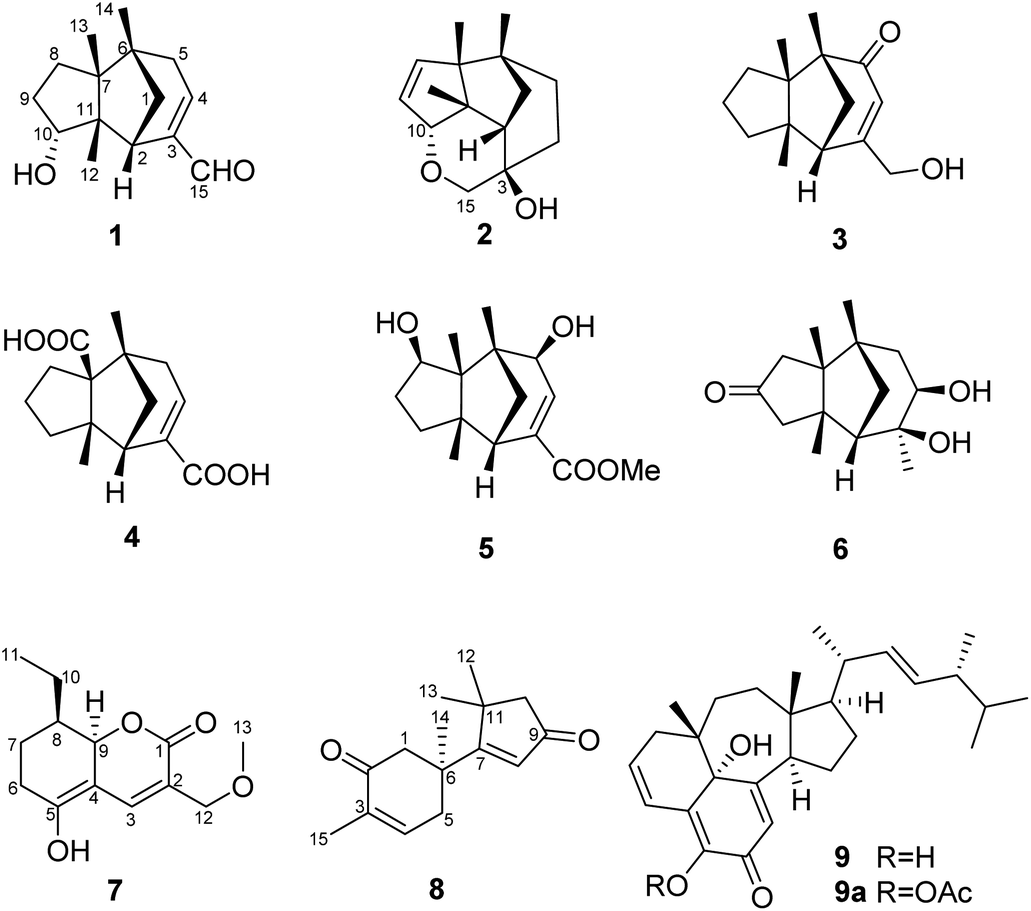 | ||
| Fig. 1 Structures of compounds 1–9, and 9a. | ||
Results and discussion
Compound 1, colourless needles (MeOH), had a molecular formula of C15H22O2 deduced by HREIMS analysis. The 1D NMR data (Tables 1 and 2) displayed signals for three methyl singlets, four sp3 methylenes, a trisubstituted double bond, two sp3 methines (one oxygenated carbon δC 77.6), an aldehyde group (δC 193.0), and three sp3 quaternary carbons. The data of 1 showed similarity to those of (+)-5-hydroxybarbatenal,19 a gymnomitrane-type sesquiterpene isolated from the root of Beilschmiedia tsangii. The data also suggested a hydroxy substituted at C-10 in 1 rather than C-5 in (+)-5-hydroxybarbatenal. This change was supported by the mutual 1H–1H COSY correlations between H-9 (δH 1.76, 1.61) and H-10 (δH 3.67), along with the HMBC correlations from Me-12 (δH 1.06) to C-10 (δC 77.6) (Fig. 2). The relative configuration of 1 was determined to be 2S*,6R*,7R*,10R*,11S* in accordance with the diagnostic ROESY correlations of H-1β/H3-13/H3-14, and H3-12/H-2/H-10 (Fig. 3). The absolute configuration of 1 was unequivocally established as 2S,6R,7R,10R,11S by single crystal X-ray diffraction analysis (Fig. 4) with the Flack parameter = 0.09(6). Hence, the structure of 1 was established as shown in Fig. 1, and named as 10α-hydroxy-gymnomitr-3-en-15-al.| No. | 1b | 2a | 3b | 4a | 5a | 6b |
|---|---|---|---|---|---|---|
| a Measured in CD3OD.b Measured in CDCl3. | ||||||
| 1 | 2.05, dd (11.8, 3.5), 1.37, d (11.8) | 1.77, dd (10.0, 2.5), 1.80, dd (10.0, 2.5) | 2.01, dd (11.5, 4.5), 1.87, d (11.5) | 2.79, dd (11.0, 5.0), 1.42, d (11.0) | 1.67, dd (11.8, 4.5), 1.60, d (11.8) | 1.59, ddd (12.0, 5.0, 2.8), 2.11, d (12.0) |
| 2 | 2.59, d (3.5) | 1.61, t (2.5) | 2.11, d (4.5) | 2.61, d (5.0) | 2.51, d (4.5) | 1.85, d (5.0) |
| 4 | 6.73, t-like (2.5) | 1.86, overlapped, 1.48, ddd (16.8, 7.6, 5.2) | 6.04, br. s | 6.80, dd (4.0, 3.0) | 6.88, d (4.0) | 3.31, dd (11.2, 6.3) |
| 5 | 2.55, dd (22.0, 2.5), 2.24, dd (22.0, 2.5) | 1.84, overlapped, 1.32, ddd (16.8, 13.3, 8.0) | 2.49, dd (21.0, 4.0), 2.16, dd (21.0, 3.0) | 3.95, d (4.0) | 1.82, dd (14.0, 6.3), 1.46, dd (14.0, 11.2) | |
| 8 | 1.50, m, 1.23, m | 5.65, d (5.8) | 1.46, m, 1.17, overlapped | 2.22, ddd (14.0, 9.5, 8.0), 1.64, ddd (14.0, 6.0, 4.0) | 3.74, overlapped | 2.61, d (19.7), 1.89, d (19.7) |
| 9 | 1.76, m, 1.61, m | 5.60, dd (5.8, 2.1) | 1.53, m, 1.43, m | 1.60, m, 1.54, m | 1.74, m | |
| 10 | 3.67, t-like (5.0) | 4.11, d (2.1) | 1.59, overlapped, 1.36, m | 1.69, ddd (14.0, 7.0, 4.0), 1.25, m | 1.27, m, 1.14, overlapped | 3.00, d (19.8), 2.13, d (19.8) |
