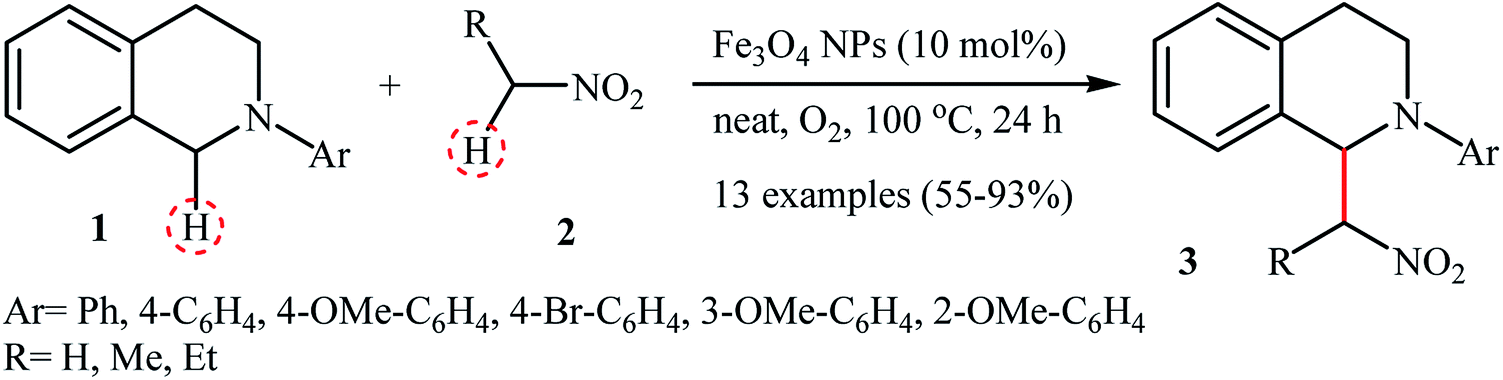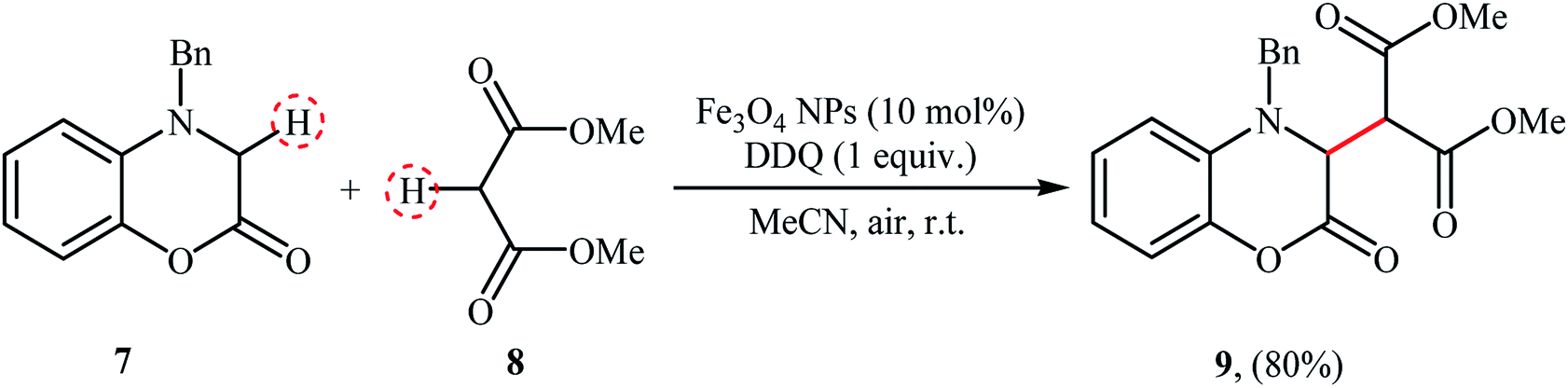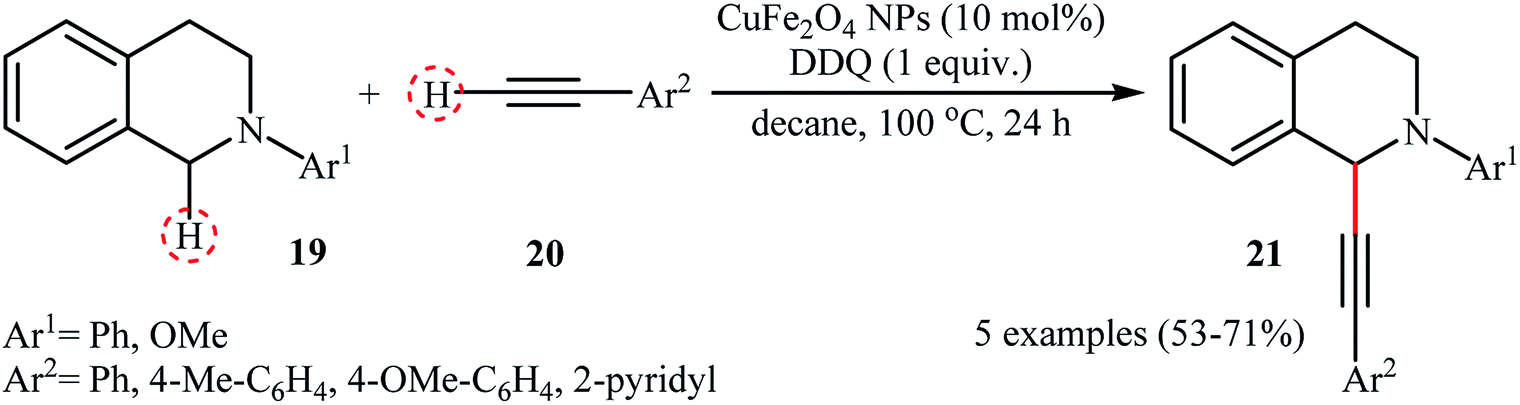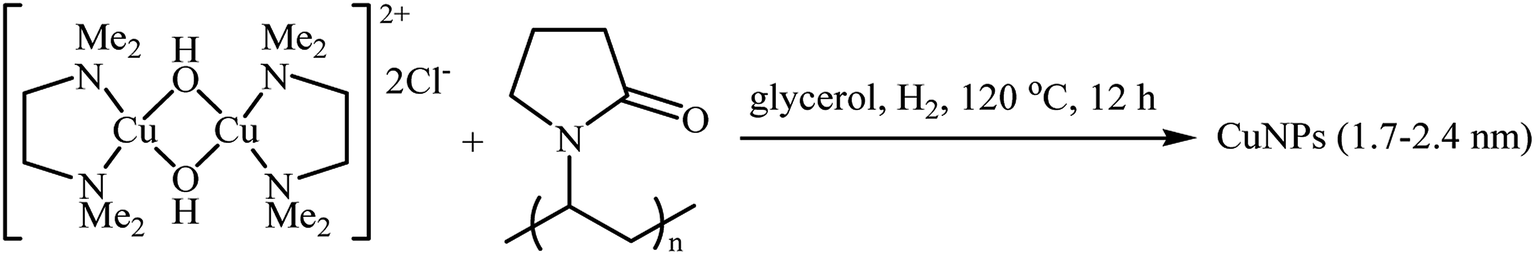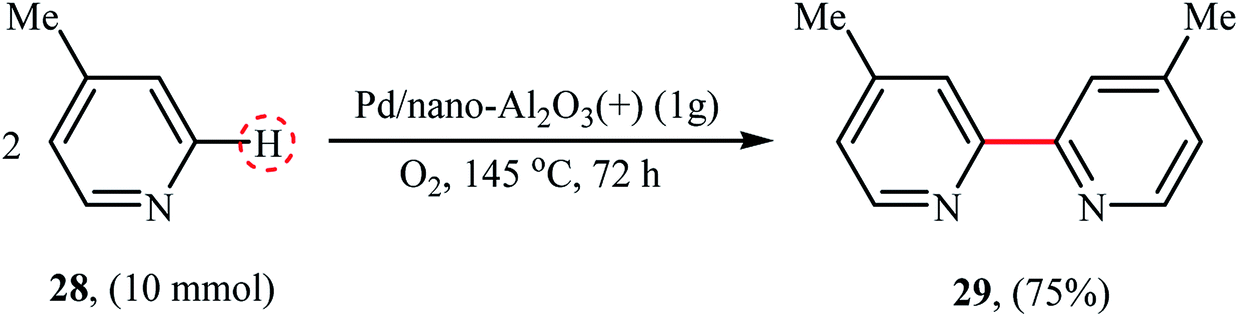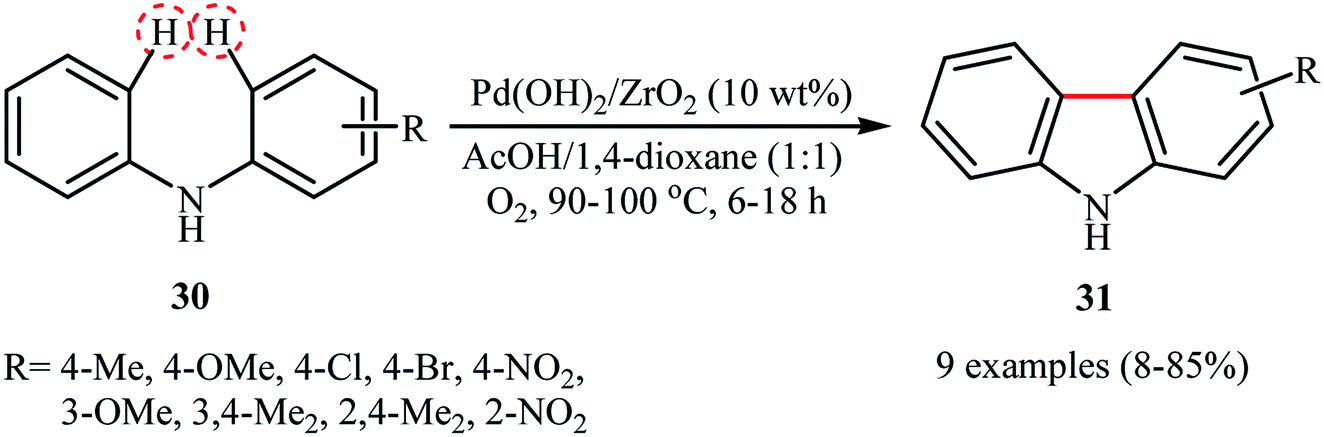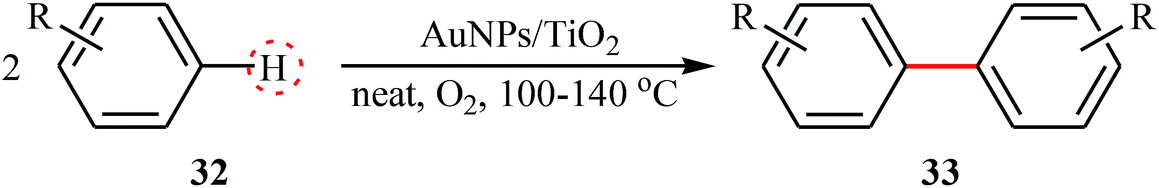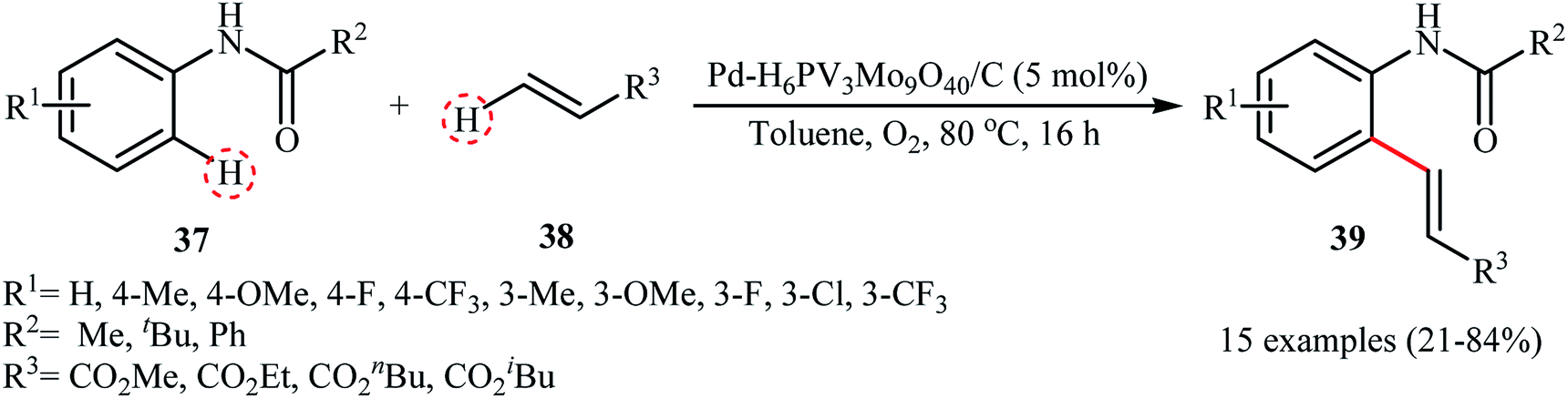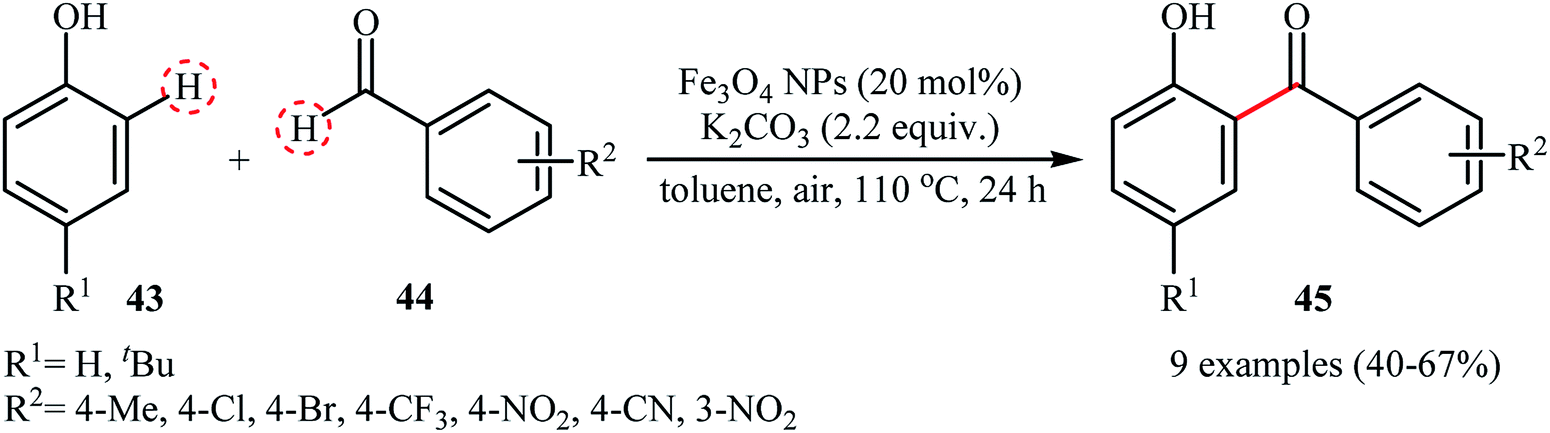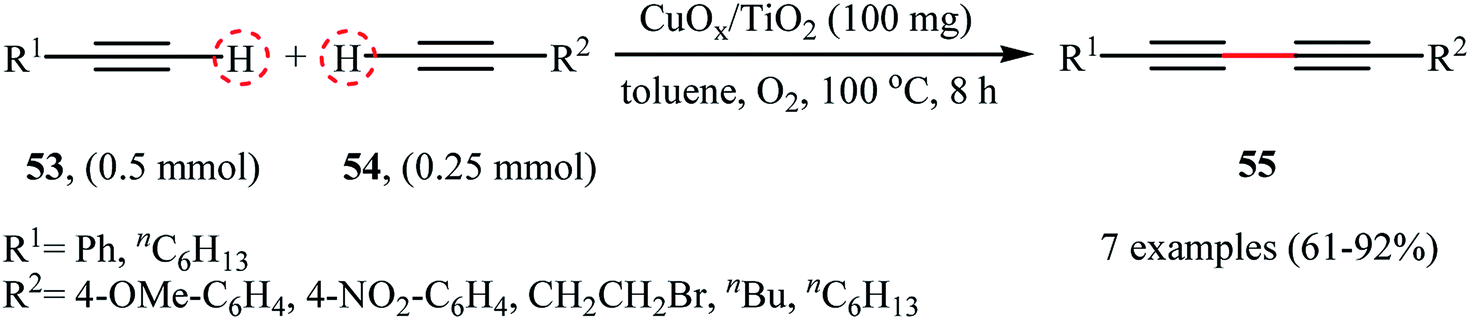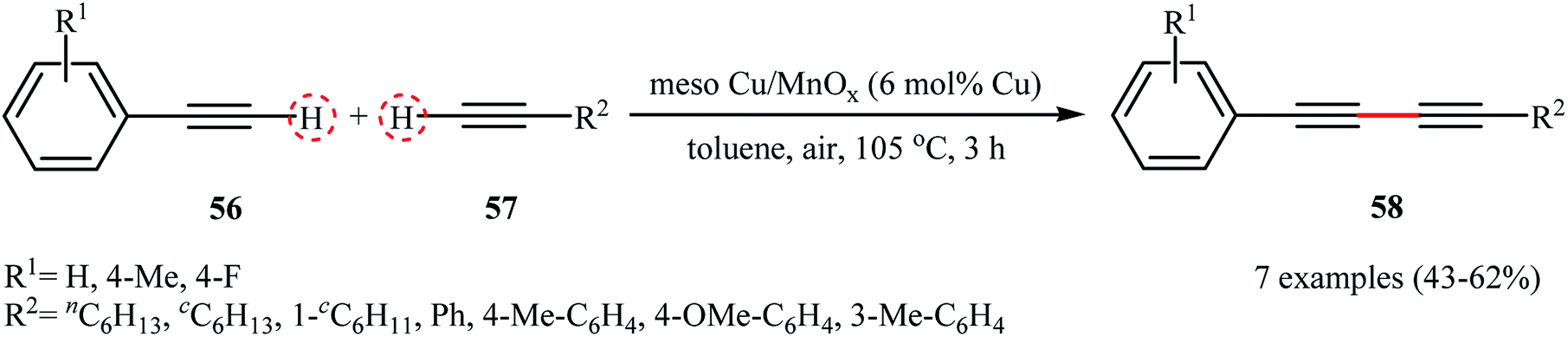DOI:
10.1039/C9RA08752D
(Review Article)
RSC Adv., 2019,
9, 41684-41702
A walk around the application of nanocatalysts for cross-dehydrogenative coupling of C–H bonds
Received
24th October 2019
, Accepted 28th November 2019
First published on 16th December 2019
Abstract
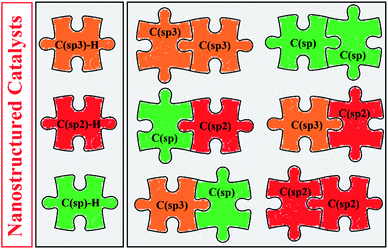
Cross-dehydrogenative coupling reactions between two unmodified C–H bonds are one of the most attractive and fundamental strategies for the construction of C–C bonds. As these reactions avoid pre-functionalization and de-functionalization of the substrates, they are cleaner, safer, and faster than traditional cross-coupling reactions. After the introduction of the modern area of cross-dehydrogenative coupling in 2003, many efforts have been devoted to the development of more efficient and selective catalytic systems for these appealing reactions. Among the different types of catalytic systems that have been investigated, nanostructured metal catalysts are highly attractive in view of their high catalytic performance, easy separability and good reusability. The purpose of this review is to focus on the application of nanocatalysts for cross-dehydrogenative coupling of C–H bonds with particular emphasis on the mechanistic aspects of the reactions. Specifically, we have structured this review based on the type of C–C bonds. Thus, the review is divided into six major sections: (i) C(sp3)–C(sp3) coupling; (ii) C(sp3)–C(sp2) coupling; (iii) C(sp3)–C(sp) coupling; (iv) C(sp2)–C(sp2) coupling; (v) C(sp2)–C(sp) coupling; and (vi) C(sp)–C(sp) coupling.
