
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric Diels–Alder reaction of 3-(acyloxy)acryloyl oxazolidinones: optically active synthesis of a high-affinity ligand for potent HIV-1 protease inhibitors†
Arun K. Ghosh *ab,
Alessandro Grilloa,
Satish Kovelaa and
Margherita Brindisi‡
a
*ab,
Alessandro Grilloa,
Satish Kovelaa and
Margherita Brindisi‡
a
aDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, Indiana 47907, USA. E-mail: akghosh@purdue.edu
bDepartment of Medicinal Chemistry and Molecular Pharmacology, Purdue University, 560 Oval Drive, West Lafayette, Indiana 47907, USA
First published on 17th December 2019
Abstract
We describe here our investigation of the asymmetric Diels–Alder reaction of chiral 3-(acyloxy)acryloyl oxazolidinones as dienophiles in various Lewis-acid promoted reactions with cyclopentadiene. The resulting highly functionalized cycloadducts are useful intermediates for the synthesis, particularly for the optically active synthesis of 6-5-5 tricyclic hexahydro-4H-3,5-methanofuro[2,3-b]pyranol (3) with five contiguous chiral centers. This stereochemically defined crown-like heterocyclic derivative is an important high affinity ligand for a variety of highly potent HIV-1 protease inhibitors. Among the various dienophiles and Lewis acid-mediated reactions surveyed, 3-(4-methoxybenzoyl)acryloyl oxazolidinone as the dienophile and diethylaluminum chloride as the Lewis-acid provided the desired endo product with excellent diastereoselectivity. The cycloaddition was carried out in multi-gram scale and the cycloadduct was efficiently converted to alcohol 3 with high enantiomeric purity. The optically active ligand was then transformed into potent HIV-1 protease inhibitor 2.
Introduction
The protein X-ray structure-based molecular design has become an integral part of modern drug discovery and development processes.1,2 The power and innovation of these strategies are particularly notable in the design and synthesis of HIV-1 protease inhibitors (PIs).3,4 HIV-1 PI drugs are critical components of current combined antiretroviral therapy (cART) which led to marked reductions of HIV-related morbidity and mortality in patients with access to cART regimens.5,6 Our structure-based design and synthesis led to the creation of numerous novel PIs incorporating a range of cyclic ether or polyether templates inherent to bioactive natural products.4,7 Conceptually, these PIs were designed to make extensive hydrogen bonding interactions with the backbone atoms throughout the active site of HIV-1 protease.7,8 In the latest FDA approved PI, darunavir (DRV, Fig. 1), we incorporated a stereochemically-defined bicyclic bis-tetrahydrofuran (bis-THF) as the P2 ligand.9,10 DRV is highly active against multidrug-resistant HIV-1 variants with a dual mechanism of action as it potently inhibits biologically active dimeric HIV-1 protease and prevents dimerization of protease monomers.11,12 DRV emerged as the most widely used PI for treating HIV/AIDS patients.13,14 However, DRV-resistant HIV-1 variants are reported in patients and emergence of such HIV-1 variants may result in treatment failure.15,16 For long-term effectiveness of cART regimens, development of novel antiretroviral agents with better potency, greater specificity and higher genetic barrier to the emergence of drug-resistant variants is critically important.To further improve properties of PIs, we have generated a range of structurally intriguing PIs with exceptional potency and clinical potential.4,7 Based upon X-ray structures of DRV-bound HIV-1 protease, our design effort particularly focused on enhancing both backbone binding and van der Waals