| 12 | 1.06. s | 1.11, s | 1.17, s | 1.21, s | 1.12, s | 1.22, s |
| 13 | 0.94, s | 0.97, s | 0.98, s | 1.05, s | 1.01, s | |
| 14 | 0.99, s | 0.94, s | 1.07, s | 1.13, s | 0.93, s | 0.90, s |
| 15 | 9.45, s | 3.43, d (10.0), 3.27, d (10.0) | 4.35, br. dd (17.0, 6.0), 4.25, br. dd (17.0, 6.0) | 1.38, s | ||
| MeO- | 3.74, s | |||||
| No. | 1b | 2a | 3b | 4a | 5a | 6b | 7b | 8b |
|---|---|---|---|---|---|---|---|---|
| a Measured in CD3OD.b Measured in CDCl3. | ||||||||
| 1 | 42.0, CH2 | 38.0, CH2 | 47.1, CH2 | 43.7, CH2 | 38.3, CH2 | 37.4, CH2 | 178.5, C | 50.4, CH2 |
| 2 | 42.1, CH | 53.9, CH | 49.0, CH | 47.1, CH | 46.5, CH | 57.9, CH | 125.1, C | 197.8, C |
| 3 | 147.6, C | 69.7, C | 169.5, C | 137.7, C | 140.0, C | 72.6, C | 152.7, CH | 135.4, C |
| 4 | 151.0, CH | 31.5, CH2 | 122.3, CH | 140.3, CH | 139.0, CH | 69.5, CH | 124.2, C | 142.3, CH |
| 5 | 41.9, CH2 | 32.7, CH2 | 205.5, C | 42.2, CH2 | 71.6, CH | 44.6, CH2 | 166.3, C | 38.4, CH2 |
| 6 | 44.5, C | 43.6, C | 60.4, C | 47.0, C | 47.6, C | 45.4, C | 28.0, CH2 | 43.2, C |
| 7 | 55.6, C | 64.5, C | 55.3, C | 71.6, C | 57.0, C | 50.1, C | 22.2, CH2 | 190.4, C |
| 8 | 32.7, CH2 | 142.3, CH | 37.9, CH2 | 31.6, CH2 | 76.2, CH | 48.4, CH2 | 40.2, CH | 130.0, CH |
| 9 | 35.2, CH2 | 130.2, CH | 26.9, CH2 | 28.3, CH2 | 35.4, CH2 | 219.6, C | 62.6, CH | 207.1, C |
| 10 | 77.6, CH | 88.8, CH | 39.2, CH2 | 40.1, CH2 | 33.6, CH2 | 50.5, CH2 | 23.7, CH2 | 55.2, CH2 |
| 11 | 61.3, C | 52.4, C | 56.9, C | 61.6, C | 59.3, C | 49.7, C | 11.9, CH3 | 45.1, C |
| 12 | 19.6, CH3 | 27.4, CH3 | 28.1, CH3 | 30.6, CH3 | 27.7, CH3 | 30.9, CH3 | 66.2, CH2 | 29.2, CH3 |
| 13 | 24.4, CH3 | 24.7, CH3 | 22.8, CH3 | 178.7, C | 20.0, CH3 | 24.5, CH3 | 29.4, CH3 | |
| 14 | 24.2, CH3 | 20.0, CH3 | 17.6, CH3 | 24.7, CH3 | 15.9, CH3 | 23.3, CH3 | 27.6, CH3 | |
| 15 | 193.0, CH | 74.2, CH2 | 65.6, CH2 | 170.7, C | 169.1, C | 27.7, CH3 | 15.4, CH3 | |
| MeO- | 52.5, CH3 | 59.1, CH3 | ||||||
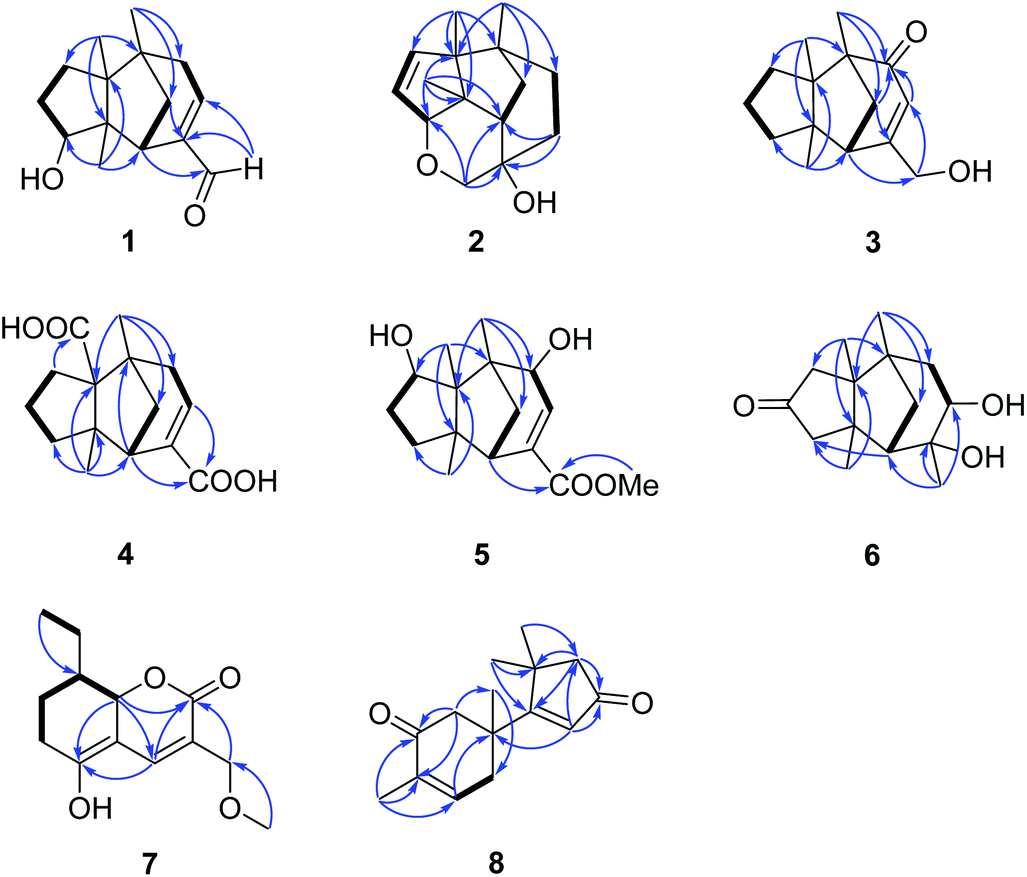 | ||
| Fig. 2 Selected HMBC and 1H–1H COSY correlations of 1–8. | ||
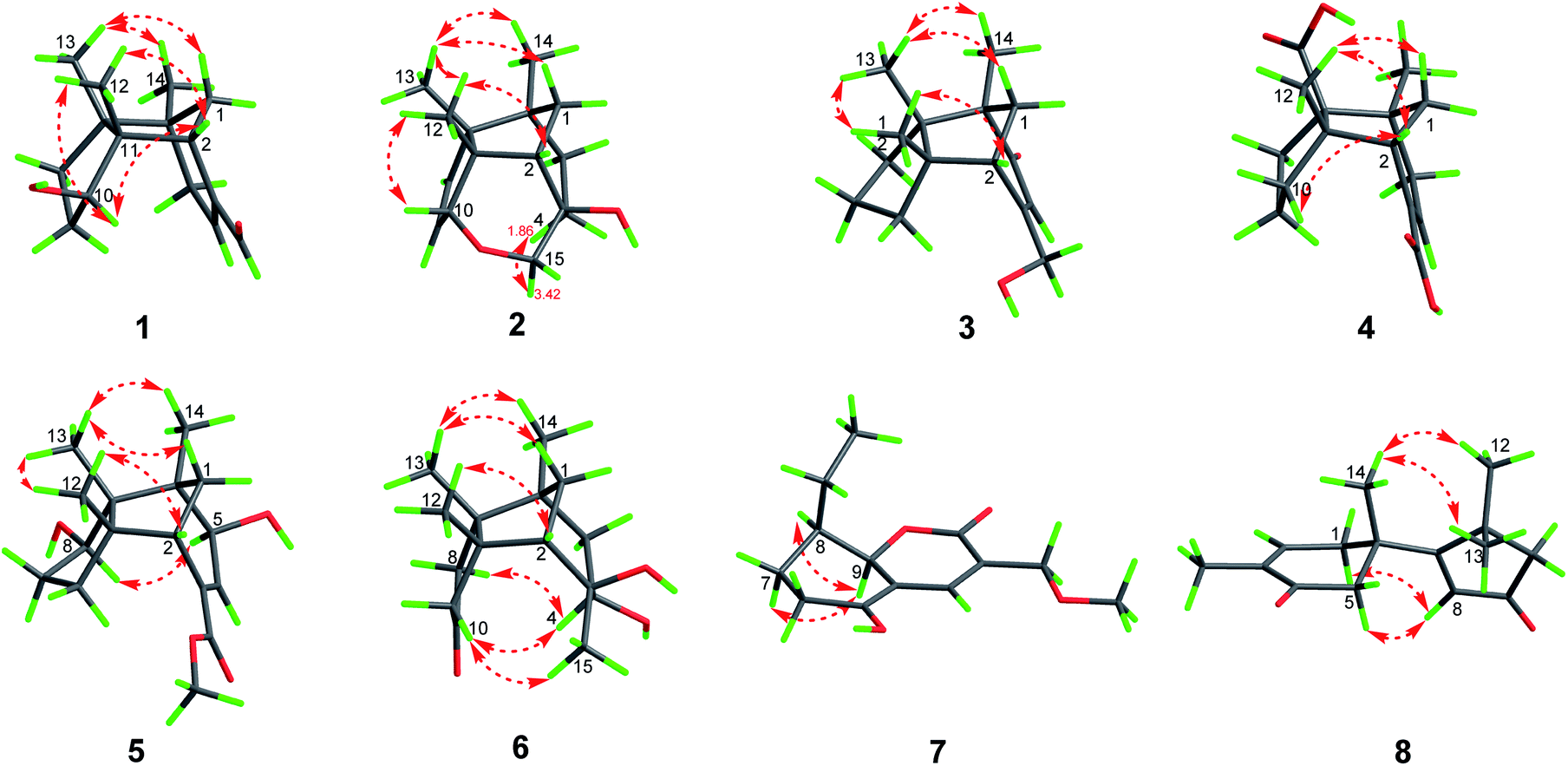 | ||
| Fig. 3 Selected ROESY correlations of 1–8. | ||
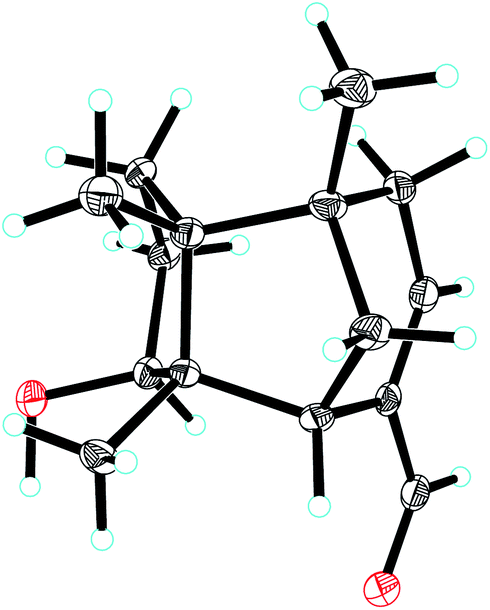 | ||
| Fig. 4 ORTEP drawing of compound 1. | ||