 Shahrzad Abdolmohammadi | Shahrzad Abdolmohammadi was born in Iran, in 1976. She received her BSc degree in chemistry from Alzahra University (AU), Tehran, Iran, in 1999, her MSc degree in organic chemistry from the Tarbiat_Modarres University (TMU), Tehran, Iran under the supervision of Professor Issa Yavari in 2002 and her PhD degree in organic chemistry from the Tehran University (TU), Tehran, Iran, under the supervision of Professor Hooshang Pirelahi and Professor Saeed Balalaie, in 2008. She is associate professor in East Tehran Branch, Islamic Azad University, Tehran, Iran. Her research interests include organic synthesis, heterocyclic synthesis, multicomponent reactions, nanocatalysis, organocatalysis, and synthetic methodology. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his BS degree in pure chemistry from university of Tabriz, Tabriz, Iran, and his MS degree in organic chemistry from Tehran university, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as Professor in organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
Carbon–carbon (C–C) bond formation represents a key step in the synthesis of organic materials and provides the foundation for constructing more complicated organic molecules from the simpler ones.1 The use of transition-metal-catalyzed cross-coupling reactions for the fabrication of C–C bonds has increased tremendously over the past few decades.2 Among the various C–C cross-coupling reactions, Suzuki, Sonogashira, Heck, Hiyama, Stille, Negishi, and Kumada–Corriu reactions find maximum application not only in academic laboratories but also in industrial processes.3 However, these reactions require at least one pre-functionalized partner to generate the desired products and therefore suffer from non-availability of starting materials and/or hazardous waste streams. Cross-dehydrogenative coupling reactions between two unmodified C–H bonds are the most atom-economical and sustainable synthetic alternatives to traditional coupling procedures since they avoid pre-functionalization and de-functionalization of the substrates and formally they loss non-toxic H2 as only by-product.4 In view of the high importance of this branch of cross-coupling reactions, many researchers have been working to explore novel and more efficient catalytic systems that can operate at lower loading and temperatures.
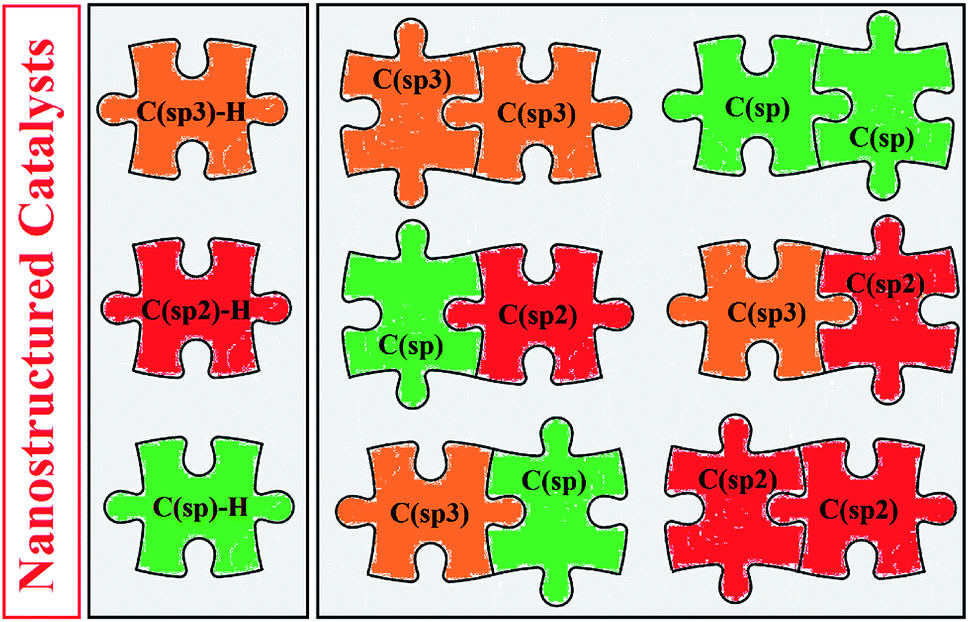
Over the past few years, nanoparticles (NPs) have gained much attention in both organic and inorganic synthesis as easily separable, reusable and environmentally sustainable catalysts.5 The high surface area per unit mass and reactive morphology of nanoparticles made them very successful heterogeneous catalysts in various organic transformations.5,6 In this context, a number of nanostructured catalytic systems have been developed in recent years which effectively promoted cross-dehydrogenative coupling reactions. To the best of our knowledge, a comprehensive review has not appeared on the application of nanocatalysts for cross-dehydrogenative coupling of C–H bonds in the literature thus far. In continuation of our preceding reviews on cross-coupling reactions, heterocyclic synthesis7 and application of nanocatalysts in organic synthesis,5b,c,6 in this review, we will highlight the most representative reports on the C–C bond forming reactions through nanoparticles catalyzed cross-dehydrogenative coupling of C–H bonds until September 2019 (Fig. 1), by hoping that it will be beneficial to the development of novel and truly efficient catalytic systems for this hot and fast growing research topic.
 |
| | Fig. 1 Nanoparticles catalyzed cross-dehydrogenative coupling of C–H bonds. | |
2. Constructing C(sp3)–C(sp3) bonds
Transition-metal-catalyzed oxidative cross-coupling of two C(sp3)–H bonds has emerged as the most attractive and also the most challenging strategy for the construction of C(alkyl)–C(alkyl) bonds.8 The difficulty arises due to their sluggish transmetalation and thus the reactive organometallic intermediates are susceptible to side reactions.9 Therefore, many researchers have been working to explore the more effective catalytic systems for this appealing synthetic strategy. In this context, several nanostructured catalysts have been developed and successfully utilized.
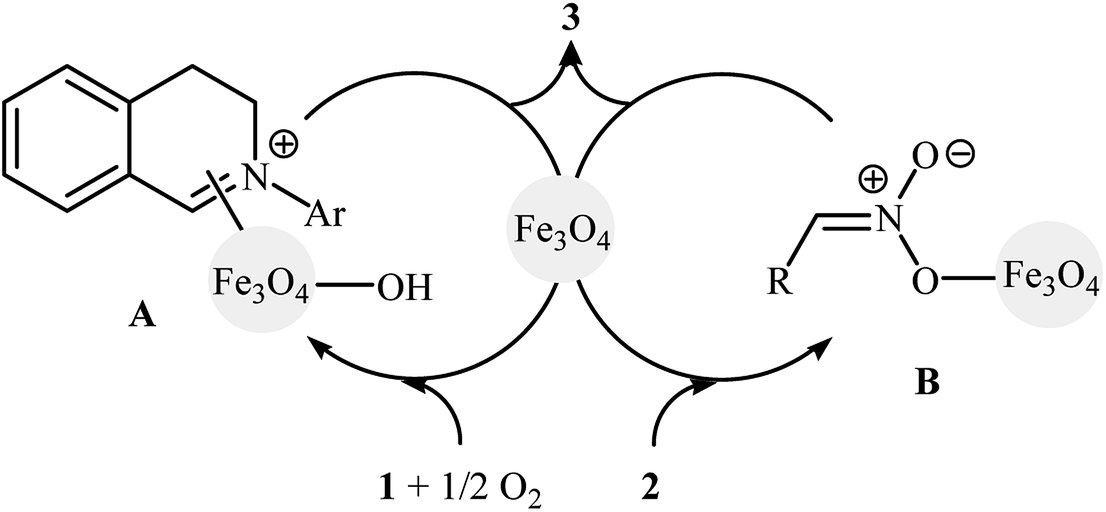
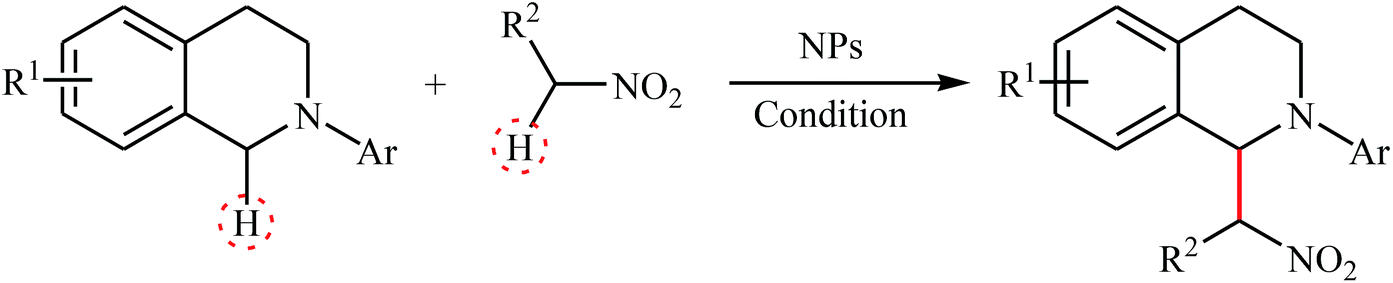
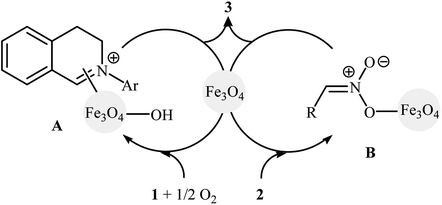
In 2010, Song and Li along with their co-workers studied the oxidative aza-Henry reactions using magnetite nanoparticles (Fe3O4 NPs) as the catalyst.10 Hence, a variety of β-nitroamine derivatives 3 were smoothly obtained in moderate to excellent yields through the reaction of corresponding N-aryl-1,2,3,4-tetrahydroisoquinolines 1 with nitroalkanes 2via oxidative functionalization of α-C(sp3)–H bonds adjacent to nitrogen atoms (Scheme 1). However, 1-phenylpyrrolidine did not work well in the reaction and therefore no other pyrrolidines were examined in the protocol. Noteworthy, nitroalkanes playing a dual role in this coupling reaction; the substrate and the solvent. Extension to oxidative Mannich reaction between tetrahydroisoquinolines and acetone were attempted and the corresponding coupling products were isolated in moderate yields. It is notable that the nanocatalyst can be easily separated from the final reaction mixture by means of an external magnet, washing with ethyl acetate, and air-dried, and then be reused for at least nine times with tangible decrease in its catalytic activity. The authors proposed reaction mechanism for this oxidative coupling reaction is illustrated in Scheme 2. The reaction starts with the oxidation of tertiary amine 1 by O2 in the presence of Fe3O4 nanoparticles, leading to the generation of the iminium cation A. Meanwhile, deprotonation of nitroalkane 2 under the reaction condition leads to intermediate B. Finally, coupling of the two intermediates A and B affords the desired product 3 and regenerates the Fe3O4 nanoparticles.
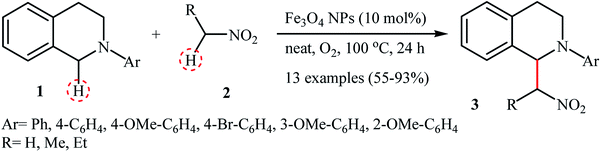 |
| | Scheme 1 Fe3O4 NPs-catalyzed oxidative aza-Henry reaction.10 | |
 |
| | Scheme 2 Proposed mechanism for the Fe3O4 NPs-catalyzed cross-dehydrogenative coupling of tertiary amines 1 with nitroalkanes 2.10 | |
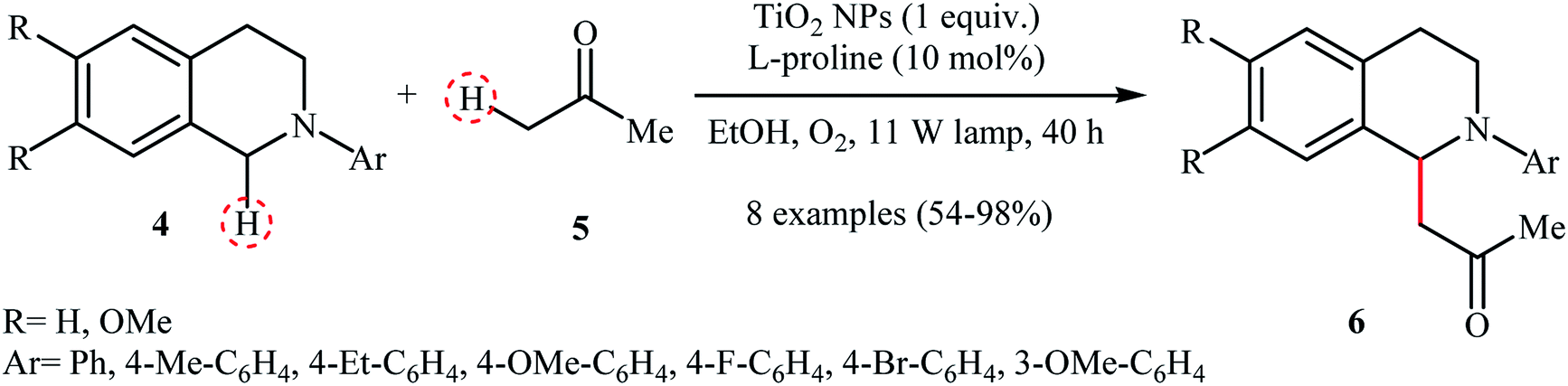
A similar example of the oxidative aza-Henry reaction by a nanocatalyst was disclosed by Rueping and co-workers in 2012, when they reported about the use of commercially available TiO2 nanoparticles (Evonik-Degussa Aeroxide P25) to promote the oxidative coupling of a series of N-aryl-1,2,3,4-tetrahydroisoquinolines with nitromethane in EtOH under irradiation of visible light.11 The catalytic system was also effective for the oxidative Mannich reaction of tetrahydroisoquinoline derivatives 4 with acetone 5 to provide the target amino-ketones 6 in moderate to almost quantitative yields (Scheme 3).
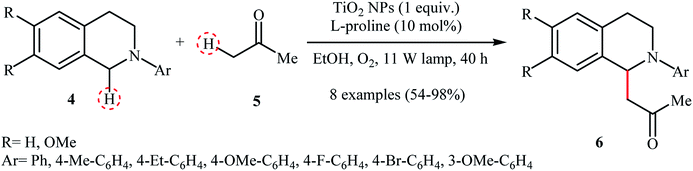 |
| | Scheme 3 Rueping's synthesis of amino-ketones 6.11 | |
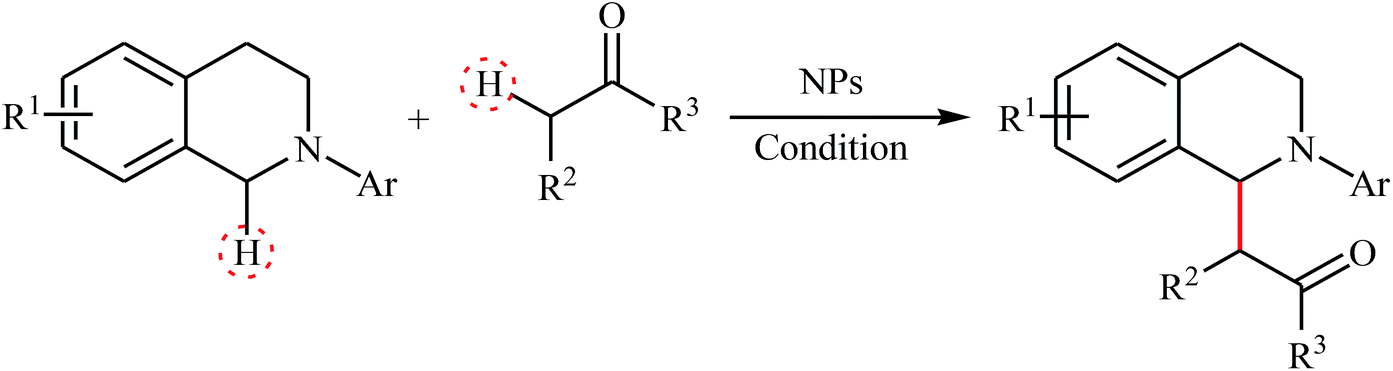
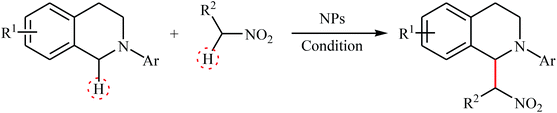
Concurrently, Wu's research team showed that graphene-supported RuO2 nanoparticles (G-RuO2 NPs) were able to promote the aza-Henry reaction of 2-aryl-substituted tetrahydroisoquinolines with nitroalkanes in the absence of any ligand or additive in the most environmentally benign solvent, water.12 Significantly, the nanocomposite has been made in situ from water-soluble sulfonic-group-functionalized graphene and RuCl3 hydrate. The nanocomposite has been characterized by using various analyses such as transmission electron microscopy (TEM), energy-dispersive spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS). The size of nanoparticles was found to be about 2 nm by TEM technique. A comparison of the catalytic performance of the G-RuO2 NPs, RuCl3·nH2O and RuO2·nH2O in the reaction of 1,2,3,4-tetrahydro-2-phenylisoquinoline with nitromethane revealed that the former was more efficient under the same condition. Moreover, G-RuO2 NPs catalyst could be easily recycled by filtration. Drawing inspiration from these works, several nanomaterial-based catalytic systems have been developed which showed high catalytic performance in the oxidative aza-Henry (Table 1) and/or oxidative Mannich reactions (Table 2) of tetrahydroisoquinolines. These include: CuFe2O4 NPs,13 AuNPore,14 Fe3O4@SiO2-bipy-[AuCl2][AuCl4],15 WO3–W1,16 sponge AuNPs,17 MNPs@eosin Y,18 and Ru@DAFO@ASMNPs.19 It is worth mentioning that some of these nanocatalyts were also successfully utilized in the cross-dehydrogenative coupling of tertiary amines with various pronucleophiles such as malononitrile, dialkyl malonate, H-phosphonate diesters, trimethylsilyl cyanide, and indole derivatives. However, the scope of tertiary amines are generally limited to the tetrahydroisoquinoline derivatives and therefore expanding the substrate scope of these reactions would be interesting.