interactions of PIs. Recently, we reported the design and synthesis of a new class of PIs incorporating a 6-5-5 ring-fused crown-like tetrahydropyrano-tetrahydrofuran as the P2 ligand with a hydroxyethyl sulfonamide transition-state mimic.17,18 Of particular importance, inhibitor 2 exhibited exceptional enzyme inhibitory activity, antiviral activity with IC50 values in the picomolar level and maintained picomolar antiviral activity against a panel of highly multidrug-resistant HIV-1 variants. Like DRV, PI 2 exhibited dual mechanism of inhibition as it very potently inhibited dimerization of HIV-1 protease as well as inhibition of dimeric enzyme. Furthermore, inhibitor 2 showed extremely high genetic barrier to the emergence of drug-resistant variants and also exhibited improved brain penetration in rats.18
The 6-5-5 ring contained in the crown-THF ligand is a critically important structural element of inhibitor 2. Based upon the X-ray structure of 2-bound HIV-1 protease, the bicyclic acetal oxygens formed enhanced hydrogen bonding interactions with the backbone NHs of Asp29 and Asp30.18 Furthermore, the methylene bridge and the extra methylene group on the 6-membered ring showed favorable van der Waals interactions with hydrophobic groups in the S2 subsite compared to the bis-THF ligand of DRV.18,19 The crown-THF ligand contains a three fused ring system with five contiguous chiral centers. As shown in Scheme 1, our previously disclosed optically active synthesis of the crown-THF ligand involved an enantioselective Diels–Alder reaction of vinyl boronate (5) and cyclopentadiene in the presence of a chiral oxazaborolidine catalyst (6) developed by Corey and Mukherjee.19–21 The boronate functionality cycloadduct 7 provided nice access to the hydroxyl group for the synthesis of our ligand.19 For an alternative synthesis of optically active 6-5-5 fused crown-THF ligand, we planned to examine asymmetric Diels–Alder reactions of cyclopentadiene with chiral 3-(acyloxy)acryloyloxazolidinone derivatives. The corresponding cycloadduct would provide direct access to functionalized cyclic templates in a diastereoselective fashion for application in synthesis. Highly diastereoselective Diels–Alder reactions of simple 3-alkyl substituted acryloyl oxazolidinone dienophiles are well precedented.22 However, Diels–Alder reactions of heteroatom-containing dienophiles, such as 3-acetoxyacryloyl oxazolidinone and their synthetic potentials have not been fully investigated. Narasaka and co-workers reported that similar dienophiles are much less reactive.23,24 However, Sibi and co-workers reported efficient Diels–Alder reactions of 3-(acetoxy)acrylates using chiral Lewis acid catalysts providing cycloadducts with high enantioselectivity.25 For our medicinal chemistry application, we planned to carry out Diels–Alder reactions of cyclopentadiene and chiral 3-acyloxyacryloyl oxazolidinone derivatives as dienophiles in a stereopredictable manner. Herein, we report the results of these studies where we investigated Diels–Alder reactions of cyclopentadiene with a variety of 3-acyloxyacryloyl oxazolidinone derivatives (8) as dienophiles. We have examined a range of Lewis acids under a variety of reaction conditions. Reaction of p-methoxybenzoate derivative in the presence of diethylaluminum chloride provided the endo adduct with high diastereoselectivity and excellent yields. The reaction provided multigram quantity of the desired cycloadduct 9 which was converted to 6-5-5-fused (3S,7aS,8S)-hexahydro-4H-3,5-methanofuro[2,3-b]pyranol (3) in high optical purity. This optically active alcohol was converted to the highly potent HIV-1 PI 2.