Compound 2 was obtained as colourless crystals (MeOH). Its molecular formula was determined to be C15H22O2 by HREIMS analysis with the ion peak at m/z 234.1617 (calcd for C15H22O2 234.1620), indicating the presence of five degrees of unsaturation. The planar structure of 2 was elucidated by interpretation of its NMR spectra. The 1D NMR spectra of 2 presented three methyls (δH/δC: 0.94/20.0, 0.97/24.7, 1.11/27.4), four methylenes (δC 31.5, 32.7, 38.0, 74.2), two sp3 methines (δC 53.9, 88.8), two olefinic carbons (δC 130.2, 142.3), and four sp3 quaternary carbons (δC 43.6, 52.4, 64.5, 69.7) (Tables 1 and 2). The aforementioned data indicated that 2 is a sesquiterpene derivative harbouring a gymnomitrane skeleton analogous to that of 1. In addition to the tricyclic system and one double bond, the remaining one degree of unsaturation revealed the existence of an additional ring. Analysis of the 2D NMR spectra of 2 helped to establish its structure unambiguously. The HMBC correlations from H3-13 (δH 0.97) to C-8 (δC 142.3) revealed that the double bond was located between C-8 and C-9. Additionally, the HMBC correlations from H-4 (δH 1.86, 1.48) to C-3 (δC 69.7) and C-15 (δC 74.2), and from H-15 (δH 3.43, 3.27) to C-10 (δC 88.8) suggested an ether linkage between C-10 and C-15, and a hydroxy group substituted at C-3 (Fig. 2). The key ROESY correlations between H3-12 (δH 1.11)/H-10 (δH 4.11), together with biosynthetic comparison with the structure of 1 and the structure inflexibility of 2, the absolute configuration of 2 was assigned to be 2S,3R,6R,7R,10R,11S. The above assignment indicated the structure of 2 as a unique boat-like molecule bearing a rigid structure unprecedented in the gymnomitrane family, it was given the name 10α,15-epoxy-gymnomitr-8-en-3β-ol.
Compound 3 was isolated as a white amorphous powder. The molecular formula was deduced from the sodium adduct ion peak at m/z 257.1514 (calcd for C15H22O2Na, 257.1512) as C15H22O2 in the HRESIMS analysis. The 13C NMR spectrum (Table 2) showed signals for 15 carbons, including three methyls, five methylenes (one was oxygenated δC 65.6), one sp3 methines, three sp3 quaternary carbons, and an α,β-unsaturated ketone group (δC 169.5, 122.3, 205.5). The data shared striking similarity with those of the known compound ganosinensine isolated from G. sinense,13 indicating that 3 was also a gymnomitrane-type sesquiterpene. The structural difference between ganosinensine and 3 was that the C-9 in 3 remained unoxygenated, supported by the 1H–1H COSY correlations of H-8 (δH 1.46, 1.17)/H-9 (δH 1.53, 1.43)/H-10 (δH 1.59, 1.36) (Fig. 2) and HRESIMS data. The configuration of 3 was identical with that of 1 by analysis of the ROESY spectrum (Fig. 3). Thus, compound 3 was determined as shown in Fig. 1, and named as 15-hydroxy-gymnomitr-3-en-5-one.