Table 1 Nanoparticles catalyzed oxidative aza-Henry reactions of tetrahydroisoquinolines with nitroalkanes
|
|
| Entry |
Catalyst |
Conditions |
Number of examples |
Yield (%) |
Ref. |
| 1 |
CuFe2O4 NPs |
Neat, O2, 100 °C, 24 h |
7 |
73–92 |
13
|
| 2 |
AuNPore |
Neat, O2, 80 °C, 24 h |
8 |
70–99 |
14
|
| 3 |
[Fe3O4@SiO2-bipy-AuCl2][AuCl4] |
MeOH, air, 60 °C, 8 h |
15 |
74–91 |
15
|
| 4 |
WO3–W1 |
Neat, O2, r.t., 24 h |
8 |
70–91 |
16
|
| 5 |
Sponge AuNPs |
H2O, TBHP, 80 °C, 24 h |
10 |
66–98 |
17
|
| 6 |
MNPs@eosin Y |
DMSO, air, light, 12 h |
9 |
80–92 |
18
|
| 7 |
Ru@DAFO@ASMNPs |
MeOH, air, light, r.t., 12–30 h |
6 |
72–89 |
19
|
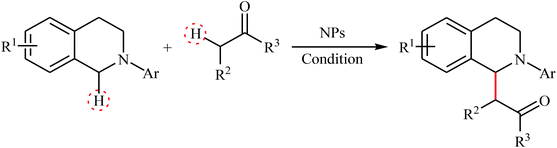
Table 2 Nanoparticles catalyzed oxidative Mannich reactions of tetrahydroisoquinolines with ketones
|
|
| Entry |
Catalyst |
Conditions |
Number of examples |
Yield (%) |
Ref. |
| 1 |
[Fe3O4@SiO2-bipy-AuCl2][AuCl4] |
AcOH, 4A MS, air, 60 °C, 12 h |
12 |
72–86 |
15
|
| 2 |
WO3–W1 |
MeOH, proline, O2, light, 24 h |
8 |
70–82 |
16
|
| 3 |
Ru@DAFO@ASMNPs |
MeCN, proline, air, light, 15–24 h |
11 |
69–88 |
19
|
The versatile magnetite nanoparticles were also exploited for the cross-dehydrogenative coupling reaction between 4-benzyl-3,4-dihydro-2H benzo[b]1,4 oxazin-2-one 7 and dimethyl malonate 8.20 The reaction was carried out in the presence of a stoichiometric amount of DDQ as oxidant in MeCN at room temperature and provided the coupling product 9 in high yield of 80% (Scheme 4). After completion of the reaction, the catalyst was easily recycled by means of an external magnet and reused seven times with a slight decrease in activity.
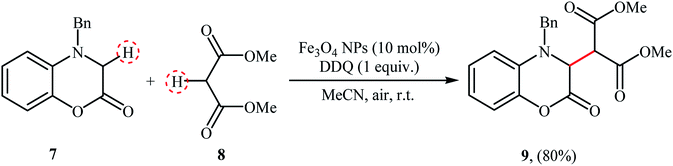 |
| | Scheme 4 Fe3O4-catalyzed coupling of 3,4-dihydro-1,4-benzoxazin-2-one 7 with malonic ester 8.20 | |
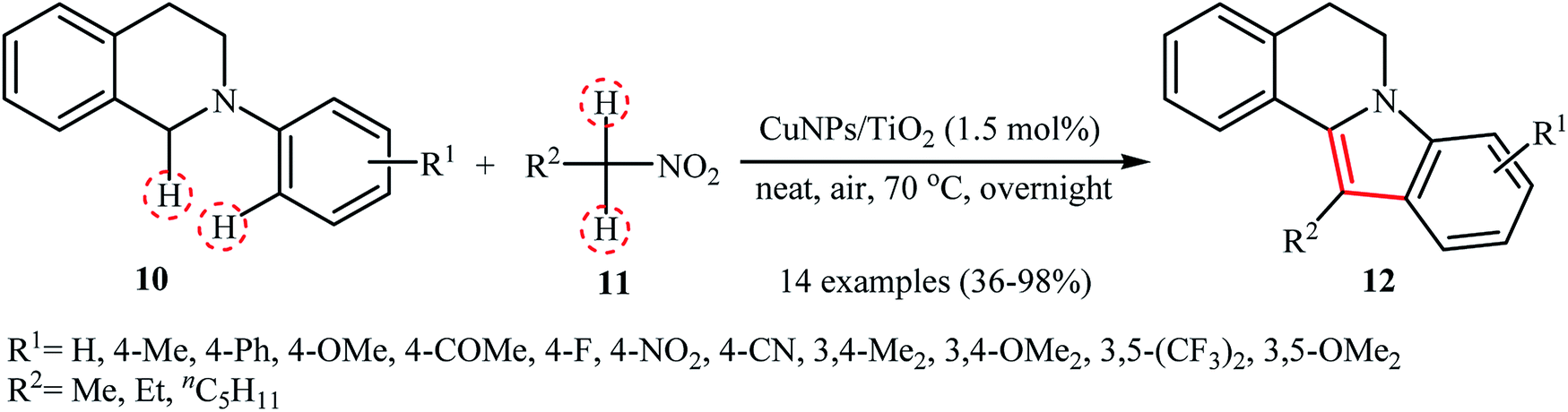
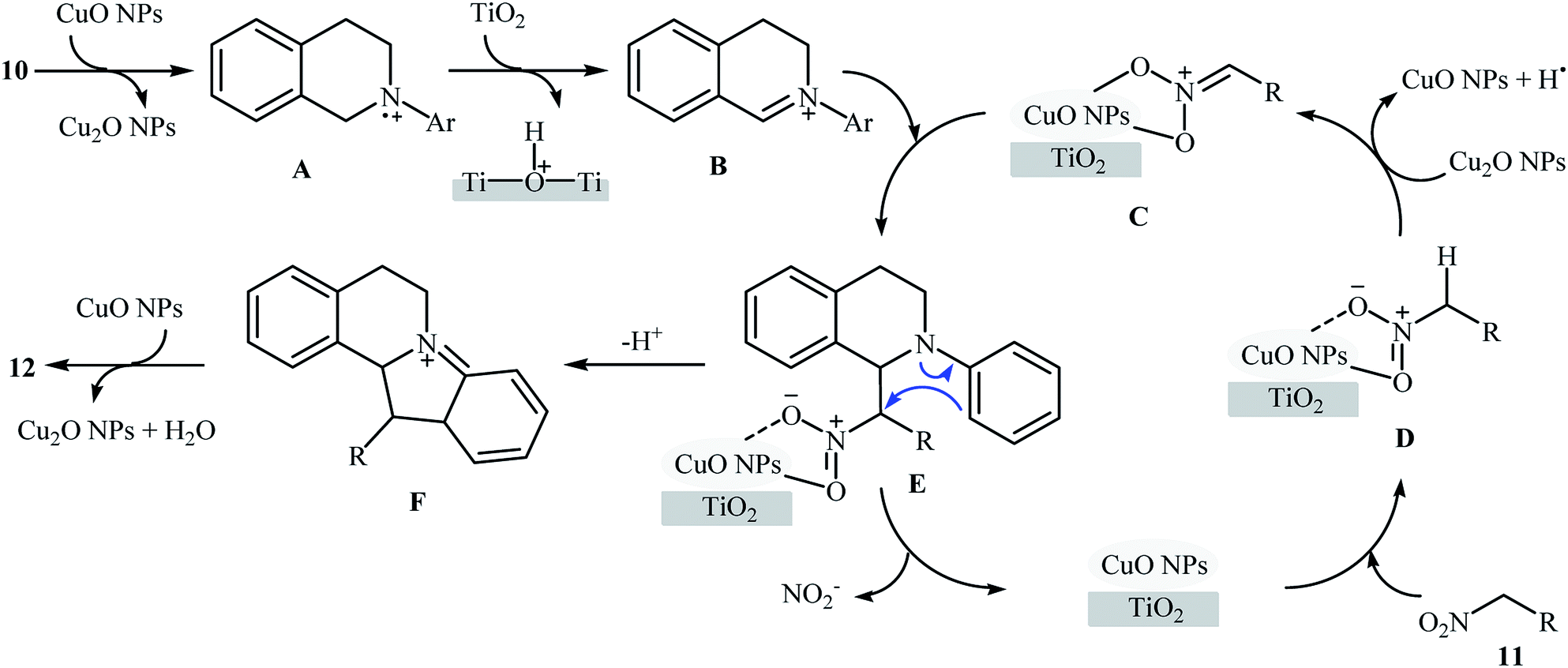
Recently, García and Alonso reported an interesting and effective synthetic route to 5,6-dihydroindolo[2,1-a]isoquinolines 12 through cross-dehydrogenative coupling of N-aryl-1,2,3,4-tetrahydroisoquinolines 10 with nitroalkanes 11 using copper nanoparticles supported on titanium oxide (Cu NPs/TiO2) as catalyst (Scheme 5).21 The best conversion efficiency was obtained for the reactions containing 1.5 mol% of catalyst at 70 °C under solvent-free conditions. Under optimized conditions, both electron-rich and electron-deficient tetrahydroisoquinoline derivatives were tolerated well and gave the target products in almost fair to excellent yields. However, nitro-substituted tetrahydroisoquinolines were incompatible in this reaction. Of note, the presence of both titanium oxide support and copper nanoparticles were crucial for the success of this transformation. No desired product was found in the absence of any of them. Moreover, the yields in this system were strongly dependent on the temperature, either increase or decrease of the temperature was decreased the reaction efficiency. Mechanistically, a free radical process was likely involved in this reaction (Scheme 6), which could be completely inhibited by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), a radical scavenger.
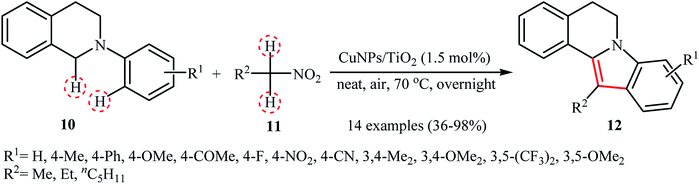 |
| | Scheme 5 Synthesis of dihydroindoloisoquinolines 12 through the CuNPs/TiO2-catalyzed cross-dehydrogenative coupling of tetrahydroisoquinolines 10 and nitroalkanes 11.21 | |
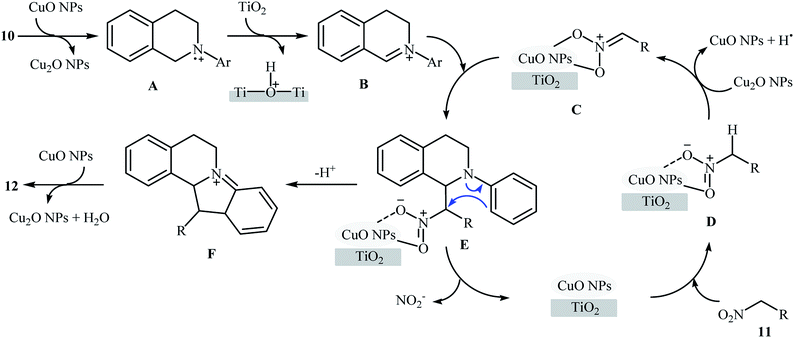 |
| | Scheme 6 Proposed mechanism for the formation of dihydroindoloisoquinolines 12.21 | |
3. Constructing C(sp3)–C(sp2) bonds
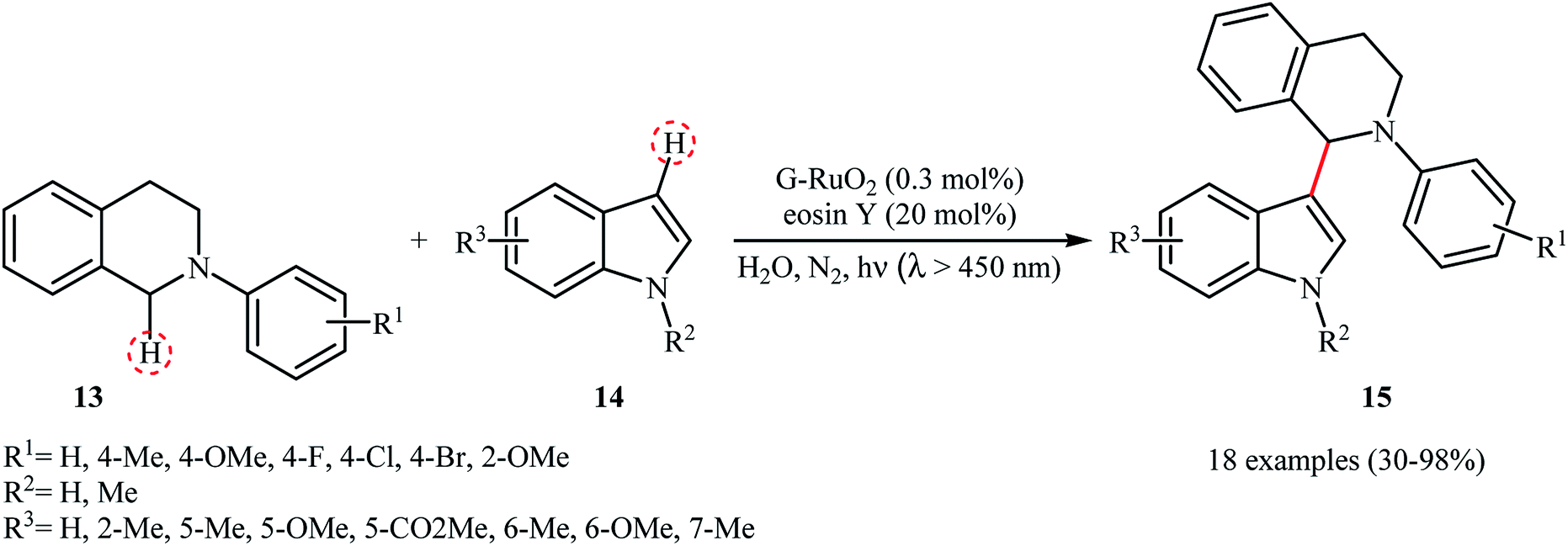
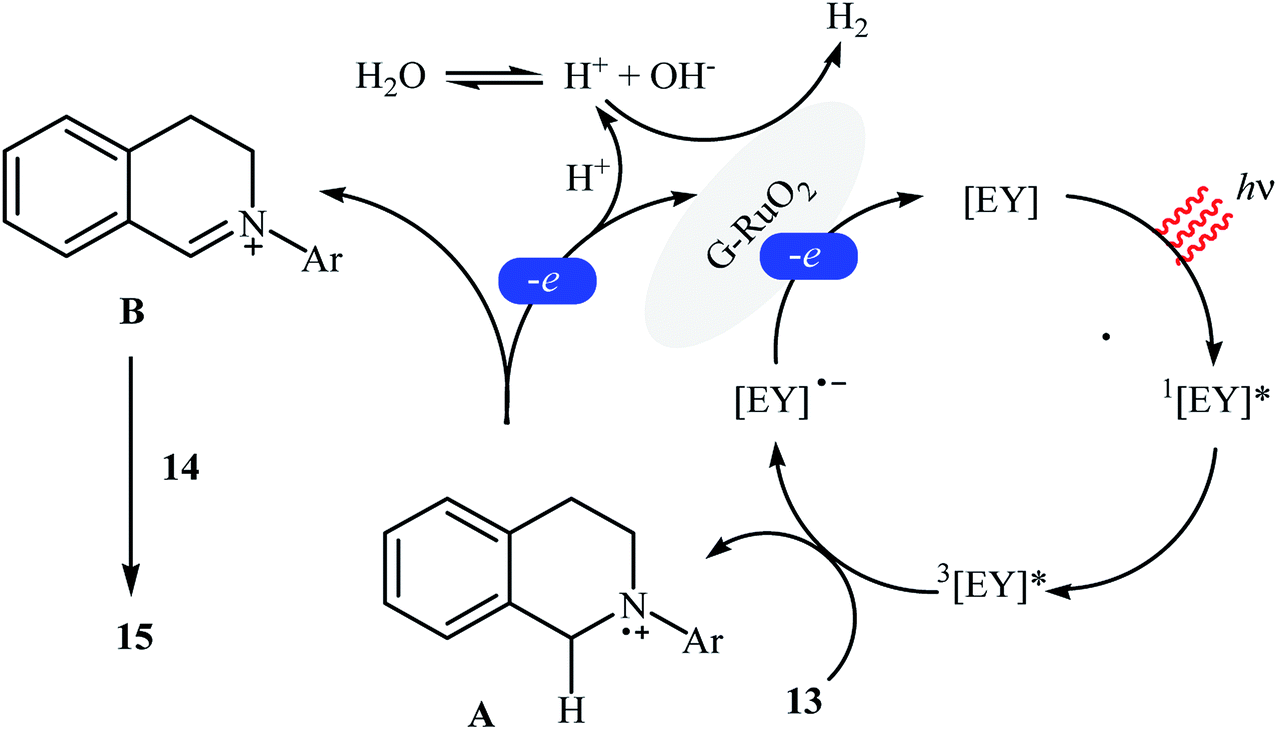
The first example of a photocatalytic cross-dehydrogenative coupling reaction employing two different C–H bonds was described in 2013 by Wu et al., who showed that the treatment of N-aryl-1,2,3,4-tetrahydroisoquinolines 13 with functionalized indoles 14 in the presence of a combination of eosin Y and graphene-supported RuO2 nanocomposite (G-RuO2 NPs) in water under irradiation of visible light (λ > 450 nm) afforded the C3-alkylated indoles 15 with yield ranging from 30% to 98% (Scheme 7).22 The reaction tolerated a number of important functional groups such as fluoro, chloro, bromo, methoxy, and ester functionalities and promised its potential applications for further manipulation of the final products. However, the reaction failed in the case of a cyano-substituted substrate. Unfortunately, the recyclability/reusability of the catalyst was not explored in this study. To gain insights into the mechanism for this oxidative coupling reaction, kinetic isotope effect experiment was conducted, which revealed that the dissociation of proton from amine was involved in the rate-determining step. In accord to the presumed mechanistic pathway, the reaction starts with the formation of cation radical species Avia a single-electron transfer from tertiary amine 13 to the excited organophotocatalyst [eosin Y]*. Next, this intermediate A converts into the iminium ion intermediate B through a deprotonation and oxidation sequential process. Subsequently, the nucleophilic addition of indole 14 to this intermediate gives rise to the cross-coupling product 15. On the other hand, the formed radical anion [eosin Y]˙− is restored to its ground state by G-RuO2 in water (Scheme 8). Shortly afterwards, Jin and co-workers studied the similar oxidative coupling of tertiary amines with indoles using zero-valent nanoporous gold (AuNPore) as the catalyst and O2 as an oxidant in refluxing MeOH.14 Thereby, the corresponding α-indolyl-substituted amines were obtained in moderate to good yields after 24 hours (45–77% for 4 examples). Later, Yan demonstrated that Ru nanoparticles (Ru-NP-1; thickness of 1.4 nm) are also suitable catalysts for the cross-dehydrogenative coupling of N-aryl-1,2,3,4-tetrahydroisoquinolines with indoles.23 Thus, by using 8 mol% of catalyst in binary solvent H2O/MeOH in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at room temperature, the desired coupling product were obtained in high yields (up to 98%); however, acidic condition and long reaction time (27–110 h) were necessary. The recyclability of the catalyst was examined, showing a little but noticeable loss in catalytic activity (from 97% in the first run to 77% in the sixth run).