 | ||
| Fig. 1 Structures of HIV-1 protease inhibitors 1 and 2 and key building blocks 3 and 4. | ||
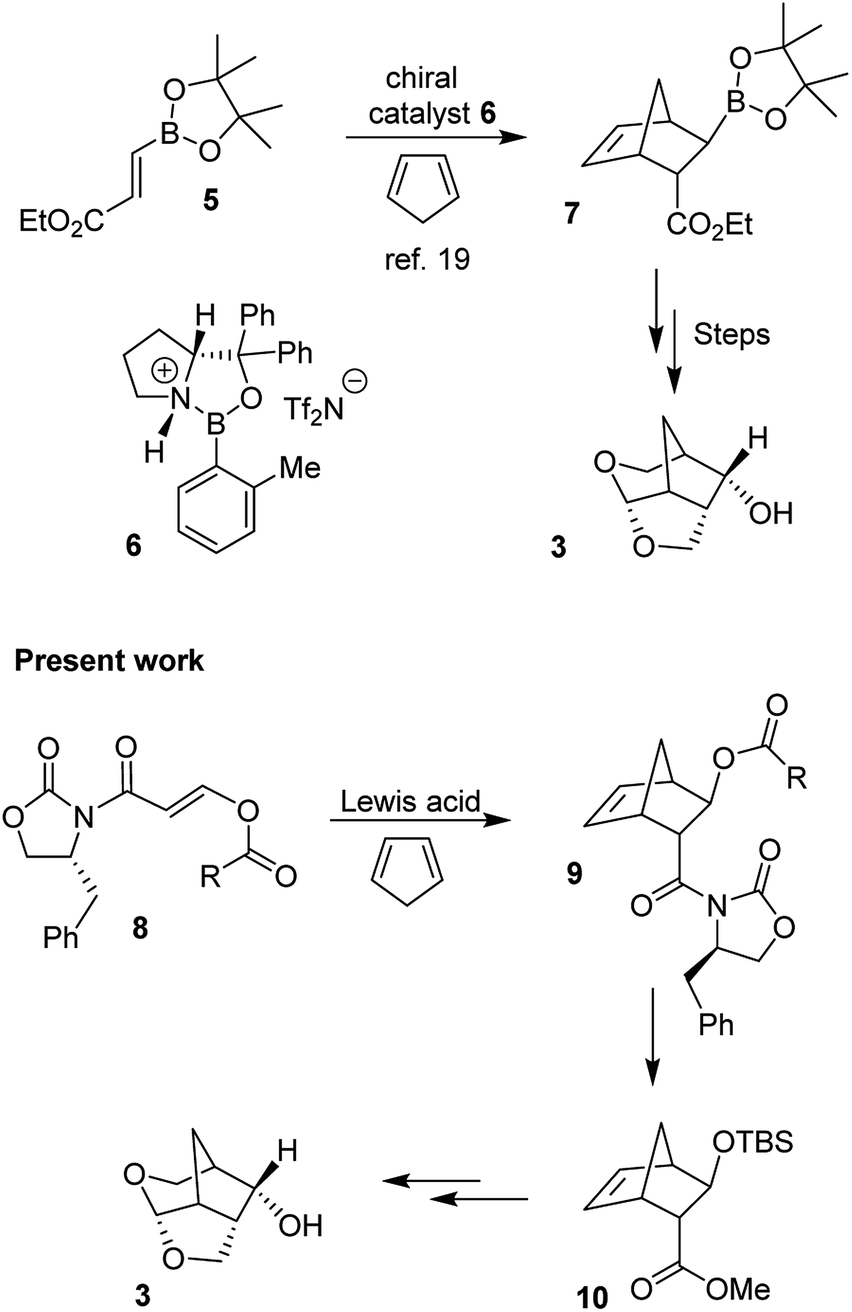 | ||
| Scheme 1 An asymmetric Diels–Alder route to optically active crown-THF ligand for HIV-1 protease inhibitors. | ||
Results and discussion
We synthesized a range of chiral 3-(acyloxy)acryloyl oxazolidinones for our studies as shown in Scheme 2. Commercially available propiolic acid (11) was reacted with oxalyl chloride in CH2Cl2 in the presence of DMF at 0 °C to 23 °C. The resulting 3-chloroacryloyl chloride was treated with the lithium salt of (S)-4-benzyl-2-oxazolidinone 12 to afford 3-(chloro)acryloyl oxazolidinone 13 in 78% yield. Reaction of 13 with various acids in the presence of N-methylmorpholine furnished 