The HRESIMS analysis of 4 suggested the molecular formula was C15H20O4 (m/z 263.1291 [M − H]−, calcd for C15H19O4, 263.1289), appropriate for six degrees of unsaturation. The 1H and 13C NMR data (Tables 1 and 2) showed representative chemical shifts for a gymnomitrane skeleton: two methyl singlets (δH 1.13, 1.21; δC 24.7, 30.6), three sp3 quaternary carbons (δC 47.0, 61.6, 71.6), two carbonyl groups (δC 170.7, 178.7), one tri-substituted double bond (δC 140.3, 137.7; δH 6.80). The HMBC correlations from H-2 (δH 2.61) to C-3 (δC 137.7) and C-4 (δC 140.3) allowed the assignment of C-3/C-4 as a double bond (Fig. 2). The gymnomitrane scaffold and the double bond accounted for four out of six degrees of unsaturation. The remaining two degrees of unsaturation were assigned to two carboxylic groups at C-13 and C-15, which was supported by the HMBC correlations from H-8 (δH 2.22) to C-13 (δC 178.7), and from H-2 (δH 2.61) and H-4 (δH 6.80) to C-15 (δC 170.7) (Fig. 2). The relative stereochemistry of 4 is identical to that of 1 by analysis of the ROESY spectrum (Fig. 3). Hence, 4 was named gymnomitr-3-ene-13,15-dioic acid.
The 1D NMR data of 5 (Tables 1 and 2) exhibited four methyl singlets (one methoxy group), three sp3 methylenes, three sp3 methines (two oxygenated), one tri-substituted double bond, three sp3 quaternary carbons, and one carbonyl group. The data displayed similarity to those of 1–4, suggesting the gymnomitrane skeleton of 5. Compared to compound 1, two hydroxy groups substituted at C-5 and C-8 in 5, rather than a hydroxy group at C-10 in 1. The changes were supported by the HMBC correlations from H3-13 (δH 1.05) to C-8 (δC 76.2), H3-14 (δH 0.93) to C-5 (δC 71.6), and H3-12 (δH 1.12) to the methylene C-10 (δC 33.6) (Fig. 2). Furthermore, the HMBC correlation from the methoxy singlet at δH 3.74 to the carbonyl group δC 169.1 suggested the presence of a methyl ester group at C-15 in compound 5 (Fig. 2). The above assignments are consistent with the molecular formula C16H24O4, which was determined by HRESIMS data. The relative configurations of C-5 and C-8 were assigned as S* and R*, respectively, by the key ROESY correlations between H-5 (δH 3.95) and H-8 (δH 3.74) (Fig. 3). The absolute configuration of 5 was unequivocally settled by single crystal X-ray diffraction analysis as 2S,5S,6R,7S,8R,11S (Fig. 5). Thus, compound 5 was identified as methyl 5β,8β-dihydroxy-gymnomitr-3-en-15-oate.
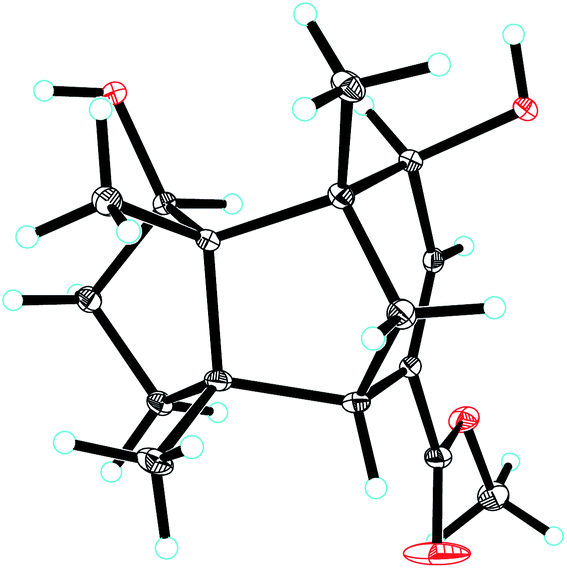 | ||
| Fig. 5 ORTEP drawing of compound 5. | ||
Compounds 6 and 7 were isolated as an inseparable mixture. The NMR spectra of the mixture presented complicated but clear signals which could be assigned to two sets with the aid of HSQC, HMBC, and 1H–1H COSY spectra. The first group of signals designated to 6 included four methyl singlets (δH 0.90, 1.01, 1.22, 1.38; δC 23.3, 24.5, 30.9, 27.7), four methylenes, two methines (one attached to an oxygen atom), and five quaternary carbons (one carbonyl and one was oxygenated) (Tables 1 and 2). The above-mentioned data are agreement with a gymnomitrane skeleton substituted by two hydroxy groups at C-3 (δC 72.6), C-4 (δC 69.5), and a carbonyl group at C-9 (δC 219.6C), which were supported by the HMBC correlations from H3-15 (δH 1.38) to C-3 and C-4, and from H-8 (δH 2.61, 1.89), H-10 (δH 3.00, 2.13) to C-9 (δC 219.6) (Fig. 2). The assignments were in accordance with the molecular formula of C15H24O3 generated by HREIMS ion peak at m/z 252.1724 [M]+ (calcd for 252.1725). As for the relative configuration of 6, both of the hydroxy groups at C-3 and C-4 were assigned to be β orientated by the diagnostic ROESY correlations between H-8α (δH 2.61) and H-4 (δH 3.31), H-10α (δH 2.98) and H-4, and between H3-15 (δH 1.38) and H-10α (Fig. 4). Thus, compound 6 was established to be 3β,4β-dihydroxy-gymnomitr-9-one.
The other group of signals included a methyl triplet (δH 1.01; δC 11.9), a methoxy group (δH 3.44; δC 59.1), four methylenes (one oxygenated), three methines (one oxygenated, one olefinic), and four sp2 quaternary carbons (Tables 2 and 3). The data displayed characteristic signals similar to those of xylaolide A.20 Analysis of the 2D NMR spectra of 7 revealed that C-12 was substituted by a methoxy group in 7 instead of a hydroxy group in xylaolide A, which was supported by the HMBC correlation from the methoxy proton (δH 3.44) to C-12 (δC 66.2) (Fig. 2). Other parts of the structure, along with the relative configuration, were consistent to those of xylaolide A by analysis of the ROESY spectrum (Fig. 3). The above assignments accounted for a molecular formula of C13H18O4, in accord with the HRESIMS result (m/z 261.1098 [M + Na]+, calcd for C13H18O4Na, 261.1098). Therefore, compound 7 was named as 12-O-methyl-xylaolide A.
| No. | 7 | 8 |
|---|---|---|
| 1 | 2.87, d (15.8), 2.59, d (15.8) | |
| 3 | 7.77, s | |
| 4 | 6.64, br. dd (6.0, 3.0) | |
| 5 | 2.83, dd (17.6, 2.4), 2.50, dd (17.6, 2.4) | |
| 6 | 2.59, overlapped | |
| 7 | 1.79, m, 1.71, overlapped | |
| 8 | 1.44, m | 5.93, s |
| 9 | 4.87, d (3.2) | |
| 10 | 1.70, overlapped, 1.42, overlapped | 2.36, s (2H) |
| 11 | 1.01, t (7.0) | |
| 12 | 4.32, d (12.4), 4.29, d (12.4) | 1.38 s |
| 13 | 1.40, s | |
| 14 | 1.37, s | |
| 15 | 1.78, s | |
| MeO- | 3.44, s |
Compound 8, a colourless oil, had the molecular formula C15H20O2 determined by HREIMS analysis at m/z 232.1460 [M]+ (calcd C15H20O2, 232.1463), requiring six degrees of unsaturation. The 1D NMR data presented signals for four methyl singlets (δH 1.37, 1.38, 1.40, 1.78; δC 27.6, 29.2, 29.4, 15.4), three methylenes, two sp3 quaternary carbons, and two pairs of α,β-unsaturated ketone groups (δH 6.64, δC 142.3, 135.4, 197.8; δH 5.93, δC 130.0, 190.4, 207.1) (Tables 2 and 3). The 1D NMR data accounted for four degrees of unsaturation, implying the existence of two rings in 8. The planar structure of 8 was established by analysis of the 2D NMR data. The key HMBC correlations from H3-14 (δH 1.37) to C-1 (δC 50.4), C-5 (δC 38.4), and C-6 (δC 43.2), and from H-1 (δH 2.87, 2.59) to C-2 (δC 197.8), and from H3-15 (δH 1.78) to C-2, C-3 (δC 135.4), and C-4 (δC 142.3), along with the 1H–1H COSY correlation between H-4 (δH 6.64) and H-5 (δH 2.83, 2.50) (Fig. 2), allowed the completion of a six-membered ring. Moreover, the characteristic HMBC correlations from H3-12 (δH 1.38), H3-13 (δH 1.40) to C-7 (δC 190.4), C-10 (δC 55.2), and C-11 (δC 45.1), and from H-8 (δH 5.93) to C-7 and C-9 (δC 207.1) (Fig. 2) enabled the accomplishment of a five-membered ring. Besides, the HMBC correlation from H3-14 to C-7 suggested that the two rings are connected via C-6–C-7 (Fig. 2). Therefore, the planar structure of 8 was determined as shown in Fig. 1. Notably, compound 8 represents an unusual sesquiterpene skeleton in nature, holding structural similarity to that of the cuparane-type sesquiterpenes, and we herein designate the skeleton of 8 as 14(7→6)-cuparane.