:1 ratio at room temperature, the desired coupling product were obtained in high yields (up to 98%); however, acidic condition and long reaction time (27–110 h) were necessary. The recyclability of the catalyst was examined, showing a little but noticeable loss in catalytic activity (from 97% in the first run to 77% in the sixth run).
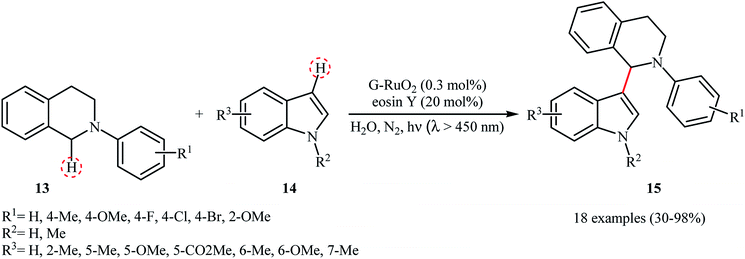 |
| | Scheme 7 Wu's synthesis of C3-alkylated indoles 15.22 | |
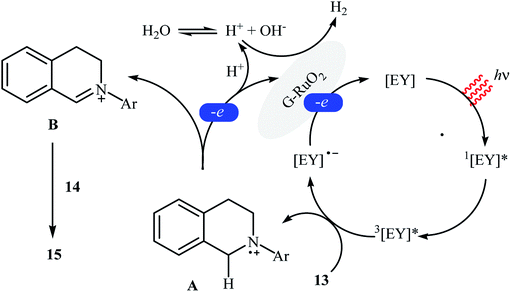 |
| | Scheme 8 Suggested mechanism for the reaction in Scheme 7.22 | |
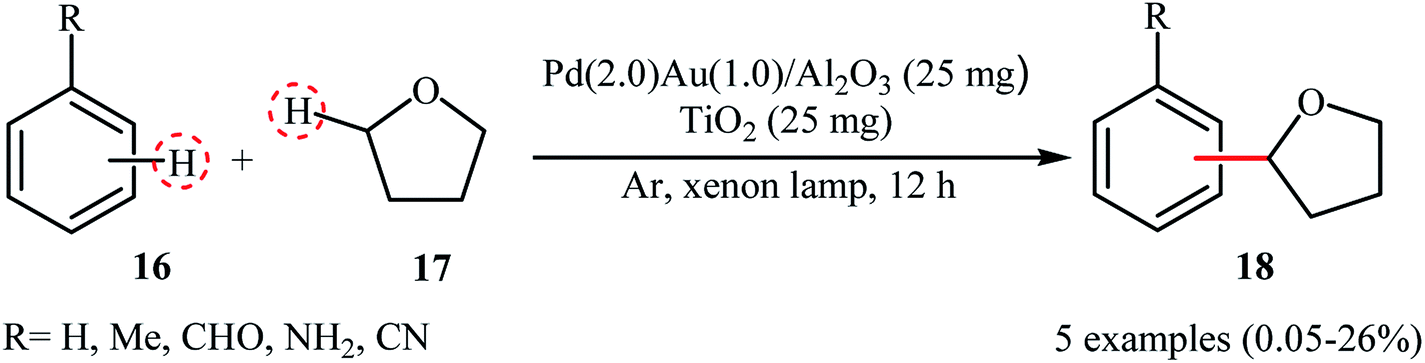
Recently, Yoshida and colleagues disclosed photocatalytic direct cross-dehydrogenative coupling reaction between simple arenes 16 and tetrahydrofuran 17 by using a novel blended catalyst consisting of a TiO2 photocatalyst and an Al2O3-supported PdAu bimetallic catalyst (3–4 nm).24 The reactions were done under the irradiation of xenon lamp and solvent-free conditions without consuming any oxidizing agent or other additional chemicals; however, only trace amounts of the corresponding α-arylated ethers 18 were obtained (Scheme 9). The conversion and selectivity of the target cross-coupling products heavily depended on the ratio of the two catalytic components, the best results were observed when Pd(2.0)Au(1.0)/Al2O3 was used as the catalyst. Noteworthy, compared to the conventional Pd-loaded TiO2 photocatalyst and the monometallic Pd/Al2O3 and Au/Al2O3 catalysts, the hybrid catalyst showed superior catalytic activity. In the bimetallic catalyst, electron density being transferred from the Pd atoms to Au atoms and thus the Pd atoms became electron deficient and the Au atoms became electron rich on the support, as indicated by XANES (X-ray absorption near edge structure) analyses of the monometallic and bimetallic samples.
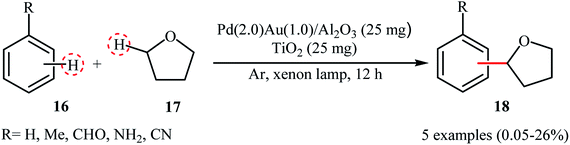 |
| | Scheme 9 Dehydrogenative cross-coupling between arenes 16 and tetrahydrofuran 17 reported by Yoshida.24 | |
4. Constructing C(sp3)–C(sp) bonds
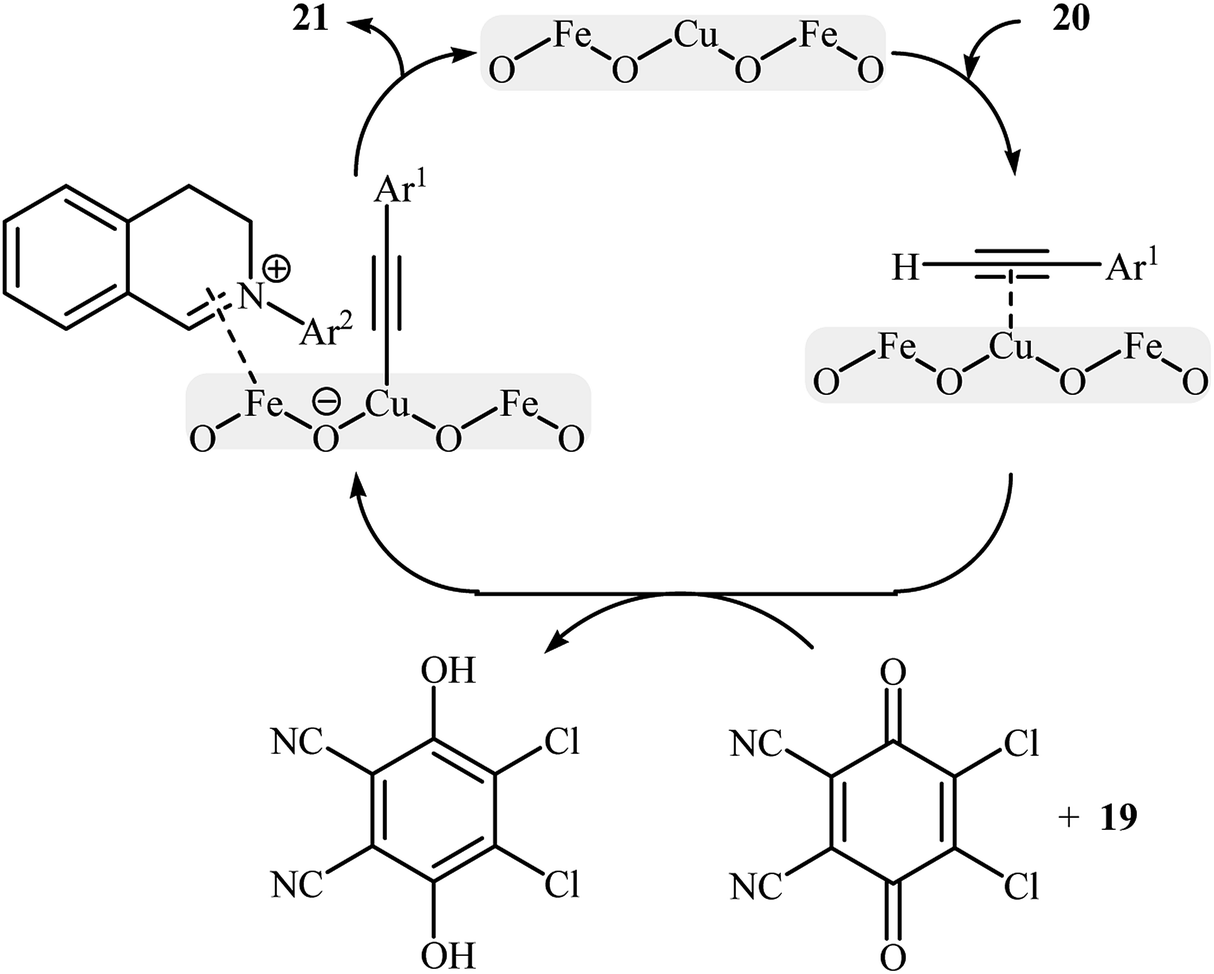
Li-Moores and co-workers were among the first to realize cross-dehydrogenative coupling reactions between sp3-hybridized and sp-hybridized C–H bonds using metal nanoparticles as catalysts.13 In 2013, they discovered that a magnetic nanocatalyst in combination with an oxidant effects the direct α-alkynylation of 2-aryl-1,2,3,4-tetrahydroisoquinolines 19 with aromatic alkynes 20, a reaction that has been subject of number of papers in recent years. The best conversion efficiency was obtained for the reactions containing CuFe2O4 (10 mol%) and DDQ (1 equiv.) in decane at 100 °C. Under optimized conditions, both aromatic and heteroaromatic terminal alkynes were tolerated well and gave the desired coupling products 21 in moderate to good yields (Scheme 10). It is worthwhile to mention that Fe3O4 NPs completely failed to promote this transformation. This result clearly showed that the catalytic activity of nano-CuFe2O4 is due to the presence of copper atoms that able to coordination to the C–C triple bonds and formation of active copper acetylides (Scheme 11). In a similar approach, the Ramón group reported the synthesis of a library of α-alkynylated tetrahydroisoquinoline derivatives utilizing a catalytic amount of iron oxide-supported copper(II) oxide nanoparticles (Fe3O4–CuO NPs; 2–4 nm) in a 1:2 mixture of choline chloride and ethylene glycol.25 Of note, other metal oxides impregnated on magnetite such as PdO–Fe3O4, Ru2O3–Fe3O4, NiO–Fe3O4, CoO–Fe3O4, OsO2–Fe3O4, Ag2O–Fe3O4, Au2O3–Fe3O4, IrO2–Fe3O4, PtO/PtO2–Fe3O4, WO3–Fe3O4 were also found to promote this reaction, albeit at lower efficiencies. As compared to the heterogeneous Fe3O4–CuO catalyst, poorer results were obtained by the CuO and a mixture of CuO and Fe3O4. These results indicated that the metal oxide and support in the catalyst play synergistic roles in activating both C(sp3)–H and C(sp)–H bonds.
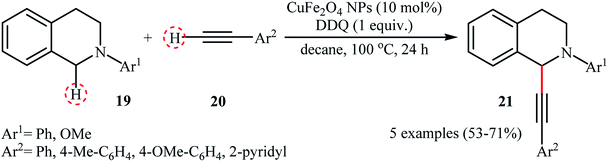 |
| | Scheme 10 CuFe2O4 NPs-catalyzed α-alkynylation of 2-aryl-1,2,3,4-tetrahydroisoquinolines 19 with aromatic alkynes 20 reported by Ramón.25 | |
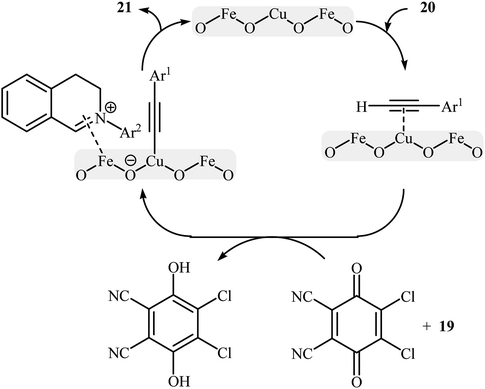 |
| | Scheme 11 Proposed mechanism for the reaction in Scheme 10.25 | |
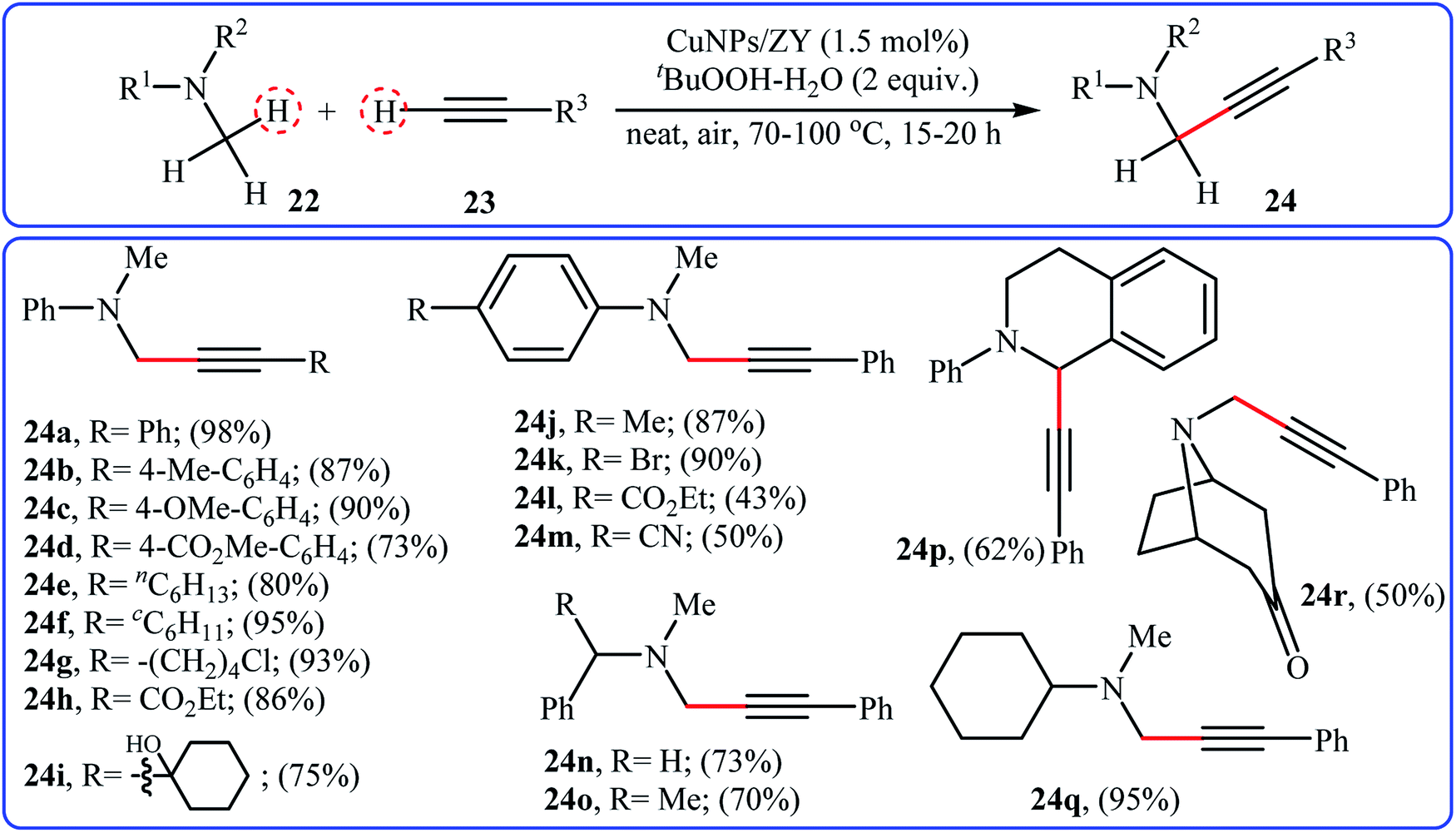
In 2015, a related α-alkenylation of tertiary amines with terminal alkynes was reported by Alonso and co-workers.26 They showed that in the presence of only 1.5 mol% of zeolite Y-supported Cu nanoparticles as the catalyst and tert-butyl hydroperoxide (tBuOOH) as an oxidant, reaction of a variety of tertiary amines 22, including aromatic, benzylic and aliphatic ones with a wide range of aromatic and aliphatic terminal alkynes 23 under the open air furnished the corresponding α-alkenylated products 24 in moderate to excellent yields (Scheme 12). This synthetic strategy tolerated a number of common functional groups, including chloro, bromo, hydroxy, cyano, ether, ester, and ketone functionalities, and exhibited very high level of regioselectivity. Noteworthy, the reaction can be performed on a gram scale without a significant decrease in yield. Additionally, the catalyst can be reused several times, without loss of reactivity. It should be pointed out that replacing zeolite Y with other supports such as activated carbon, mont K-10, MgO, TiO2 decreased reaction efficiencies. Later, Jain and Singh along with their co-workers disclosed the alkenylation of benzylic tertiary amines employing Cu6Se4.5 NPs/TBHP combination as a catalytic system under solvent-free condition; however, lower product yields were obtained and higher temperature (100 °C) and an inert atmosphere were required.27
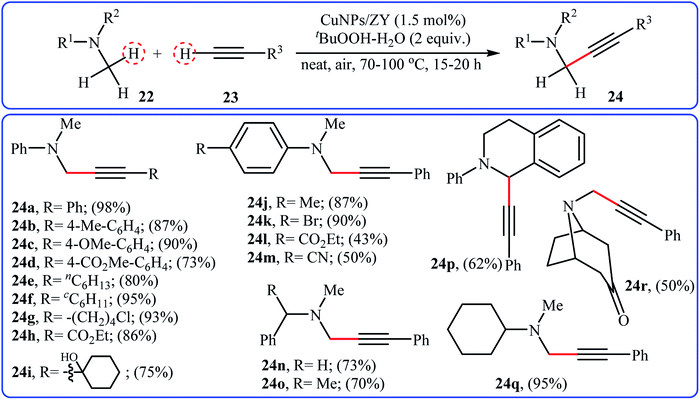 |
| | Scheme 12 Cross-dehydrogenative coupling of tertiary amines 22 and terminal alkynes 23 catalyzed by CuNPs/ZY.26 | |
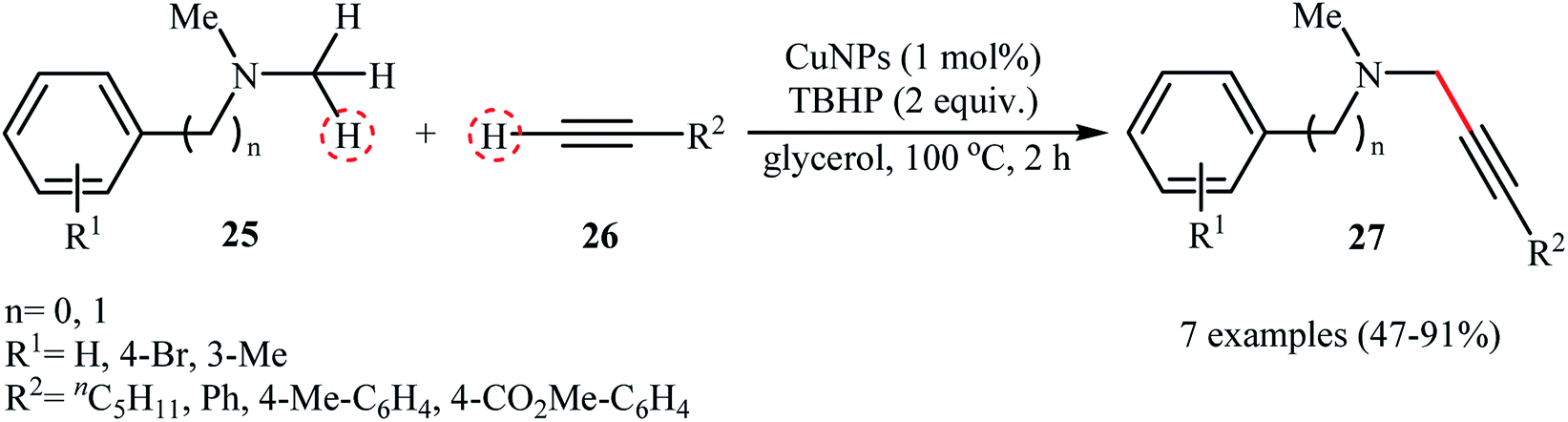
Recently, the group of Favier-Gómez's reported the synthesis of ultrafine zero-valent copper nanoparticles (NPs) with mean diameters from about 1.7 to 2.4 nm by decomposition of [Cu(κ2-N,N,N′,N′-TMEDA)(μ-OH)]2Cl2 (TMEDA = tetramethylethylenediamine) under hydrogen gas atmosphere (3 atm), in the presence of polyvinylpyrrolidone (PVP) as stabilizer in glycerol (Scheme 13).28 The catalytic activity was evaluated in the oxidative coupling of tertiary amines 25 with terminal alkynes 26 in the presence of TBHP as an oxidant in glycerol. The obtained results from this study showed that the desired coupling products 27 were formed in moderate to excellent isolated yields (Scheme 14). The best results were obtained for aromatic tertiary amines. The prepared Cu NPs was also successfully applied as a catalyst for one-pot construction of synthetically important propargylic amines,29 through the A3 coupling reaction of amines, aldehydes, and terminal alkynes.