3-(acyloxy)acrylamide dienophiles 8a–f for asymmetric Diels–Alder reactions. For example, reaction of 13 with acetic acid and N-methylmorpholine in CH2Cl2 at 23 °C for 24 h provided 3-(acetoxy)acryloyl oxazolidinone 8a in 73% yield.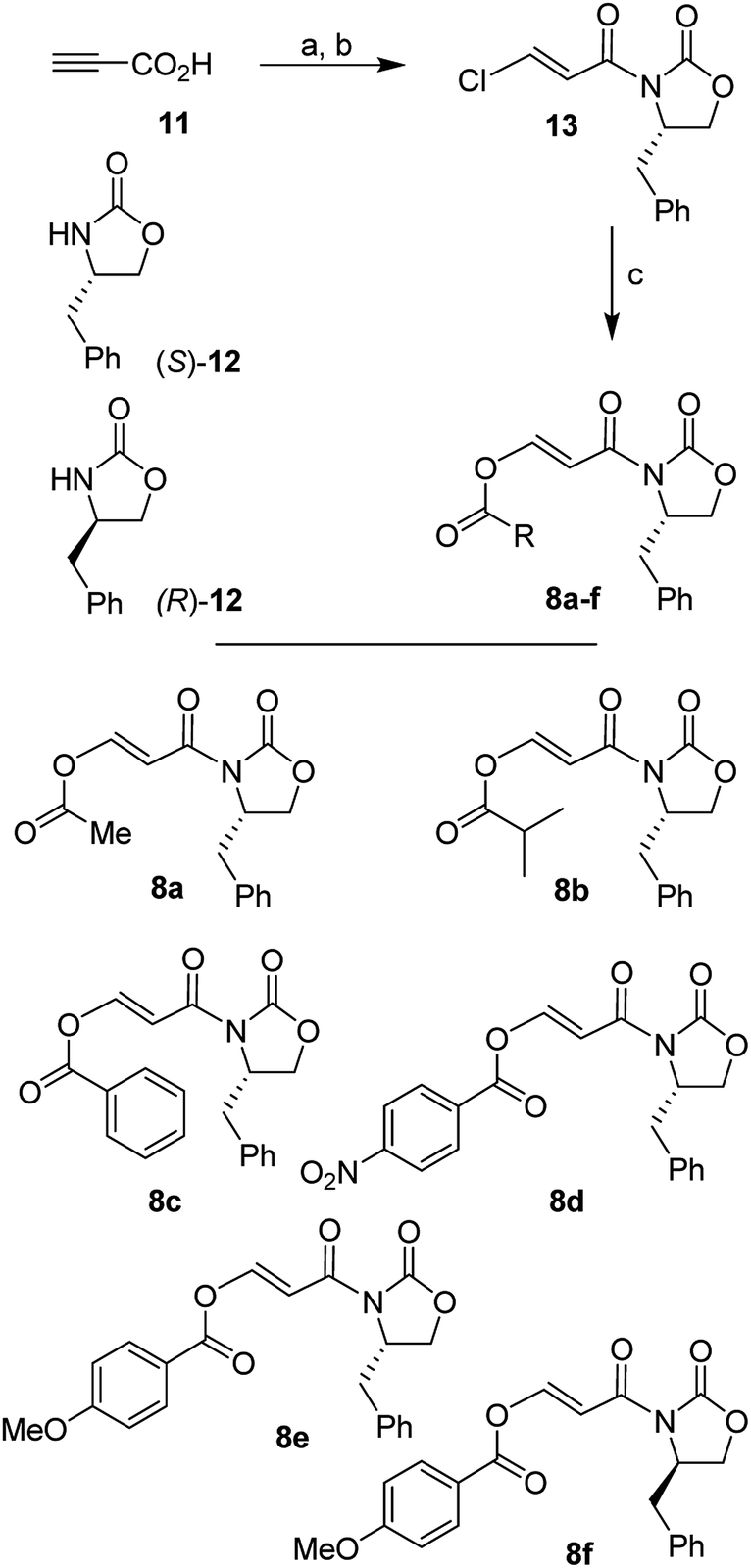 | ||
| Scheme 2 Synthesis of 3-(acyloxy)acryloyl oxazolidinones. Reagents and conditions. (a) (COCl)2, dry DMF, CH2Cl2, from 0 °C to 23 °C, 3 h; (b) n-BuLi, (S)-12 or (R)-12, dry THF, from −78 °C to 0 °C, 1 h (78%); (c) appropriate carboxylic acid, N-methylmorpholine, CH2Cl2, 23 °C, 24 h (67–84%). | ||
Similarly, other oxazolidinone derivatives 8b–8f were prepared (67–84% yields). Dienophile 8f with (R)-4-benzyl-2-oxazolidinone was prepared in 83% yield for the synthesis of optically active ligand alcohol 3.