The steric hindrance between the five- and six-membered rings made it possible to establish the absolute configuration of C-6 by computational methods. The possible conformers of (6S)-8 were generated by the MMFF94s force field conformation search, the geometries with population higher than 1% were further optimized by density functional theory method at the B3LYP/6-31G(d,p) level on Gaussian 09 program package21 to give eight predominant conformers within relative energies 3 kcal mol−1. These eight conformers were subjected to theoretical calculation of ECD spectra by using the time-dependent DFT method at the B3LYP/6-31G(d,p) in air. As shown in Fig. 6, the calculated ECD spectra for (6S)-8 displayed a similar curve with the experimental CD. Thus, the absolute configuration of 1 was determined as 6S. Compound 8 was trivially named as lingzhidienone.
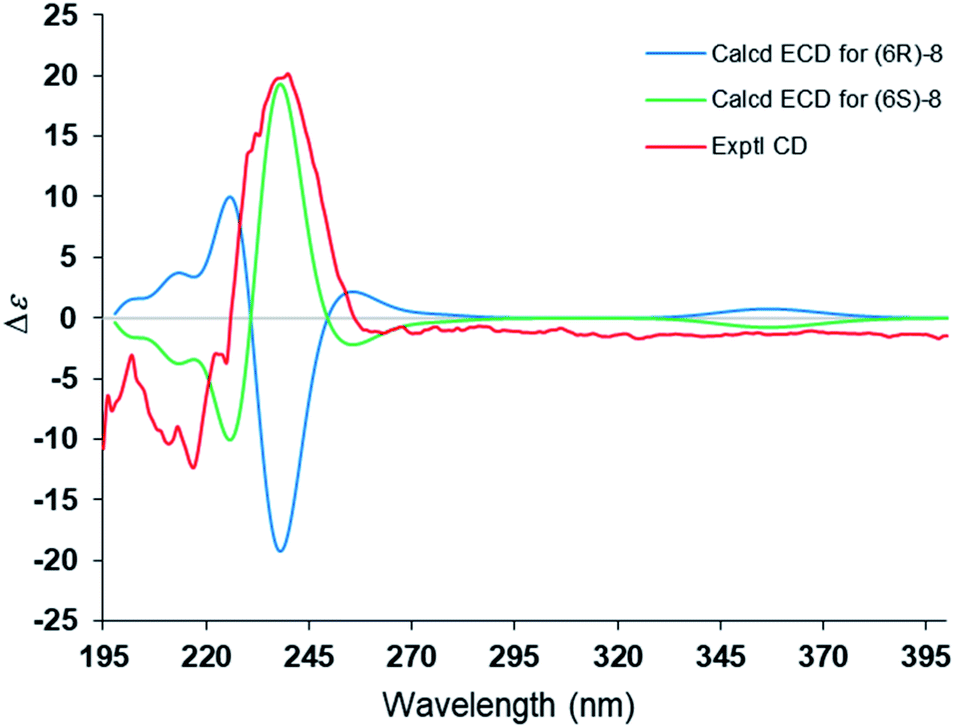 | ||
| Fig. 6 Comparison of the experimental CD and calculated ECD spectra of compound 8. | ||
Compound 9, named ganotheaecolin A,22 a known isolate reported more recently was encountered in this study. Ganotheaecolin A represented a novel type of steroid with a unique naphtha[1,8-ef]azulene ring system, and it was suggested that 9 exhibited cell differentiation-stimulating activity by a dose-dependent manner and reach a maximum effect at 10 μM. With this fascinating molecule in hand, we evaluated the cytotoxicity of 9 and its acetylated product 9a (5-O-acetyl ganotheaecolin (A) against the five human cancer cell lines HL-60, MCF-7, SW480, A549, and SMMC-7721. Interestingly, compound 9 displayed weak cytotoxicity against five human cancer cell lines with IC50 values of 18.0–32.3 μM (Table 4), while 9a was devoid of activity (IC50 > 40 μM). The results suggested that the 5-OH of 9 was indispensable for its cytotoxicity.
| Sample | HL-60 | A-549 | SMMC-7721 | MCF-7 | SW480 |
|---|---|---|---|---|---|
| IC50 (μM) | |||||
| 9 | 18.8 | 32.3 | 25.1 | 18.1 | 22.3 |
| DDP | 4.7 | 27.6 | 16.0 | 34.8 | 16.1 |
| Taxol | <0.008 | <0.008 | <0.008 | <0.008 | <0.008 |
The gymnomitrane and 14(7→6)-cuparane scaffolds appear to be biogenetically related. As shown in Scheme 1, the hypothetical biosynthetic pathways of the gymnomitranes (1–6, compound 2 as the illustrative example) and 14(7→6)-cuparane (8) are described. First of all, the farnesyl pyrophosphate (FPP) undergoes cascade 1,6- and 7,11-cyclization reactions and a 1,4-hydride shift from C-6 to C-10 to give the key intermediate cuparane skeleton. The cuparane yields the gymnomitrane and 14(7→6)-cuparane skeleton via two ways. One way is the synchronous methyl and hydride migrations to produce the 14(7→6)-cuparane scaffold. The other way is two methyl migrations, i.e. 14(7→6) and 13(11→7), in combination with a 2,11-cyclization reaction to produce the gymnomitrane scaffold. Further enzymatic oxidations and dehydration reactions yield 2 and 8.