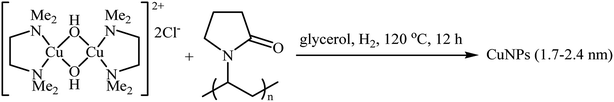 |
| | Scheme 13 Preparation of CuNPs reported by Favier and Gómez.28 | |
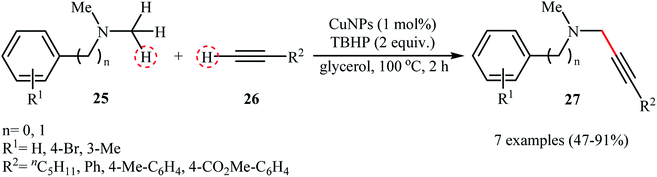 |
| | Scheme 14 Copper(0) nanoparticles catalyzed coupling of C(sp3)–H bonds with C(sp)–H bonds.28 | |
5. Constructing C(sp2)–C(sp2) bonds
5.1. C(aryl)–C(aryl) bonds
Biaryls are the core structure of many natural products and commercialized drugs such as imatinib, losartan, felbinac, febuxostat, and telmisartan.30 Additionally, they are widely found in agrochemicals, liquid crystals, and ligands.30,31 In light of the widespread biological activities of functionalized biaryls, numerous synthetic methods have been developed for their preparation which the majority of them relying on the known C–C cross-coupling reactions, including Ullmann, Suzuki, Hiyama, Negishi, stille, and Kumada–Corriu reactions.5b,c,30–34 Unfortunately, all of these methods relied on the use of pre-functionalized coupling partners, which limit their range of applications. An alternative protocol for the construction of the titled scaffolds involves the oxidative coupling between two C(sp2)–H bonds.35 Although several efficient catalytic systems have been developed, the application of nanoparticles as catalysts for this reaction is a new research area and thus scarcely studied.
In 2008, the first biaryl synthesis by double C–H bond coupling over heterogeneous metal nanoparticles was reported by Neal and Weaver.36 Several nano alumina-supported palladium catalysts were tested for the synthesis of 4,4′-dimethyl-2,2′-bipyridine 29 from 4-methyl pyridine 28 and Pd/nano-Al2O3(+) catalysts prepared via the precipitation method afforded the highest yield (Scheme 15). Interestingly, the catalytic activity was very dependent on the catalyst preparation methods, the catalyst prepared via the wet impregnation method exhibited poor activities in this reaction compared to the one prepared via the precipitation method. Additionally, the catalysts prepared using another alumina nanoparticles (e.g., nano-Al2O3(−) and γ-Al2O3) did not exhibit significant activities in the reaction and the commercial Pd/Al2O3 was found to be almost inactive catalyst.
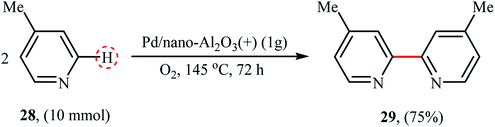 |
| | Scheme 15 Pd/nano-Al2O3(+)-catalyzed homo-coupling of 4-methyl pyridine 28.36 | |
Six years later, Ishida and Tokunaga along with their co-workers describe that ZrO2-supported Pd(OH)2 nanoparticles (1.8 ± 0.9 nm) can be used as effective heterogeneous catalysts for the C(aryl)–C(ary) bonds formation via double C(aryl)–H bond functionalization.37 10 wt% of catalyst has been utilized in the oxidative intramolecular coupling of diarylamines 30 in a 1:1 mixture of AcOH/1,4-dioxane under oxygen atmosphere to give the corresponding carbazoles 31 in poor to high yields, ranging from 8% to 85% (Scheme 16). The results demonstrated that the electronic character of the substituents on the aryl rings had a high impact on the facility of the reaction. Generally, the electron-rich diarylamines afforded better yields compared to the electron-deficient ones. Moreover, the outcome of the reaction was strongly dependent on the substitution pattern of the starting material. For example, 3-methoxydiphenylamine was more reactive than 4-methoxydiphenylamine. The authors described this fact by high electron density of m-methoxydiphenylamine at the o,p-positions which accelerate the attack by electron-deficient Pd(II) species. It should be mentioned that the catalyst were also found to be highly efficient for the synthesis of dibenzofurans from the corresponding diarylethers.
 |
| | Scheme 16 Synthesis of carbazoles 31 from the corresponding diarylamines 30 through the double C(aryl)–H bond functionalization.37 | |
Similar to PdNPs-catalyzed double C(aryl)–H bond coupling reactions, a few AuNPs-catalyzed reactions were also reported in literature. In 2014, Serna and Corma reported the successful use of AuNPs/TiO2 in the waste-free preparation of biaryls 33 from the corresponding aryl molecules 32 using molecular oxygen as the sole oxidant under solvent-free conditions (Table 3).38,39 Noteworthy, the particle size of supported gold strongly influences the activity of catalyst. The highest catalytic activity was observed for gold crystallites of ∼3 nm diameter.
Table 3 AuNPs/TiO2-catalyzed oxidative C–H/C–H homocoupling of aromatic compounds 32
|
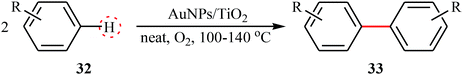
|
| Entry |
R |
Au/arene (mol ratio × 100) |
TOFa |
TONb |
Selectivity (%) |
|
Turnover frequency (TOF).
Turnover number (TON).
|
| 1 |
H |
0.022 |
382 |
230 |
99 |
| 2 |
Me |
0.026 |
176 |
206 |
98 |
| 3 |
1,5-Me2 |
0.030 |
18 |
71 |
98 |
| 4 |
1,2,6-Me3 |
0.034 |
6 |
8 |
98 |
| 5 |
Cl |
0.032 |
368 |
88 |
99 |
| 6 |
NO2 |
0.035 |
176 |
175 |
99 |
| 7 |
OH |
0.027 |
85 |
336 |
80 |
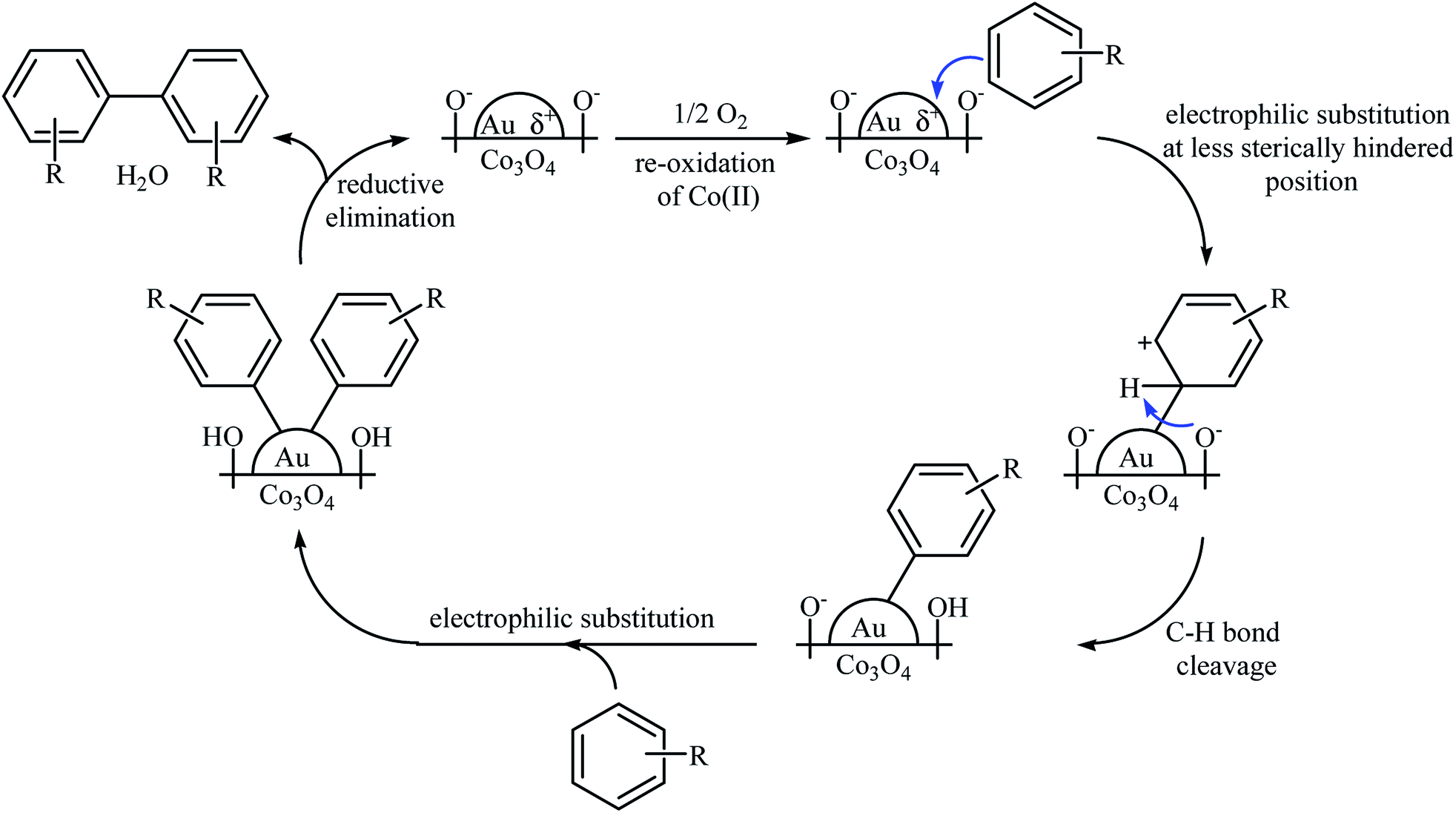
Another (Au NP)-based catalyst, i.e., Au/Co3O4, was also found to be active towards the oxidative coupling of simple arenes.40 The reactions were occurred in AcOH under 1.5 MPa pressure of O2 at 100–150 °C. Although a series of functionalized biaryls were obtained in good yields, the procedure was not general and several substrates (e.g., 1,4-di-tert-butylbenzene, methyl benzoate, dimethyl terephthalate, 1-(trifluoromethyl)benzene) failed to participate in this reaction or afforded only a trace amount of the target products. Based on a series of control experiments, the authors suggested that the reaction proceeds by an electrophilic aromatic substitution pathway as depicted in Scheme 17.
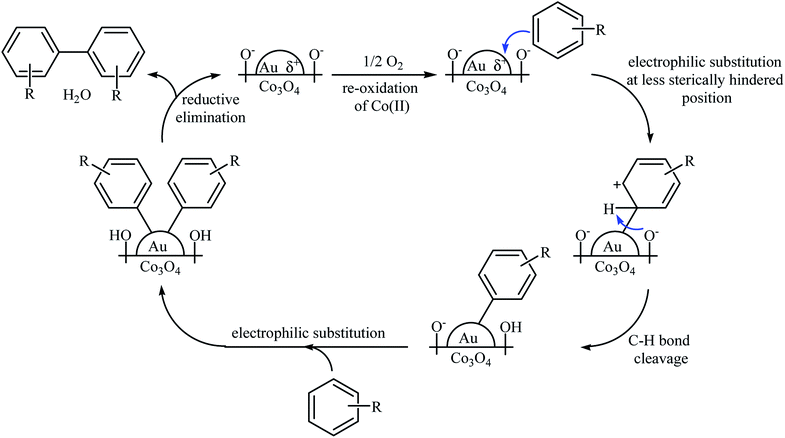 |
| | Scheme 17 Plausible reaction mechanism for the oxidative coupling of aromatic compounds over Au/Co3O4.40 | |
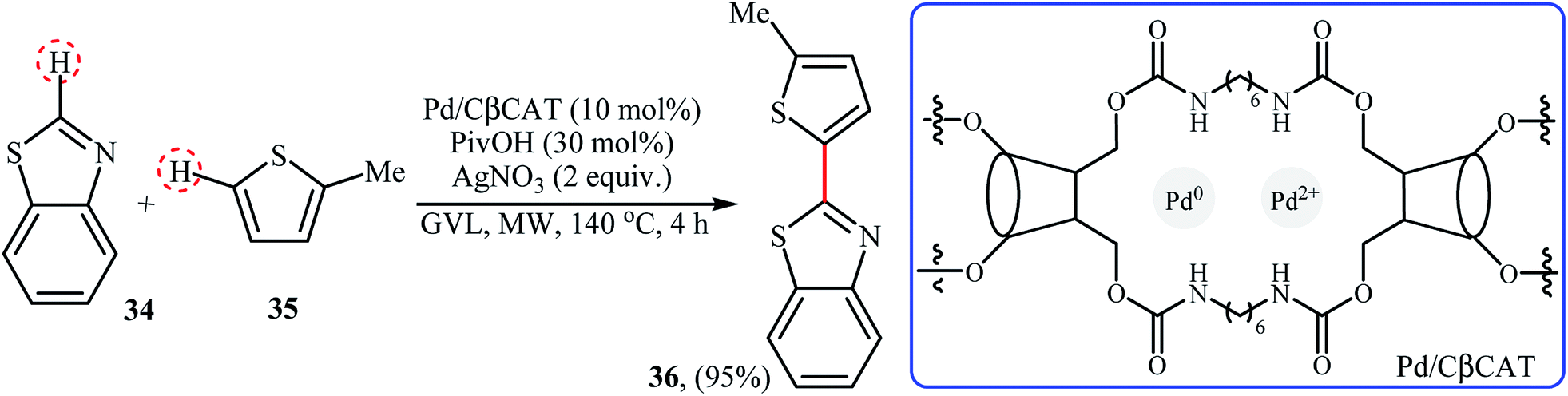
Very recently, Cravotto and co-workers prepared Pd cross-linked β-cyclodextrin (Pd/CβCAT) as a green heterogeneous catalyst for the MW-assisted cross-dehydrogenative coupling of arenes.41 In the presence of 10 mol% of catalyst, 30 mol% of pivalic acid (PivOH), and 2 equiv. of AgNO3, benzo[d]thiazole 34 react with 2-methylthiophene 35 in γ-valerolactone (GVL) at 140 °C to provide the C-2 heteroarylated product 36 in 95% yield (Scheme 18). DMA was also found to be useful solvent for this transformation. However, other solvents like ethyl lactate, ethyl levulinate were not so effective like GVL or DMA. Unfortunately, recycling test of the catalyst indicated a strong decrease in activity after each reaction cycle, as the conversion drops dramatically from 95% to 15% in the sixth cycle.
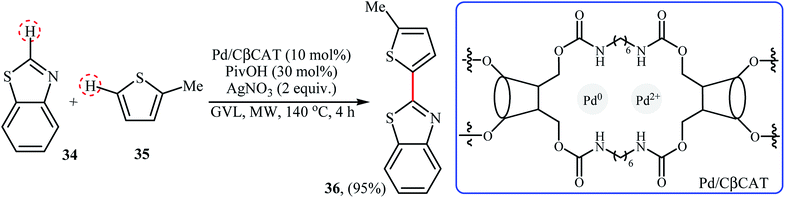 |
| | Scheme 18 Coupling of benzo[d]thiazole 34 with 2-methylthiophene 35 reported by Cravotto.41 | |
5.2. C(aryl)–C(alkenyl) bonds
The transition-metal-catalyzed dehydrogenative Heck reaction (Fujiwara–Moritani reaction) is one of the most straightforward and economical protocols for the construction of olefination products.42 Over the years, numerous excellent catalytic system have been developed for this elegant C–C bond forming reaction.43 Despite their benefits, usually they require the addition of co-catalysts, ligands, and/or additives, which may limit the application of this interesting page of C–C bond formation more or less. In 2011, Ying and colleagues developed a highly active and reusable palladium-polyoxometalate nanocatalyst with the formula Pd–H6PV3Mo9O40/C for the efficient oxidative olefination of anilides 37 with acrylates 38, using O2 as the terminal oxidant to produce substituted anilides 39 under relatively mild conditions (Scheme 19).44 The results revealed that the process strongly depended on the electronic nature of the anilides, with the best yields were obtained with unsubstituted or with monomethylated substrates. The same catalytic system was also applied to the oxidative C–H/N–H cross-coupling of various acrylates with secondary amines, and the corresponding products were obtained in high yields (up to 90%). The presence of polyoxometalate was crucial for the oxidative C–C and C–N coupling reactions since it served as a reoxidation catalyst under O2. The catalyst could be recycled several times with only a minor loss in its activity and without any change in the size of the Pd NPs as confirmed by their TEM images (Fig. 2).