Since Et2AlCl was utilized as an effective Lewis acid for Diels–Alder reactions of various alkyl substituted N-acryloyl-2-oxazolidinones by Evans and co-workers,26–28 we first examined Diels–Alder reactions of cyclopentadiene with 3-(acetoxy)acryloyl oxazolidinone 8a with varying amounts of Et2AlCl. The results are shown in Table 1. Diels–Alder reaction of dienophile 8a and cyclopentadiene in CH2Cl2 in the presence of 1.4 equiv. of Et2AlCl at −78 °C for 2 h resulted in exclusively endo-adduct in 36% yield with very high diastereoselectivity (by 1H-NMR and 13C-NMR analysis). No exo product was detected by 1H-NMR. As outlined in Scheme 3, only a single endo-diastereomer 9a was observed after purification of products by chromatography over silica gel (entry 1). The reactions with 2 and 3 equivalents of Et2AlCl at −78 °C for 24 h provided cycloadducts in 50% and 76% yields, respectively. The endo-diastereoselectivity remained unchanged (entries 2 and 3). When the cycloaddition was carried out at −78 °C to 23 °C for 24 h with 2 equivalents of Et2AlCl, cycloadduct was obtained in 71% yield, however, endo-diastereoselectivity was slightly reduced to 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (entry 4). The stereochemical outcome of the Diels–Alder reaction can be rationalized based upon bidentate chelated model prepared by Evans and co-workers.27,28 As shown in Scheme 3, bidentate chelation of Et2AlCl with adjacent substrate carbonyl groups in 3-(acetoxy)acryloyl oxazolidinone 8a leads to chelated intermediate 15. The cycloaddition proceeded through the Cα-Si-face of the dienophile 8a (R = CH3) since the Cα-Re-face approach of diene would develop significant steric hindrance between the bulky benzyl side chain and cyclopentadiene. The endo-diastereomer 9a is the only observable product after silica gel chromatography. The use of TiCl4 as the Lewis acid at −78 °C for 24 h only provided trace of cycloadducts with endo-diastereoselectivity of 90:10 (entries 5 and 6). We have also examined Yb(OTf)3 as the Lewis acid at 23 °C for 24 h, however, no cycloadduct was formed under these conditions (entry 7). Among various Lewis acids surveyed, Et2AlCl provided the best result with 3-(acetoxy)acryloyl oxazolidinone 8a. Reaction of dienophile 8b containing sterically hindered isobutyrate in the presence of 