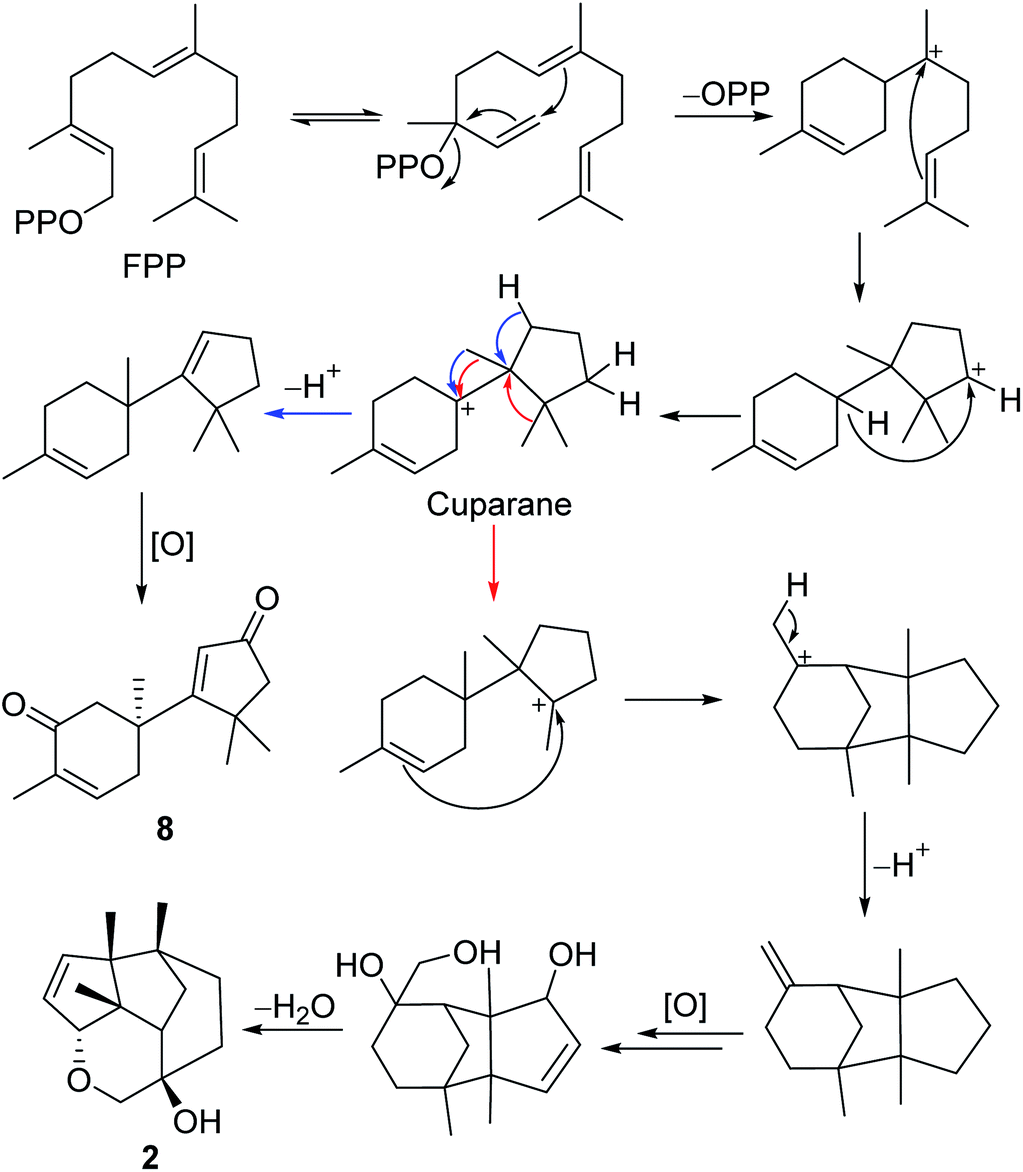 | ||
| Scheme 1 Proposed biosynthetic pathway of 2 and 8. | ||
All the compounds except 6 and 7 were evaluated for their inhibitory activity against NO production. However, none of them showed remarkable inhibitory activities of NO production in murine monocytic RAW 264.7 macrophages at the concentration of 25 μM.
Compounds 1–5, 8, 9, and 9a were screened for their cytotoxicity against five human cancer cell lines (HL-60, A-549, SMMC-7721, MCF-7, SW480). Compounds 1–5, 8, and 9a were inactive in the cytotoxicity assay (IC50 > 40 μM). Only compound 9 displayed inhibitory activity against the five human cancer cell lines with IC50 values ranging from 18.0 to 32.3 μM (Table 4), indicating that 5-OH is essential for its activity.
Conclusions
In summary, chemical investigation on the edible and medicinal mushroom Ganoderma lingzhi afforded six new gymnomitrane-type sesquiterpenoids (1–6), an unusual sesquiterpenoid (8), and a new polyketide (7) as well as a known steroid (9). The absolute configurations of the isolates were unambiguously determined via single crystal X-ray diffraction analysis, and ECD calculation. The biosynthetic pathway of 1–6 and 8 were also discussed. Among the structures, 2 is a tetracyclic boat-like rigid molecule with a 10,15-epoxy group. Compound 8 possessed an unusual 14(7→6)-cuparane scaffold. It is noteworthy that the gymnomitrane-type sesquiterpenoids have rarely been encountered from fungal origin6,14,23 and this work represents the first report of clustered gymnomitranes from Ganoderma, or even from fungi. This work also provides evidence consistent with the genome sequence data of Ganoderma. Although no promising biological assay results are reported in this study, this does not mean that the sesquiterpenoids should be neglected in the development of quality criteria for Ganoderma. More work on identifying the constituents of Ganoderma should be put into practice in the course of preclinical studies prior to clinical use.Experimental section
General experimental procedures
Optical rotations were measured by a JASCO P-1020 digital polarimeter (Horiba, Kyoto, Japan). A UV-2401PC UV-visible recording spectrophotometer (Shimadzu, Kyoto, Japan) was used to record the ultraviolet (UV) spectra. A Chirascan circular dichroism spectrometer (Applied Photophysics Limited, Leatherhead, Surrey, UK) was used to recorded the CD spectra. 1D and 2D NMR spectra were obtained on Bruker Avance III 600 MHz or Ascend 800 MHz spectrometers (Bruker Corporation, Karlsruhe, Germany). An Agilent 6200 Q-TOF MS system (Agilent Technologies, Santa Clara, CA) was used to acquire the HRESIMS data. A Waters AutoSpec Premier P776 MS system (Waters Corporation, Milford, MA) was used to acquire the HREIMS data. An APEX II DUO spectrophotometer (Bruker AXS GmbH, Karlsruhe, Germany) was applied for performing the single crystal X-ray diffraction experiments. Column chromatography (CC) were run on Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) and silica gel (Qingdao Haiyang Chemical Co., Ltd, Qingdao, China). A Büchi Sepacore System (pump manager C-615, pump modules C-605, and fraction collector C-660) (Büchi Labortechnik AG, Flawil, Switzerland) was used to perform medium pressure liquid chromatography (MPLC), equipped with a column (400 mm × 7.4 mm i.d., 40–75 μm, flow rate 40 mL min−1) filled with Chromatorex C-18 (Fuji Silysia Chemical Ltd., Kasugai, Japan) RP-C18 silica gel. An Agilent 1260 liquid chromatography system (Agilent) equipped with an ODS column (Zorbax SB-C18, 150 mm × 9.4 mm i.d., 5 μm, flow rate 10 mL min−1) was used for preparative high performance liquid chromatography (prep-HPLC).Fungal material
The fruiting bodies of G. lingzhi were purchased from the herbal trading market of Kunming Luosiwan International Trade City in 2015, and identified by Prof. Yu-Cheng Dai (Beijing Forestry University), an expert in the field of mushroom taxonomy. A voucher specimen (No. HFC 20150518) of G. lingzhi was deposited in the Herbarium of Ethnic Medicinal Plants of South-Central University for Nationalities (SCUEC).Extraction and isolation
The air-dried and powdered fruiting bodies of G. lingzhi (10.0 kg) was extracted with 95% ethanol three times (three days each). The extract was evaporated under reduced pressure and partitioned between ethyl acetate and water four times to give a crude extract (400 g). The crude extract was subject to silica gels with a stepwise gradient of petroleum ether/acetone (from 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 1:1, v/v, totally 20 L) to afford five fractions (A–E).