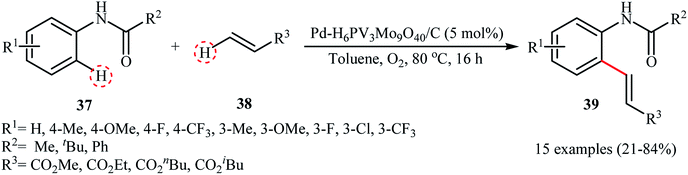 |
| | Scheme 19 PdNPs-catalyzed dehydrogenative Heck reaction.44 | |
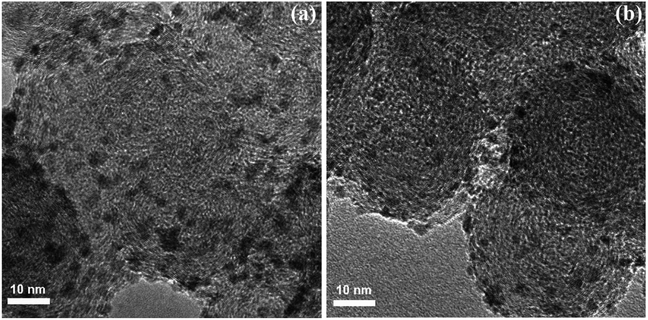 |
| | Fig. 2 TEM images of Pd-PV3Mo9/C (a) before and (b) after the first recycle of the C–C coupling reaction.44 | |
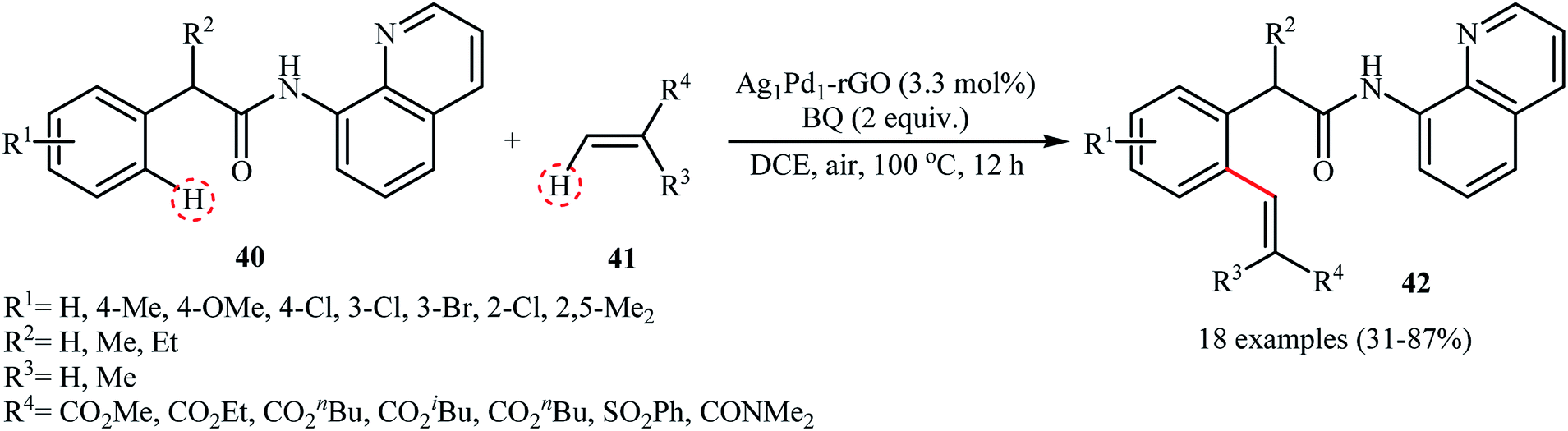
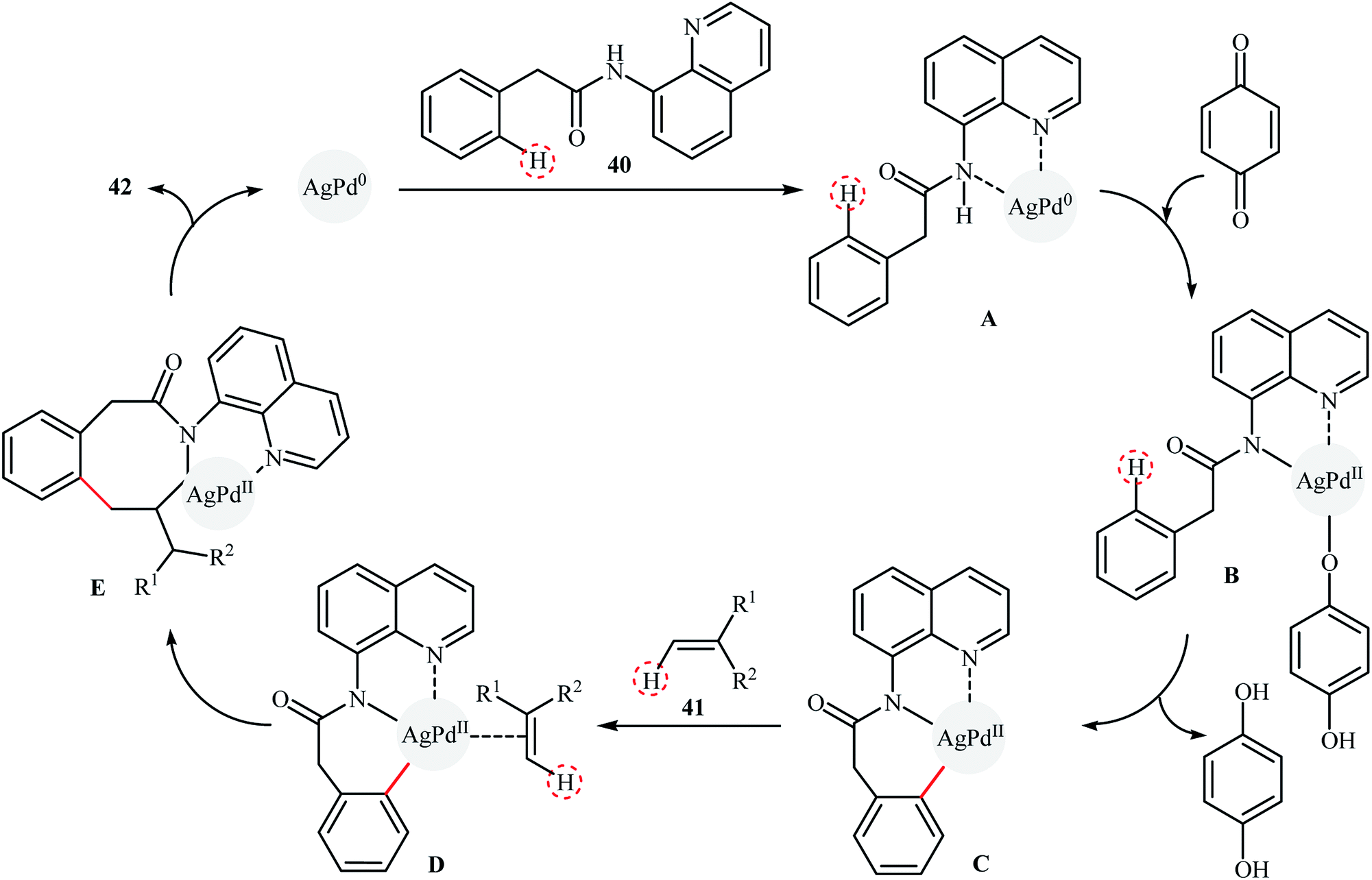
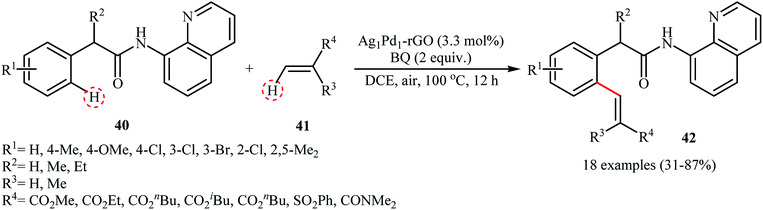
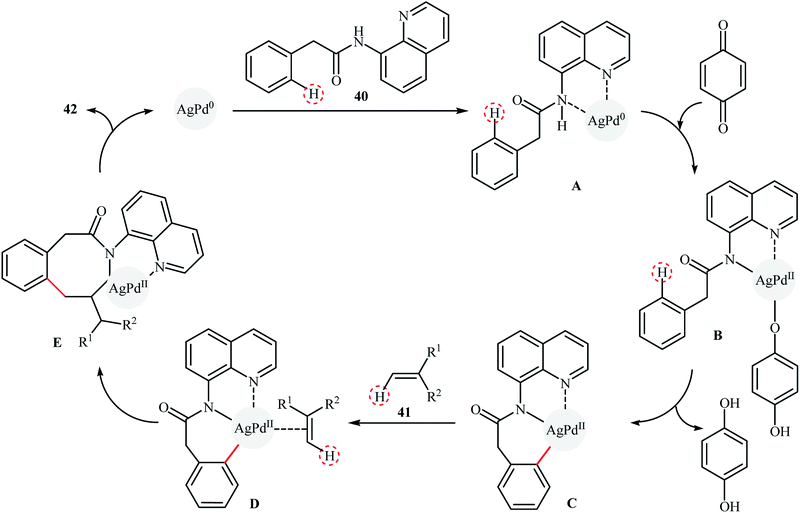
Recently, silver–palladium alloy nanoparticles anchored on reduced graphene oxide (Ag1Pd1-rGO) was used as efficient and recyclable catalyst for the chelation-assisted ortho C–H bond olefination of aromatic amides 40 with acrylates 41 by Hu et al.45 The reactions take place in the presence of benzoquinone (BQ) as an oxidant in DCE at 100 °C under an air atmosphere and afforded the target coupling products 42 in moderate to high yields within 12 h (Scheme 20). It is worth mentioning that the use of two-fold of acrylate for an extended time (24 h) led to di-olefinated products in good to excellent yields. Noteworthy, the catalyst could be reused for five consecutive runs, with only negligible loss of activity. The authors found that Ag and Pd atoms in the alloy nanoparticles play synergistic roles in the catalytic activity. Investigation revealed that Pd-rGO had very low catalytic activity than Ag1Pd1-rGO and Ag-rGO was catalytically inert for this C–C coupling reaction. Mechanistic investigations revealed that the transformation possibly proceeds though the following key steps (Scheme 21): (i) coordination of the Pd atoms of the catalyst to the two nitrogen atoms of the amide 40 to produce intermediate A; (ii) oxidation of A by BQ and air to afford intermediate B; (iii) coordination of the in situ-formed Pd(II) species with the intramolecular aminoquinoline along with the loss of the hydroquinone to form cyclopalladated intermediate C; (iv) coordination of intermediate C to the olefin 41 to give intermediate D; (v) insertion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond into the C–Pd bond to yield intermediate E; and (vi) β-hydride elimination of the alkylpalladium intermediate E to form the expected products 42.
C bond into the C–Pd bond to yield intermediate E; and (vi) β-hydride elimination of the alkylpalladium intermediate E to form the expected products 42.
 |
| | Scheme 20 Direct C–H bond olefination of amides 40 with acrylates 41 catalyzed by Ag1Pd1-rGO.45 | |
 |
| | Scheme 21 The possible mechanism of Ag1Pd1-rGO-catalyzed olefination of aromatic amides 40.45 | |
5.3. C(aryl)–C(aldehyde) bonds
The wide importance of diaryl ketones as intermediates in the synthesis of valuable industrial products such as pharmaceuticals, agrochemicals, fragrances, flavors, and dyes,46 stimulates researchers for develop more efficient and simple methods for their preparation. Among numerous known methodologies for diaryl ketone synthesis, the most green and straightforward procedure is cross-dehydrogenative-coupling reactions between arenes and arenecarbaldehydes.47 In 2013, Ramani and co-workers reported for the first time the usefulness of metal nanoparticles as catalysts for the oxidative cross-coupling of simple arenes with aromatic dehydes.48 They used the Fe3O4 magnetic nanoparticles (10–30 nm) to promote the ortho-benzoylation of phenols 43 with aromatic aldehydes 44 utilizing K2CO3 as an inexpensive base and air as the terminal oxidant (Scheme 22). The reaction proved to be efficient on a variety of electron-rich and electron-poor aldehydes, affording exclusively the ortho-benzoylated products 45. However, the reaction did not work with electron-poor phenols. Interestingly, when 2-substituted (NO2, Cl, Br) arenecarbaldehydes were treated with phenols under the standard condition, biologically important xanthones were obtained in good yields (55–72% for 6 examples) in one step via a double C–H bond coupling. Shortly afterwards, Gerbino's research team improved the efficiency of this reaction by performing the process in the presence of a low loading (0.9 mol%) of CuNPs supported on silica coated maghemite (CuNPs/MagSilica) as a magnetically retrievable catalyst.49
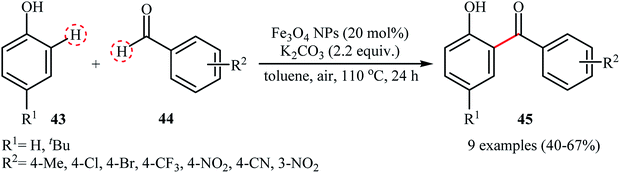 |
| | Scheme 22 Synthesis of ortho-hydroxybenzophenones 45 catalyzed by Fe3O4 NPs.48 | |
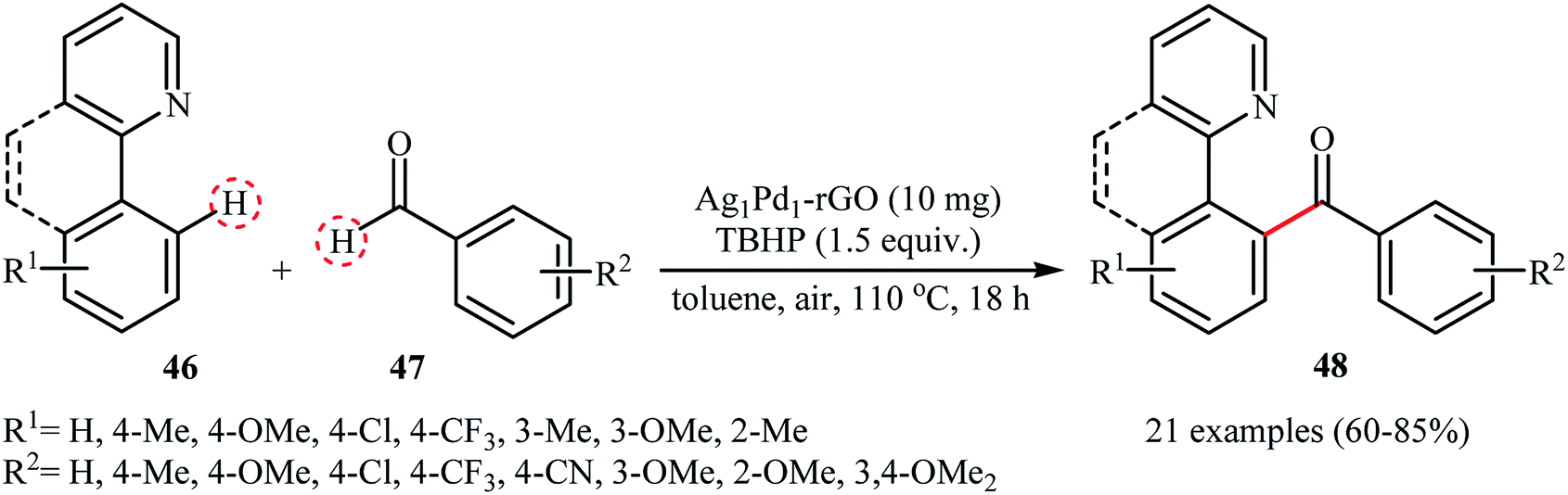
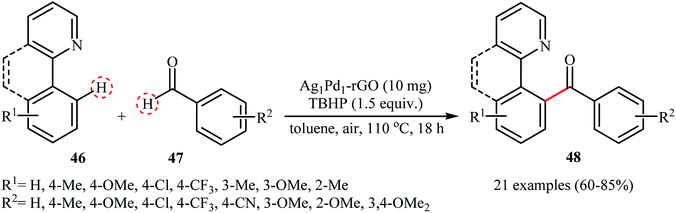
Recently, Liu and Zhang used their Ag1Pd1 nanoparticle-reduced graphene oxide nanocomposite as a catalyst for the acylation of 2-arylpyridines 46 with aldehydes 47.50 The reaction was conducted using tert-butyl hydroperoxide (TBHP) as an oxidant under an air atmosphere and afforded the diaryl ketones products 48 in good to high yields (Scheme 23). Applying this method, aliphatic aldehydes could also be arylated in moderate yields. Of note, the catalyst could be reused at least for five times with only 9% decline in the activity. The authors proposed mechanism for this transformation is analogous to the one depicted for cross-dehydrogenative Heck reaction in Scheme 21.
 |
| | Scheme 23 Ag1Pd1-rGO-catalyzed cross-dehydrogenative coupling of 2-arylpyridines 46 with aromatic aldehydes 47.50 | |
6. Constructing C(sp2)–C(sp) bonds
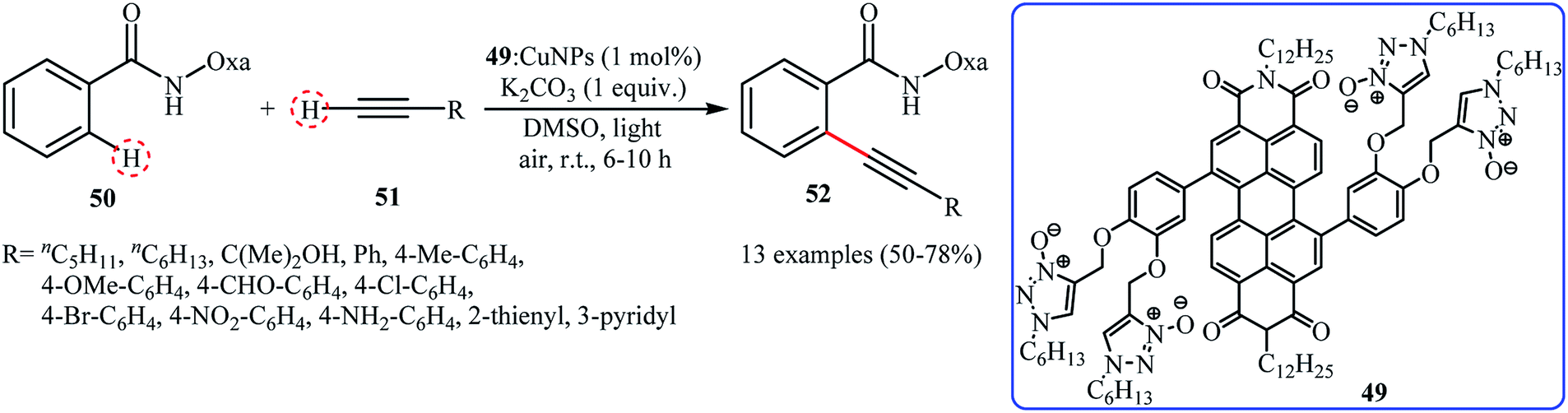
In 2016, Kaur, Kumar and Bhalla reported about the use of CuNPs (8–20 nm) in combination with supramolecular aggregate of triazole N-oxide appended perylene bisimide (PBI) derivative 49 (49: CuNPs) as a highly active photocatalytic system for ortho-selective mono-alkynylation of oxazoline substituted benzamide derivative 50 with terminal alkynes 51 under irradiation of visible light. The results demonstrated that the desired internal alkynes 52 were slowly formed in moderate to good yields at room temperature under an atmosphere of air (Scheme 24). Various aliphatic, aromatic and heteroaromatic terminal alkynes were utilized to establish the general applicability of this synthetic process. It should be mentioned that the recycling test showed a small loss of catalytic activity from 72% in the 1st run to 56% in the 4th run. To the best of our knowledge, this is only reported example on the utilization of nanocatalysts in direct C(sp2)–C(sp) bonds forming reactions.