1.5 equivalents of Et2AlCl at −78 °C resulted in 58% yield of cycloadduct (entry 8). Reaction of dienophile 8c with benzoate derivative in the presence of 1.5 equiv. of Et2AlCl at −78 °C for 8 h provided endo adduct 9c (R = Ph) in 70% yield and excellent diastereoselectivity (entry 9). Dienophile 8d with a p-nitrobenzoate derivative also furnished endo-cycloadduct 9d (R = p-NO2-Ph) in 73% yield under similar reaction conditions (entry 10). The use of one equivalent of SnCl4 as the Lewis acid with 8d at 0 °C for 48 h provided cycloadduct 9d in 23% yield and endo-diastereoselectivity was excellent (entry 11). To establish stereochemical identity conclusively, as shown in Scheme 4, Diels–Alder adduct 9d was treated with 10 mol% Sm(OTf)3 in MeOH at 23 °C for 12 h to provide the corresponding methyl ester in 93% yield. This was recrystallized from ethyl acetate. Subsequent single crystal X-ray crystallographic analysis supported stereochemical assignment of 4-nitrobenzoyl ester 17 as shown in the ORTEP drawing.29,30
:3 (entry 4). The stereochemical outcome of the Diels–Alder reaction can be rationalized based upon bidentate chelated model prepared by Evans and co-workers.27,28 As shown in Scheme 3, bidentate chelation of Et2AlCl with adjacent substrate carbonyl groups in 3-(acetoxy)acryloyl oxazolidinone 8a leads to chelated intermediate 15. The cycloaddition proceeded through the Cα-Si-face of the dienophile 8a (R = CH3) since the Cα-Re-face approach of diene would develop significant steric hindrance between the bulky benzyl side chain and cyclopentadiene. The endo-diastereomer 9a is the only observable product after silica gel chromatography. The use of TiCl4 as the Lewis acid at −78 °C for 24 h only provided trace of cycloadducts with endo-diastereoselectivity of 90:10 (entries 5 and 6). We have also examined Yb(OTf)3 as the Lewis acid at 23 °C for 24 h, however, no cycloadduct was formed under these conditions (entry 7). Among various Lewis acids surveyed, Et2AlCl provided the best result with 3-(acetoxy)acryloyl oxazolidinone 8a. Reaction of dienophile 8b containing sterically hindered isobutyrate in the presence of 1.5 equivalents of Et2AlCl at −78 °C resulted in 58% yield of cycloadduct (entry 8). Reaction of dienophile 8c with benzoate derivative in the presence of 1.5 equiv. of Et2AlCl at −78 °C for 8 h provided endo adduct 9c (R = Ph) in 70% yield and excellent diastereoselectivity (entry 9). Dienophile 8d with a p-nitrobenzoate derivative also furnished endo-cycloadduct 9d (R = p-NO2-Ph) in 73% yield under similar reaction conditions (entry 10). The use of one equivalent of SnCl4 as the Lewis acid with 8d at 0 °C for 48 h provided cycloadduct 9d in 23% yield and endo-diastereoselectivity was excellent (entry 11). To establish stereochemical identity conclusively, as shown in Scheme 4, Diels–Alder adduct 9d was treated with 10 mol% Sm(OTf)3 in MeOH at 23 °C for 12 h to provide the corresponding methyl ester in 93% yield. This was recrystallized from ethyl acetate. Subsequent single crystal X-ray crystallographic analysis supported stereochemical assignment of 4-nitrobenzoyl ester 17 as shown in the ORTEP drawing.29,30
| Entry | Dienophile | Lewis acid (equiv.) | Temp (°C) (time, h) | Yielda (%) | Endob (dr) |
|---|---|---|---|---|---|
| a Reactions were carried out in 0.1 to 1 mmol scale. Isolated yield after silica gel chromatography.b Endo diastereomeric ratios were determined by 1H NMR; SM: starting material recovered. | |||||
| 1 | 8a | Et2AlCl (1.4) | −78 (2) | 36 | 99:1 |
| 2 | 8a | Et2AlCl (2) | −78 (24) | 50 | 99:1 |
| 3 | 8a | Et2AlCl (3) | −78 (24) | 76 | 99:1 |
| 4 | 8a | Et2AlCl (2) | −78 to 23 (24) | 71 | 97:3 |
| 5 | 8a | TiCl4 (1.4) | −78 (24) | Traces | 90:10 |
| 6 | 8a | TiCl4 (2) | −78 (24) | Traces | 90:10 |
| 7 | 8a | Yb(OTf)3 (2) | 23 (24) | SM | — |
| 8 | 8b | Et2AlCl (1.5) | −78 (8) | 58 | 99:1 |
| 9 | 8c | Et2AlCl (1.5) | −78 (8) | 70 | 99:1 |
| 10 | 8d | Et2AlCl (1.5) | −78 (8) | 73 | 99:1 |
| 11 | 8d | SnCl4 (1) | 0 (48) | 23 | 99:1 |
| 12 | 8e | Et2AlCl (1.5) | −78 (8) | 70 | 99:1 |
| 13 | 8f | Et2AlCl (1.6) | −78 (8) | 98 | 99:1 |
| 14 | 8f | Et2AlCl (1) | −78 to 0 (48) | 15 | 90:10 |
| 15 | 8f | EtAlCl2 (1) | −78 (48) | SM | — |
| 16 | 8f | SnCl4 (1) | 0 (24) | 27 | 99:1 |
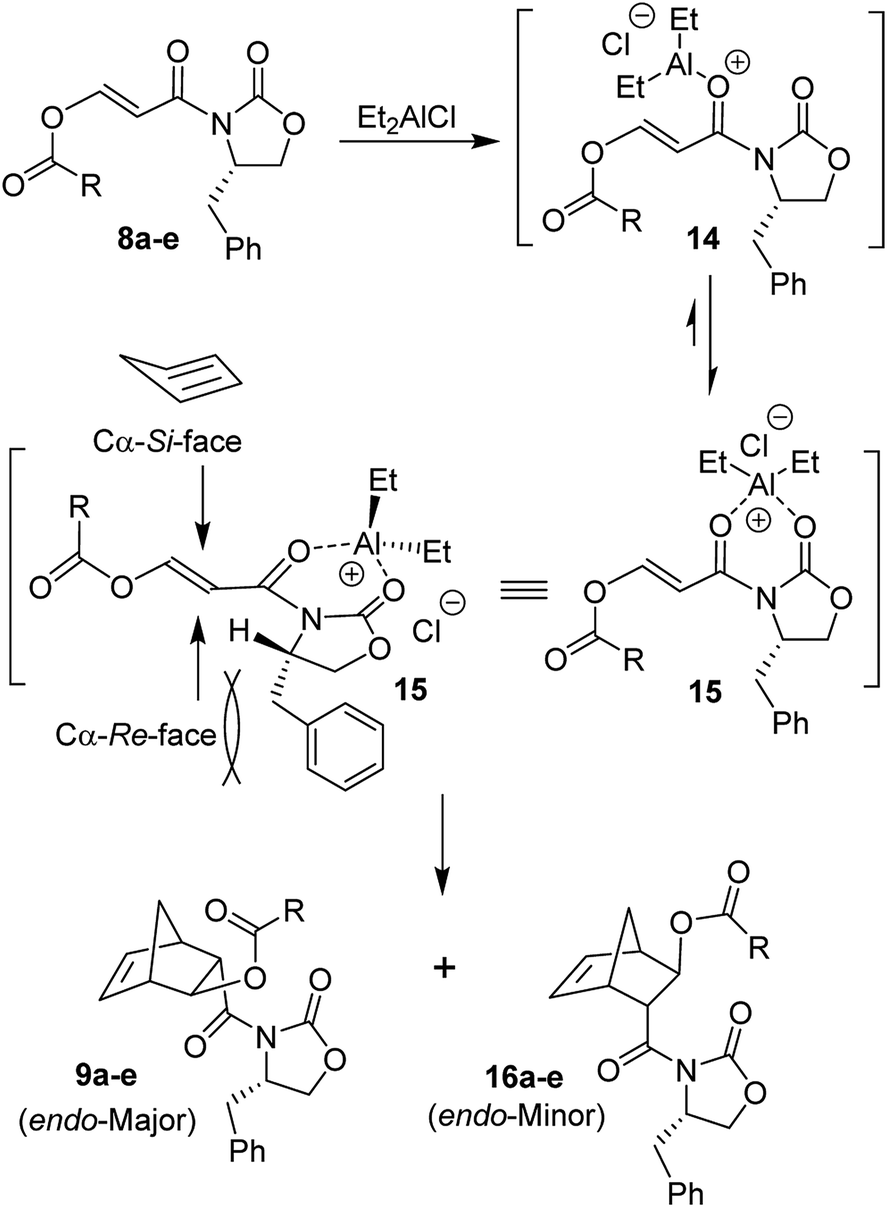 | ||
| Scheme 3 Diels–Alder reactions and preference for diastereofacial selectivity. | ||
 | ||
| Scheme 4 Diels–Alder reaction of dienophile 8d and ORTEP drawing of compound 17. Reagents and conditions. (a) cyclopentadiene, Et2AlCl (1 M in hexane), dry CHCl2, −78 °C, 8 h (73%); (b) Sm(OTf)3, MeOH, 23 °C, 12 h (93%). | ||