:1 → 1:1, v/v, totally 20 L) to afford five fractions (A–E).
Fraction B (45 g) was subjected to MPLC with a solvent gradient system of MeOH/H2O (from 80:20 → 100:0, v/v, 35 mL min−1) to obtain twenty-two subfractions (B1 to B22). Subfraction B16 (40 mg) was further purified by Sephadex LH-20 (MeOH) and then B16d was separated by prep-HPLC using a gradient elution (MeCN/H2O, 30:70 → 50:50, v/v, 30 min, 7 mL min−1) to yield compound 2 (0.7 mg, tR = 8.5 min). Subfraction B1 (22 mg) was applied to Sephadex LH-20 (MeOH) to afford four minor fractions (B1a to B1d). Compound 8 (2.2 mg, MeCN/H2O, 10:90 → 35:65, v/v, 30 min, 7 mL min−1, tR = 23.2 min) was obtained from subfraction B1c via prep-HPLC. Subfraction B17 (65 mg) was further purified by prep-HPLC using a gradient elution (MeCN/H2O, 60:40 → 90:10, 30 min, 7 mL min−1) to yield compound 9 (32 mg, tR = 27.2 min).
Fraction C (27 g) was subjected to MPLC with a gradient solvent system of MeOH/H2O (80:20 → 100:0, v/v, 35 mL min−1) to obtain twenty-two subfractions (C1 to C15). Subfraction C2 (45 mg) was further purified by Sephadex LH-20 (MeOH), then fractions C2e, C2f and C2g were separated by prep-HPLC to afford compound 1 (0.9 mg, MeCN/H2O, 27:73 → 42:58, v/v, 20 min, 7 mL min−1, tR = 13.5 min), compounds 6/7 mixture (2.6 mg, MeCN/H2O, 15:85 → 30:70, v/v, 20 min, 7 mL min−1, tR = 16.0 min), and compound 3 (1.1 mg, MeCN/H2O, 20:80 → 40:60, 20 min, 7 mL min−1, tR = 17.2 min), respectively. Subfraction C3 (105 mg) was separated by Sephadex LH-20 (MeOH) yielding six minor fractions (C3a to C3f). Next, fraction C3e was applied on Sephadex LH-20 (Acetone) to afford five minor fractions (C3e1 to C3e5), then C3e4 were treated by prep-HPLC to afford compounds 4 (5.0 mg, MeCN/H2O, 20:80 → 40:60, v/v, 7 mL min−1, 20 min, tR = 13.5 min), 5 (10.0 mg, MeCN/H2O, 20:80 → 40:60, 20 min, 7 mL min−1, tR = 4.5 min), respectively.
ε): 241.60 (3.23); 1H NMR data (Table 1); 13C NMR data (Table 2); HREIMS m/z 234.1602 [M]+ (calcd for C15H22O2, 234.1620).ε): 244.20 (3.87); 1H NMR data (Table 1); 13C NMR data (Table 2); HRESIMS m/z 257.1514 [M + Na]+ (calcd for C15H22O2Na, 257.1512).ε): 221.50 (3.57); 1H NMR data (Table 1); 13C NMR data (Table 2); HRESIMS m/z 303.1565 [M + Na]+ (calcd for C16H24O4Na, 303.1567).ε): 230.80 (4.23); 1H NMR data (Table 3); 13C NMR data (Table 2); HREIMS m/z 232.1460 [M]+ (calcd for C15H20O2, 232.1463); CD (MeOH) λmax (Δ ε): 217 (−12.3), 240 (+20.2), 260 (−1.2).:30 → 90:10, 25 min, 7 mL min−1) to yield the corresponding 5-O-acetyl derivative. Application of this procedure afforded the new acetate 5-O-acetyl-ganotheaecolin A (9a).5-O-Acetyl-ganotheaecolin A (9a): 1H NMR (500 MHz, CDCl3): H-1 (δH 2.59, br. d, J = 18.8 Hz; δH 1.75, overlapped), H-2 (δH 6.18, ddd, J = 10.0, 5.0, 2.5 Hz); H-3 (δH 6.35, dd, J = 10.0, 2.5 Hz); H-11 (δH 2.31, ddd, J = 15.0, 15.0, 3.0; δH 1.01, ddd, J = 15.0, 4.7, 2.5); H-12 (δH 1.96, ddd, J = 15.0, 5.0, 3.0 Hz; δH 1.34, br. d, J = 15.0 Hz); H-14 (δH 3.41, dd, J = 12.5, 7.2 Hz); H-15 (δH 1.79, overlapped; δH 1.61, overlapped); H-16 (δH 1.74, overlapped; δH 1.41, overlapped); H-17 (δH 1.61, m); H-18 (δH 0.79, s, 3H); H-19 (δH 0.72, s, 3H); H-20 (δH 2.14, m); H-21 (δH 1.04, d, J = 6.7 Hz); H-22 (δH 5.20, dd, J = 15.3, 7.5 Hz); H-23 (δH 5.24, dd, J = 15.3, 7.0 Hz); H-24 (δH 1.86, m); H-25 (δH 1.47, m); H-26 (δH 0.82, d, J = 6.8 Hz); H-27 (δH 0.84, d, J = 6.8 Hz); H-28 (δH 0.92, d, J = 6.8 Hz); 13C NMR (125 MHz, CDCl3): C-1 (δC 40.0); C-2 (δC 137.3); C-3 (δC 127.0); C-4 (δC 168.9); C-5 (δC 131.9); C-6 (δC 184.5); C-7 (δC 127.0); C-8 (δC 162.3); C-9 (δC 74.2); C-10 (δC 41.4); C-11 (δC 33.1); C-12 (δC 33.8); C-13 (δC 43.5); C-14 (δC 47.8); C-15 (δC 27.0); C-16 (δC 27.2); C-17 (δC 56.7); C-18 (δC 18.7); C-19 (δC 19.8); C-20 (δC 40.5); C-21 (δC 22.0); C-22 (δC 134.9); C-23 (δC 133.0); C-24 (δC 43.5); C-25 (δC 33.2); C-26 (δC 19.8); C-27 (δC 20.2); C-28 (δC 17.8).