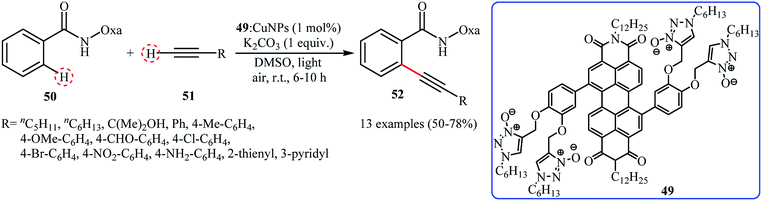 |
| | Scheme 24
Ortho-selective alkynylation of oxazoline substituted benzamide 50 with terminal alkynes 51 catalyzed by 49: CuNPs.51 | |
7. Constructing C(sp)–C(sp) bonds
1,3-Diynes are not only omnipresent in bioactive nature products,52 but also paramount important building blocks in organic synthesis.53 Without slight doubt, the most straightforward approach to the synthesis of symmetrical 1,3-diynes is the oxidative homo-coupling of two terminal alkynes, which known as the Glaser coupling.54 Although numerous efficient metal nanocatalysts have been reported for this interesting and useful reaction (Table 4),55–70 the nanometal-catalyzed hetero-coupling reactions between two different alkynes were less developed. Noteworthy, due to the competitive reaction pathways between homo-coupling and hetero-coupling reactions, synthesis of unsymmetrical 1,3-diynes is more difficult compared to the symmetrical ones.71
Table 4 Glaser homocoupling reactions catalyzed by nanosized metalsa
|
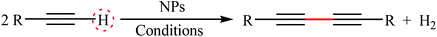
|
| Entry |
Catalyst |
Conditions |
Number of examples |
Yield (%) |
Ref. |
|
Number of examples.
|
| 1 |
CuNPs |
THF, 66 °C, 8–24 h |
11 |
65–90 |
55
|
| 2 |
Cu2O NPa |
TBAB/H2O, 60 °C, 14–24 h |
12 |
33–93 |
56
|
| 3 |
CuNPs/TiO2 |
THF, 65 °C, 2–24 h |
16 |
60–96 |
57
|
| 4 |
CuNPs/MnOx |
Toluene, 105 °C, 1–12 h |
13 |
82–98 |
58
|
| 5 |
Resin-CuNPs |
DMSO, 50 °C, 8 h |
7 |
90–95 |
59
|
| 6 |
CuNPs-HAp |
MeCN, reflux, 72 h |
8 |
21–99 |
60
|
| 7 |
Cu/Cu2ONPs@GO |
EtOH, 80 °C, 8 h |
12 |
53–99 |
61
|
| 8 |
SBA-15@DABCO-Pd |
MeCN, CuI, r.t., 5–24 h |
9 |
62–94 |
62
|
| 9 |
PdNPs@Al(OH)3 |
DMSO, NaOAc, Ag2SO4, 90 °C, 8–48 h |
11 |
50–99 |
63
|
| 10 |
AuNPs@C |
1,3-DCB, 170 °C, 18 h |
10 |
60–90 |
64
|
| 11 |
Ni-MINT |
Me2O/H2O, DABCO, 140–170 °C, 15–45 min |
6 |
51–90 |
65
|
| 12 |
AgNPs@g-C3N4 |
EtOH/H2O, MW, 80 °C, 18–20 min |
9 |
94–98 |
66
|
| 13 |
CuNPs/MagSilica |
THF, 66 °C, 2–24 h |
7 |
58–95 |
67
|
| 14 |
CuO–Fe3O4 |
Neat, tBuOK, 60 °C, 2–7 h |
14 |
20–99 |
68
|
| 15 |
Fe3O4@SiO2@APTMS@Cu(en)2 |
DMF, Na2CO3, 80 °C, 15–60 min |
4 |
86–100 |
69
|
| 16 |
Pd2Au/mpg-C3N4 |
DMF, 100 °C, 12–16 h |
7 |
71–99 |
70
|
One of the earliest reports on the utilization of nanostructured catalysts in cross-dehydrogenative coupling reactions between two different terminal alkynes was published by Corma and colleagues in 2016,72 who showed that the treatment of terminal alkyne molecules 53 with 54 in the presence of a catalytic amount of TiO2-supported CuOx NPs (∼2 nm) under an oxygen atmosphere, resulted in the formation of the corresponding unsymmetrical 1,3-diynes 55 in good to excellent yields, ranging from 61% to 92% (Scheme 25). Interestingly, both aliphatic and aromatic terminal alkynes were compatible with this challenging reaction. Concurrently, Tang and Liu along with their co-workers reported an example of unsymmetrical 1,3-diyne synthesis via the reaction of phenylacetylene with 4-methoxyphenylacetylene using 10 mol% of Cu2O NPs as catalyst in a binary solvent H2O/TBAB (tetrabutylammonium bromide) in a 1:4 ratio.56 However, the target diyne was isolated in only 30% yield along with a mixture of both possible homo-coupling products.
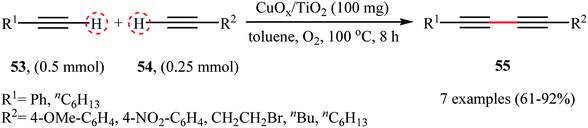 |
| | Scheme 25 CuOx/TiO2-catalyzed hetero-coupling of 53 with 54.72 | |
In the same year, the group of Rossi-Suib prepared thermally stable mesoporous copper oxide supported manganese oxide nanoparticles with Mn/Cu molar ratio 10/1 through the reaction of Cu(NO3)2·3H2O with Mn(NO3)2·4H2O in the presence of P123 (PEO20PPO70PEO20) as a pluoronic surfactant in acidic nBuOH.58 The prepared material (meso Cu/MnOx) exhibits a large surface area of over 270 m2 g−1 and uniform mesoporous size (3.0–3.4 nm) distribution. The authors demonstrated that their heterogeneous catalyst can effectively promote the oxidative coupling reactions of two different terminal alkynes 56 and 57 to furnish the corresponding 1,3-diyne products 58 in moderate yields (Scheme 26). Noteworthy, the catalyst can be readily separated by simple filtration and reused over 8 reaction cycles without a significant decrease in activity. Moreover, no change in the PXRD (powder X-ray diffraction) pattern after the 8th cycle was observed, which confirms that the catalyst can retain the crystal structure even after multiple reuse cycles.
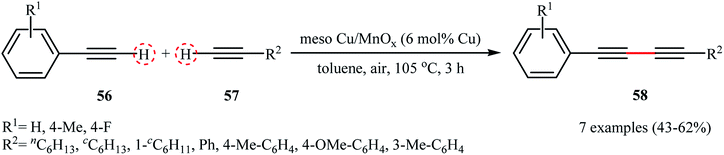 |
| | Scheme 26 Cross-dehydrogenative coupling of terminal alkynes 56 with 57 catalyzed by meso Cu/MnOx.58 | |
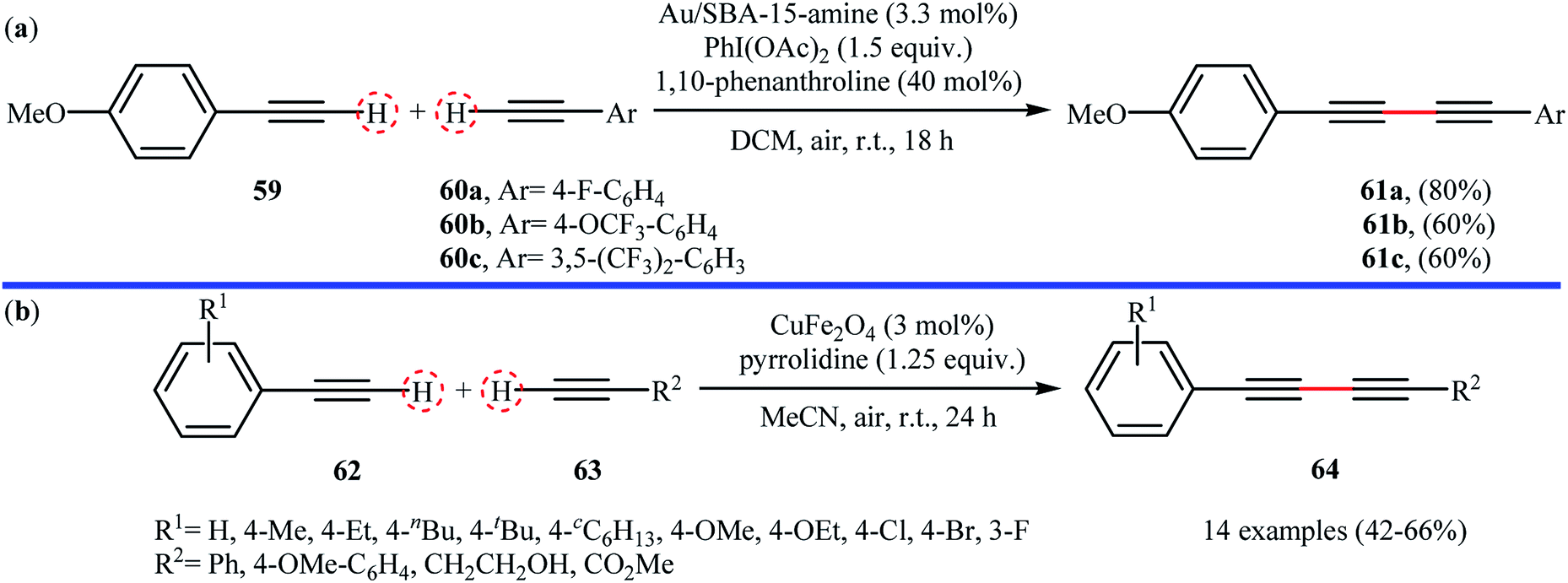
Following these reports, sub-nanometer (d = 0.8 ± 0.2 nm) gold particles supported on 3-aminopropyl-functionalized silicate SBA-15 (Au/SBA-15-amine) was successfully used as recyclable catalyst for high yielding synthesis of unsymmetrical 1,3-diynes 61 from the reaction of 4-methoxyphenylacetylene 59 with aromatic terminal alkynes 60 at room temperature.73 Better results were observed when commercially available λ3-iodane PhI(OAc)2 was used as the oxidant and 1,10-phenanthroline as additive (Scheme 27a). Under the optimized reaction conditions and by using a catalyst loading of 5 mol%, a library of symmetrical 1,3-diynes were also obtained in excellent yields (up to 98%) from the corresponding terminal alkynes. Magnetically separable nano-CuFe2O4 (with an average particle size of 30–35 nm) was also found to be active catalyst towards the oxidative cross-coupling of aromatic alkynes 62 with terminal alkynes 63 at room temperature.74 The reaction was performed under open air using pyrrolidine as a base, tolerated a number of important functional groups (e.g., F, Cl, Br, OMe, CO2Me, and OH), and provided the expected 1,3-diynes 64 in moderate yields (Scheme 27). Interestingly, the catalyst can be easily separated from the reaction mixture by using an external magnetic field, and its efficiency remains unchanged even after recycling over five runs.
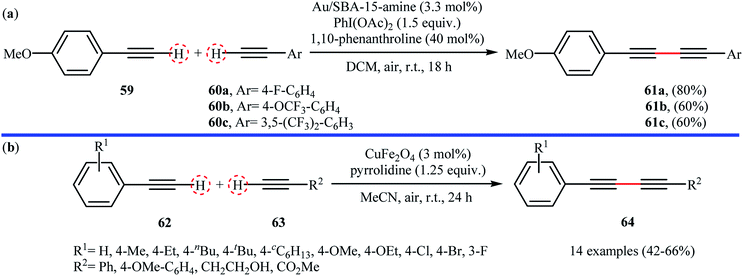 |
| | Scheme 27 (a) Oxidative cross-coupling of 4-methoxyphenylacetylene 59 with aromatic terminal alkynes 60 catalyzed by Au/SBA-15-amine;73 (b) CuFe2O4-catalyzed cross-dehydrogenative coupling of 62 with 63.74 | |
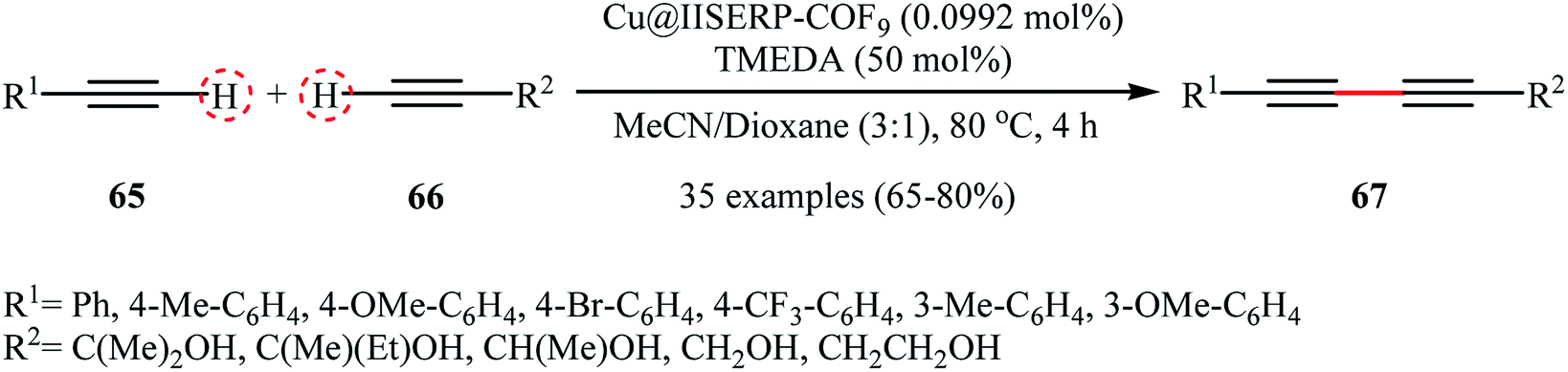
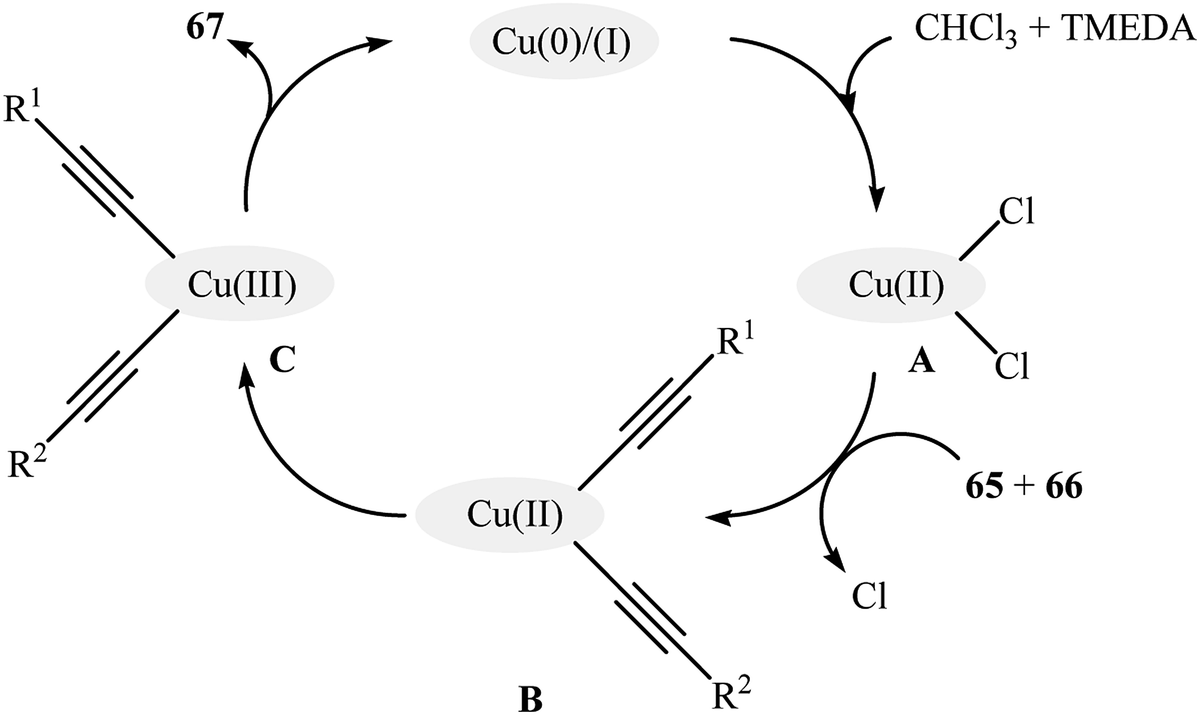
Very recently, Vinod and Vaidhyanathan's research team prepared a novel phenol-pyridyl covalent organic framework (IISERP-COF9) supported Cu/Cu2O nanoparticles (Cu@IISERP-COF9) through the condensation of 4,4′,4′′-(pyridine-2,4,6-triyl)trianiline with 2-hydroxybenzene-1,3,5-tricarbaldehyde in the binary solvent DCB-BuOH at 120 °C, followed by stirring of the resulting COF with CuCl2 in the presence of ascorbic acid in THF at 80 °C.75 The catalytic activity of the synthesized composite has been evaluated in the cross-dehydrogenative coupling of a range of aromatic terminal alkynes 65 with aliphatic alkynes 66 in the presence of tetramethylethylenediamine (TMEDA) as a base in a 3:1 mixture of CHCl3/dioxane. Good to high yields of the target unsymmetrical 1,3-diynes 67 were obtained within 4 h at 80 °C (Scheme 28). Recycling tests indicate that the catalyst can be reused in five consecutive trials with only a slight decrease in activity. A possible mechanism for the formation of 1,3-diynes 67 was given by this report (Scheme 29), which involves the initial formation of the Cu(II) species A through the oxidative addition of the carbon–halogen bond of CHCl3 to the Cu center of the catalyst, followed by its ligand exchange with the alkynes 65 and 66 to form a copper diacetylide complex B, which after oxidation in the presence of air converts to the Cu(III) complex C. Finally, the reductive elimination of this intermediate C affords the observed 1,3-diynes 67 and regenerates the catalyst.