Bioassays
Nitric oxide production in RAW 264.7 macrophages
The RPMI 1640 medium (Hyclone, Logan, UT) containing 10% FBS was used to culture the murine monocytic RAW 264.7 macrophages. The compounds were dissolved in DMSO and further diluted in medium to produce different concentrations. The culture medium and cell mixture were dispensed into 96-well plates (2 × 105 cells per well) and maintained at 37 °C under 5% CO2 in a humidified atmosphere. After preincubation for 24 h, serial dilutions of the test compounds were added into the cells, up to the maximum concentration 25 μM, then LPS was added to a concentration 1 μg mL−1 and incubation continued for 18 h. After addition of 100 μL of Griess reagent (reagent A and reagent B, Sigma, St. Louis, Mo) to 100 μL of each supernatant from the LPS-treated or LPS- and compound-treated cells in triplicate and incubation for 5 min, NO production of each cell was assessed by sample absorbance at 570 nm by a 2104 Envision Multilabel Plate Reader. L-NG-Monomethyl arginine (L-NMMA) was used as a positive control.Cytotoxicity against five human cancer cell lines. The following five human cancer cell lines were used: the HL-60 (ATCC CCL-240) human myeloid leukemia; SMMC-7721 human hepatocellular carcinoma; A-549 (ATCC CCL-185) lung cancer; MCF-7 (ATCC HTB-22) breast cancer; SW-480 (ATCC CCL-228) human colon cancer. The cell line SMMC-7721 was bought from China Infrastructure of Cell Line Resources (Beijing, China), and others were bought from American Type Culture Collection (ATCC, Manassas, VA). All cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS) (Hyclone) and maintained at 37 °C under 5% CO2 in a humidified atmosphere. Colorimetric measurements of the amount of insoluble formazan which was produced in living cells based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma, St. Louis, MO) was used to assess cell viability. In brief, each well of a 96-well cell culture plate was seeded with 100 μL of adherent cells and kept for 12 h for adherence, and then added with test compounds, however, suspended cells were seeded before added with test compounds with both the same density of 1 ×105 cells per mL every 100 μL of culture medium. After different concentrations of test compounds addition, each cancer cell line was incubated for 48 h in triplicate. Cisplatin was used as positive control. After the incubation, each well was treated with MTT (100 μg) and incubation continued for 4 h at 37 °C. After removal of the 100 μL culture medium, the cells were lysed with 20% SDS-50% DMF (100 μL). The remaining lysates were subjected to measure of optical density at 595 nm with a 96-well microtiter plate reader. The IC50 value for each compound was calculated by a published method.24
Quantum chemistry calculation details
A conformation search based on molecular mechanics with MMFF94s force fields were performed for (6S)-8 gave 10 stable conformers with distributions higher than 1%.25,26 All these conformers were further optimized by the density functional theory method at the B3LYP/6-31G(d,p) level in Gaussian 09 program package,21 leading to six ((6S)-8a–(6S)-8f) conformers within 3 kcal mol−1 energy threshold from global minimum, respectively. The predominant conformers were subjected to theoretical calculation of ECD using time-dependent density functional theory (TDDFT) at B3LYP/6-31G(d,p) level with IEFPCM model in air based on B3LYP/6-31G(d,p) optimized conformers. The calculated ECD curves for (6S)-8 were weighted using SpecDis 1.71 with σ = 0.2 eV, and UV shift 11 nm, respectively.27 The ECD curve of the enantiomer (6R)-8 was generated by SpecDis by the function “enantiomeric ECD”.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Analytical and Measuring Center, School of Pharmaceutical Sciences, South-Central University for Nationalities for MS and NMR spectra tests. This work was financially supported by National Natural Science Foundation of China (81903512, 81773590), and the Start-up Research Funds of South-Central University for Nationalities (YZZ17011).Notes and references
- M. F. Ahmad, Biomed. Pharmacother., 2018, 107, 507 CrossRef CAS PubMed.
- K. S. Bishop, C. H. J. Kao, Y. Y. Xu, M. P. Glucina, R. R. M. Paterson and L. R. Ferguson, Phytochemistry, 2015, 114, 56 CrossRef CAS.
- Y. C. Dai, L. W. Zhou, T. Hattori, Y. Cao, J. A. Stalpers, L. Ryvarden, P. Buchanan, F. Oberwinkler, N. Hallenberg, P. G. Liu and S. H. Wu, Mycol. Prog., 2017, 16, 1051 CrossRef.
- S. Baby and A. J. Johnson, Phytochemistry, 2015, 114, 66 CrossRef CAS PubMed.
- D. Cör, Ž. Knez and M. K. Hrnčič, Molecules, 2018, 23, 649 CrossRef PubMed.
- H. P. Chen, Z. Z. Zhao, Y. Zhang, X. Bai, L. Zhang and J. K. Liu, RSC Adv., 2016, 6, 64469 RSC.
- J. Wang, B. Cao, H. P. Zhao and J. Feng, Aging Dis., 2017, 8, 691 CrossRef PubMed.
- X. Peng and M. Qiu, Nat. Prod. Bioprospect., 2018, 8, 137 CrossRef CAS PubMed.
- K. D. Hsu and K. C. Cheng, Appl. Microbiol. Biotechnol., 2018, 102, 9037 CrossRef CAS PubMed.
- P. J. Zeng, Z. H. Guo, Z. Zeng, C. Hao, Y. R. Zhang, M. Zhang, Y. Liu, H. Li, J. Li and L. J. Zhang, J. Cell. Mol. Med., 2018, 22, 3278 CrossRef PubMed.
- H. P. Chen and J. K. Liu, Prog. Chem. Org. Nat. Prod., 2017, 106, 1 CAS.
- S. L. Chen, J. Xu, C. Liu, Y. J. Zhu, D. R. Nelson, S. G. Zhou, C. F. Li, L. Z. Wang, X. Guo, Y. Z. Sun, H. M. Luo, Y. Li, J. Y. Song, B. Henrissat, A. Levasseur, J. Qian, J. Q. Li, X. Luo, L. C. Shi, L. He, L. Xiang, X. L. Xu, Y. Y. Niu, Q. S. Li, M. V. Han, H. X. Yan, J. Zhang, H. M. Chen, A. P. Lv, Z. Wang, M. Z. Liu, D. C. Schwartz and C. Sun, Nat. Commun., 2012, 3, 913 CrossRef PubMed.
- J. Q. Liu, C. F. Wang, Y. Li, H. R. Luo and M. H. Qiu, Planta Med., 2012, 78, 368 CrossRef CAS PubMed.
- P. T. Binh, D. Descoutures, N. H. Dang, N. P. D. Nguyen and N. T. Dat, Nat. Prod. Commun., 2015, 10, 1911 CrossRef PubMed.
- Y. Cao, S. H. Wu and Y. C. Dai, Fungal Divers., 2012, 56, 49 CrossRef.
- C. Richter, K. Wittstein, P. M. Kirk and M. Stadler, Fungal Divers., 2015, 71, 1 CrossRef.
- Z. L. Yang and B. Feng, Mycology, 2013, 4, 1 CAS.
- Y. M. Yan, X. L. Wang, Q. Luo, L. P. Jiang, C. P. Yang, B. Hou, Z. Z. Zuo, Y. B. Chen and Y. Z. Cheng, Phytochemistry, 2015, 114, 155 CrossRef CAS PubMed.
- Y. T. Huang, H. S. Chang, G. J. Wang, C. H. Lin and I. S. Chen, Int. J. Mol. Sci., 2012, 13, 16430 CrossRef CAS PubMed.
- B. Yang, J. Dong, X. Lin, H. Tao, X. Zhou and Y. Liu, Nat. Prod. Res., 2014, 28, 967 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Q. Luo, Z. L. Yang, Y. M. Yan and Y. X. Cheng, Org. Lett., 2017, 19, 718 CrossRef CAS PubMed.
- Z. M. Chen, H. P. Chen, F. Wang, Z. H. Li, T. Feng and J. K. Liu, Fitoterapia, 2015, 102, 61 CrossRef CAS PubMed.
- L. J. Reed and H. Muench, Am. J. Epidemiol., 1938, 27, 493 CrossRef.
- H. Goto and E. Osawa, J. Am. Chem. Soc., 1989, 111, 8950 CrossRef CAS.
- H. Goto and E. Osawa, J. Chem. Soc., Perkin Trans. 2, 1993, 187 RSC.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The 1D & 2D NMR, MS, and crystallographic data of compounds 1–9. CCDC 1561836 and 1851594. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08566a |
| This journal is © The Royal Society of Chemistry 2019 |