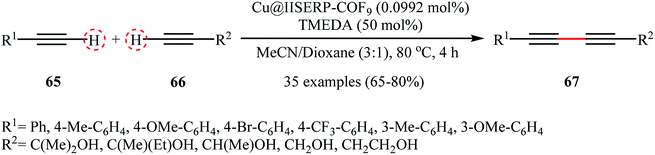 |
| | Scheme 28 Vinod–Vaidhyanathan's synthesis of unsymmetrical 1,3-diynes 67.75 | |
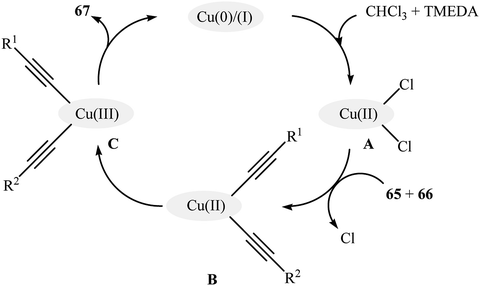 |
| | Scheme 29 Plausible mechanism for the hetero-coupling of aromatic terminal alkynes 65 with aliphatic terminal alkynes 66.75 | |
8. Conclusion
Over the past few years, the construction of C–C bonds through the direct oxidative coupling of two different C–H bonds between two reactants have attracted considerable attention as cleaner and more sustainable synthetic alternative to traditional coupling procedures which rely on the use of pre-functionalized starting materials. However, the control of regioselectivity in these reactions is a great challenge because C–H bonds are ubiquitous in organic molecules. There are two major strategies for control of selectivity: (i) directing group-based and (ii) catalyst-based controls. However, the requirement of installing and then removing the directing groups limited the application of the former strategy. As illustrated, the use of nanostructured catalysts allow the highly selective cross-dehydrogenative coupling between two C–H bonds, with the benefits of high product yield and ease of catalyst separation, recovery, and recycling. Interestingly, all the six kinds of C–C bonds [i.e., C(sp3)–C(sp3), C(sp3)–C(sp2), C(sp3)–C(sp), C(sp2)–C(sp2), C(sp2)–C(sp), C(sp)–C(sp)] were successfully fabricated by nanoparticles catalyzed oxidative coupling of the corresponding C–H bonds. Noteworthy, some of the reactions covered in this review could be easily scaled up to provide multi-gram quantities of the desired coupling products. This results clearly show the potential application of nanocatalyzed cross-dehydrogenative coupling reactions in industry. Despite the remarkable accomplishments during the past few years in this interesting research arena, many challenges still remain to be overcome: (i) most of the covered reactions in this review have been performed at high temperatures. Thus, there is a further need for investigation and discovery of novel catalytic systems, which can allow these reactions under milder conditions; (ii) the substrate scope in some reactions such as C(sp3)–H/C(sp3)–H couplings are narrow and therefore, of course, expanding of the substrate scope of these reactions are necessary; and (iii) the application of tri-metallic and multi-metallic nanoparticles in these reactions should be investigated.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors wish to thank Dr Saeideh Ebrahimiasl for editing this manuscript for English language.
References
- A. Mateska, G. Stojković, B. Mikhova, K. Mladenovska and E. Popovski, ARKIVOC, 2009, 10, 131–140 Search PubMed.
-
(a) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937–4947 RSC;
(b) C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963–4972 RSC;
(c) Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560–572 RSC;
(d) J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC.
-
A. d. Meijere and F. Diederich, Metal-catalyzed cross-coupling reactions. Weinheim, Wiley-VCH Verlag GmbH, 2004, vol. 1 Search PubMed.
-
(a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed;
(b) S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., Int. Ed. Engl., 2014, 53, 74–100 CrossRef CAS PubMed;
(c) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed.
-
(a) A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181–5203 RSC;
(b) K. Nejati, S. Ahmadi, M. Nikpassand, P. D. K. Nezhad and E. Vessally, RSC Adv., 2018, 8, 19125–19143 RSC;
(c) A. Monfared, R. Mohammadi, S. Ahmadi, M. Nikpassand and A. Hosseinian, RSC Adv., 2019, 9, 3185–3202 RSC;
(d) S. Mohammadi, M. Musavi, F. Abdollahzadeh, Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed;
(e) E. Najafi, F. Behmagham, N. Shaabani and N. Shojaei, Chem. Rev. Lett., 2019, 2, 13–20 Search PubMed;
(f) M. Chabanea and B. Dahmani, Chem. Rev. Lett., 2019, 2, 118–122 Search PubMed.
-
(a) E. Vessally, M. Babazadeh, A. Hosseinian, S. Arshadi and L. Edjlali, J. CO2 Util., 2017, 21, 491–502 CrossRef CAS;
(b) K. Didehban, E. Vessally, A. Hosseinian, L. Edjlali and E. S. Khosroshahi, RSC Adv., 2018, 8, 291–301 RSC;
(c) A. Hosseinian, S. Ahmadi, A. Monfared, P. D. Nezhad and E. Vessally, Curr. Org. Chem., 2018, 22, 1862–1874 CrossRef CAS;
(d) E. Vessally, K. Didehban, R. Mohammadi, A. Hosseinian and M. Babazadeh, J. Sulfur Chem., 2018, 39, 332–349 CrossRef CAS;
(e) S. Shahidi, P. Farajzadeh, P. Ojaghloo, A. Karbakhshzadeh and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 37–44 Search PubMed.
-
(a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef PubMed;
(b) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC;
(c) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef PubMed;
(d) A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC;
(e) A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828–33844 RSC;
(f) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 20 CrossRef PubMed;
(g) A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, J. Fluorine Chem., 2019, 220, 24–34 CrossRef CAS;
(h) S. Arshadi, S. Ebrahimiasl, A. Hosseinian, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 8964–8976 RSC;
(i) M. Hamzeloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef;
(j) S. Arshadi, A. Banaei, A. Monfared, S. Ebrahimiasl and A. Hosseinian, RSC Adv., 2019, 9, 17101–17118 RSC;
(k) A. Hosseinian, S. Arshadi, S. Sarhandi, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 289–311 CrossRef CAS;
(l) A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88–112 CrossRef CAS;
(m) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed;
(n) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed;
(o) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed.
- K. Li, Q. Wu, J. Lan and J. You, Nat. Commun., 2015, 6, 8404 CrossRef CAS PubMed.
-
J. Xie and C. Zhu, Sustainable C (sp3)–H Bond Functionalization, Springer, 2016 Search PubMed.
- T. Zeng, G. Song, A. Moores and C.-J. Li, Synlett, 2010, 2010, 2002–2008 CrossRef.
- M. Rueping, J. Zoller, D. C. Fabry, K. Poscharny, R. M. Koenigs, T. E. Weirich and J. Mayer, Chem.–Eur. J., 2012, 18, 3478–3481 CrossRef CAS PubMed.
- Q.-Y. Meng, Q. Liu, J.-J. Zhong, H.-H. Zhang, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2012, 14, 5992–5995 CrossRef CAS PubMed.
- R. Hudson, S. Ishikawa, C.-J. Li and A. Moores, Synlett, 2013, 24, 1637–1642 CrossRef CAS.
- H. E. Ho, Y. Ishikawa, N. Asao, Y. Yamamoto and T. Jin, Chem. Commun., 2015, 51, 12764–12767 RSC.
- W. Yang, L. Wei, T. Yan and M. Cai, Catal. Sci. Technol., 2017, 7, 1744–1755 RSC.
- S. Li, D. P. Shelar, C.-C. Hou, Q.-Q. Chen, P. Deng and Y. Chen, J. Photochem. Photobiol., A, 2018, 363, 44–50 CrossRef CAS.
- W. Liang, T. Zhang, Y. Liu, Y. Huang, Z. Liu, Y. Liu, B. Yang, X. Zhou and J. Zhang, ChemSusChem, 2018, 11, 3586–3590 CrossRef CAS PubMed.
- P. Li, G.-W. Wang, X. Zhu and L. Wang, Tetrahedron, 2019, 75, 3448–3455 CrossRef CAS.
- P. Rana, R. Gaur, R. Gupta, G. Arora, J. Anireddy and R. K. Sharma, Chem. Commun., 2019, 55, 7402–7405 RSC.
- J. Dong, W. Min, H. Li, Z. Quan, C. Yang and C. Huo, Adv. Synth. Catal., 2017, 359, 3940–3944 CrossRef CAS.
- I. Martín-García and F. Alonso, Chem.–Eur. J., 2018, 24, 18857–18862 CrossRef PubMed.
- Q.-Y. Meng, J.-J. Zhong, Q. Liu, X.-W. Gao, H.-H. Zhang, T. Lei, Z.-J. Li, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2013, 135, 19052–19055 CrossRef CAS PubMed.
- M. Lin, L.-X. Dai, J. Gu, L.-Q. Kang, Y.-H. Wang, R. Si, Z.-Q. Zhao, W.-C. Liu, X. Fu, L.-D. Sun, Y.-W. Zhang and C.-H. Yan, RSC Adv., 2017, 7, 33078–33085 RSC.
- A. Tyagi, A. Yamamoto and H. Yoshida, RSC Adv., 2018, 8, 24021–24028 RSC.
- X. Marset, J. M. Pérez and D. J. Ramón, Green Chem., 2016, 18, 826–833 RSC.
- F. Alonso, A. Arroyo, I. Martín-García and Y. Moglie, Adv. Synth. Catal., 2015, 357, 3549–3561 CrossRef CAS.
- S. Gupta, H. Joshi, N. Jain and A. K. Singh, J. Mol. Catal. Chem., 2016, 423, 135–142 CrossRef CAS.
- T. Dang-Bao, C. Pradel, I. Favier and M. Gómez, Adv. Synth. Catal., 2017, 359, 2832–2846 CrossRef CAS.
-
(a) E. Vessally, RSC Adv., 2016, 6, 18619–18631 RSC;
(b) E. Vessally, L. Edjlali, A. Hosseinian, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 49730–49746 RSC;
(c) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and A. Bekhradnia, RSC Adv., 2017, 7, 7079–7091 RSC;
(d) S. Arshadi, E. Vessally, L. Edjlali, R. Hosseinzadeh-Khanmiri and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–638 CrossRef CAS PubMed;
(e) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef CAS.
- J.-A. García-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766–6798 RSC.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed.
- P. E. Fanta, Synthesis, 1974, 9–21 CrossRef CAS.
- F.-X. Felpin and S. Sengupta, Chem. Soc. Rev., 2019, 48, 1150–1193 RSC.
- J. Wencel-Delord, A. Panossian, F. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC.
- I. Hussain and T. Singh, Adv. Synth. Catal., 2014, 356, 1661–1696 CrossRef CAS.
- L. M. Neal and H. E. Hagelin-Weaver, J. Mol. Catal. Chem., 2008, 284, 141–148 CrossRef CAS.
- T. Ishida, R. Tsunoda, Z. Zhang, A. Hamasaki, T. Honma, H. Ohashi, T. Yokoyama and M. Tokunaga, Appl. Catal., B, 2014, 150, 523–531 CrossRef.
- P. Serna and A. Corma, J. Catal., 2014, 315, 41–47 CrossRef CAS.
- P. Serna and A. Corma, ChemSusChem, 2014, 7, 2136–2139 CrossRef CAS PubMed.
- T. Ishida, S. Aikawa, Y. Mise, R. Akebi, A. Hamasaki, T. Honma, H. Ohashi, T. Tsuji, Y. Yamamoto and M. Miyasaka, ChemSusChem, 2015, 8, 695–701 CrossRef CAS PubMed.
- S. Tabasso, E. C. Gaudino, E. Acciardo, M. Manzoli, A. Giacomino and G. Cravotto, Molecules, 2019, 24, 288 CrossRef PubMed.
- S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC.
-
(a) X. Shang and Z.-Q. Liu, Chem. Soc. Rev., 2013, 42, 3253–3260 RSC;
(b) C. Aouf, E. Thiery, J. Le Bras and J. Muzart, Org. Lett., 2009, 11, 4096–4099 CrossRef CAS PubMed;
(c) Z. Shi, N. Schröder and F. Glorius, Angew. Chem., Int. Ed. Engl., 2012, 51, 8092–8096 CrossRef CAS PubMed;
(d) G. G. Pawar, G. Singh, V. K. Tiwari and M. Kapur, Adv. Synth. Catal., 2013, 355, 2185–2190 CrossRef CAS;
(e) S. Maity, R. Kancherla, U. Dhawa, E. Hoque, S. Pimparkar and D. Maiti, ACS Catal., 2016, 6, 5493–5499 CrossRef CAS.
- L. L. Chng, J. Zhang, J. Yang, M. Amoura and J. Y. Ying, Adv. Synth. Catal., 2011, 353, 2988–2998 CrossRef CAS.
- Q. Hu, X. Liu, G. Wang, F. Wang, Q. Li and W. Zhang, Chem. Eur. J., 2017, 23, 17659–17662 CrossRef CAS PubMed.
-
(a) J.-J. Brunet and R. Chauvin, Chem. Soc. Rev., 1995, 24, 89–95 RSC;
(b) F. Boscá and M. A. Miranda, J. Photochem. Photobiol., B, 1998, 43, 1–26 CrossRef;
(c)
J. Panten and K. Bauer, Common Fragrance and Flavor Materials: Preparation, Properties and Uses, Wiley VCH, 2006 Search PubMed.
- X. F. Wu, Chem. Eur. J., 2015, 21, 12252–12265 CrossRef CAS PubMed.
- T. Ramani, P. Umadevi, K. L. Prasanth and B. Sreedhar, Eur. J. Org. Chem., 2013, 6021–6026 CrossRef CAS.
- C. A. Menendez, F. Nador, G. Radivoy and D. C. Gerbino, Org. Lett., 2014, 16, 2846–2849 CrossRef CAS PubMed.
- Q. Hu, X. Liu, F. Huang, F. Wang, Q. Li and W. Zhang, Catal. Commun., 2018, 113, 27–31 CrossRef CAS.
- S. Kaur, M. Kumar and V. Bhalla, Green Chem., 2016, 18, 5870–5883 RSC.
-
(a) M. Villegas, D. Vargas, J. Msonthi, A. Marston and K. Hostettmann, Planta Med., 1988, 54, 36–37 CrossRef CAS PubMed;
(b) E.-A. Bae, M. J. Han, N.-I. Baek and D.-H. Kim, Arch. Pharmacal Res., 2001, 24, 297–299 CrossRef CAS PubMed;
(c) S. Purup, E. Larsen and L. P. Christensen, J. Agric. Food Chem., 2009, 57, 8290–8296 CrossRef CAS PubMed;
(d) H. Kim, J. Chin, H. Choi, K. Baek, T.-G. Lee, S. E. Park, W. Wang, D. Hahn, I. Yang and J. Lee, Org. Lett., 2013, 15, 100–103 CrossRef CAS PubMed.
- W. Shi and A. Lei, Tetrahedron Lett., 2014, 55, 2763–2772 CrossRef CAS.
-
(a) C. Glaser, Chem. Ber., 1869, 2, 422–424 CrossRef;
(b) J. Tang, H. Jiang, G. Deng and L. Zhou, Chin. J. Org. Chem., 2005, 25, 1503–1507 CAS;
(c) Z. M. Nimmo, J. F. Halonski, L. E. Chatkewitz and D. D. Young, Bioorg. Chem., 2018, 76, 326–331 CrossRef CAS PubMed.
- F. Nador, L. Fortunato, Y. Moglie, C. Vitale and G. Radivoy, Synthesis, 2009, 4027–4031 CAS.
- B. X. Tang, X. N. Fang, R. Y. Kuang, J. H. Wu, Q. Chen, S. J. Hu and Y. L. Liu, Appl. Organomet. Chem., 2016, 30, 943–945 CrossRef CAS.
- F. Alonso, T. Melkonian, Y. Moglie and M. Yus, Eur. J. Org. Chem., 2011, 2524–2530 CrossRef CAS.
- S. Biswas, K. Mullick, S.-Y. Chen, D. A. Kriz, M. Shakil, C.-H. Kuo, A. M. Angeles-Boza, A. R. Rossi and S. L. Suib, ACS Catal., 2016, 6, 5069–5080 CrossRef CAS.
- X. Zhao, X. Lu, A. Wei, X. Jia, J. Chen and K. Lu, Tetrahedron Lett., 2016, 57, 5330–5333 CrossRef CAS.
- B. Maaten, J. Moussa, C. Desmarets, P. Gredin, P. Beaunier, T. Kanger, K. Tõnsuaadu, D. Villemin and M. Gruselle, J. Mol. Catal. Chem., 2014, 393, 112–116 CrossRef CAS.
- W. Lu, W. Sun, X. Tan, L. Gao and G. Zheng, Catal. Commun., 2019, 125, 98–102 CrossRef CAS.
- H. Li, M. Yang and Q. Pu, Microporous Mesoporous Mater., 2012, 148, 166–173 CrossRef CAS.
- X. Li, D. Li, Y. Bai, C. Zhang, H. Chang, W. Gao and W. Wei, Tetrahedron, 2016, 72, 6996–7002 CrossRef CAS.
- M. Boronat, S. Laursen, A. Leyva-Pérez, J. Oliver-Meseguer, D. Combita and A. Corma, J. Catal., 2014, 315, 6–14 CrossRef CAS.
- C. Gonzalez-Arellano, A. M. Balu, R. Luque and D. J. Macquarrie, Green Chem., 2010, 12, 1995–2002 RSC.
- S. B. Patel and D. V. Vasava, ChemistrySelect, 2018, 3, 471–480 CrossRef CAS.
- F. Nador, M. A. Volpe, F. Alonso, A. Feldhoff, A. Kirschning and G. Radivoy, Appl. Catal., A, 2013, 455, 39–45 CrossRef CAS.
- J. M. Perez, R. Cano, M. Yus and D. J. Ramon, Synthesis, 2013, 45, 1373–1379 CrossRef CAS.
- F. Farzaneh, Z. Shafie, E. Rashtizadeh and M. Ghandi, React. Kinet., Mech. Catal., 2013, 110, 119–129 CrossRef CAS.
- Z. Chen, R. Shen, C. Chen, J. Li and Y. Li, Chem. Commun., 2018, 54, 13155–13158 RSC.
- H. Peng, Y. Xi, N. Ronaghi, B. Dong, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2014, 136, 13174–13177 CrossRef CAS PubMed.
- L. Liu, T. Matsushita, P. Concepción, A. Leyva-Pérez and A. Corma, ACS Catal., 2016, 6, 2211–2221 CrossRef CAS.
- B. t. Vilhanová, J. i. Václavík, L. Artiglia, M. Ranocchiari, A. Togni and J. A. van Bokhoven, ACS Catal., 2017, 7, 3414–3418 CrossRef.
- C. T. Ma, J. J. Wang, A. D. Zhao, Q. L. Wang and Z. H. Zhang, Appl. Organomet. Chem., 2017, 31, e3888 CrossRef.
- D. Chakraborty, S. Nandi, D. Mullangi, S. Haldar, C. P. Vinod and R. Vaidhyanathan, ACS Appl. Mater. Interfaces, 2019, 11, 15670–15679 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d
d