We investigated Diels–Alder reaction of dienophile 8e with 4-methoxybenzoate and (S)-4-benzyl-2-oxazolidinone as the chiral auxiliary in the presence of 1.5 equivalent of Et2AlCl to provide endo cycloadduct 9e in 70% yield as a single isomer by NMR analysis (entry 12). We then examined Diels–Alder reaction of enantiomeric dienophile 8f with (R)-4-benzyl-2-oxazolidinone for the synthesis of stereochemically defined crown-THF ligand enantiomer 3. Diels–Alder reaction of 8f was then investigated with varying equivalents of Et2AlCl at −78 °C for 8 h to furnish endo-cycloadduct 9f as a single diastereomer in 98% isolated yield with 1.6 equivalent of Et2AlCl (entry 13).
The use of one equivalent of SnCl4 at 0 °C for 24 h afforded cycloadducts in 27% yield and excellent endo-diastereoselectivity (entry 16).
For the synthesis of crown-THF ligand, we have carried out the Diels–Alder reaction with cyclopentadiene and dienophile 8f in multigram scale (5 g) (Scheme 5). This resulted in cycloadduct 9f in 98% yield. Exposure of cycloadduct 9f to Sm(OTf)3 in MeOH at 23 °C for 12 h resulted in the corresponding methyl ester which was immediately treated with K2CO3 in MeOH at 40 °C for 3 h to provide the corresponding β-hydroxy ester as a colorless oil. The resulting alcohol was treated with TBSOTf in CH2Cl2 in the presence of 2,6-lutidine at 0 °C to 23 °C for 1 h to provide the TBS-ether 10 in 61% yield over 3-steps. As described previously, the TBS-ether 10 was converted to tricyclic ligand alcohol 3.19 Alcohol 3 was converted to succinimidyl carbonate 18 by reaction with disuccinimidyl carbonate (DSC) and Et3N in CH2Cl2 at 23 °C for 24 h.31 The crude carbonate was reacted with known amine 4 in CH2Cl2 in the presence of diisopropylethylamine (DIPEA) for 18 h to provide inhibitor 2 in 69% yield over two steps. Inhibitor 2 displayed an HIV-1 inhibitory Ki value of 14 pM and antiviral IC50 value of 17 pM.17,18
 | ||
| Scheme 5 Synthesis of optically active ligand alcohol 3 and HIV protease inhibitor 2. Reagents and conditions: (a) Et2AlCl, cyclopentadiene, CH2Cl2, −78 °C, 8 h (98%); (b) Sm(OTf)3, MeOH, 23 °C, 12 h (93%); (c) K2CO3, MeOH, 40 °C, 3 h (99%); (d) TBSOTf, 2,6-lutidine, 0 °C to 23 °C, 1 h (66%); (e) DSC, Et3N, CH2Cl2, 23 °C, 24 h; (f) amine 4, mixed carbonate 18, 23 °C, 18 h (69% over two steps). | ||
Conclusions
In summary, we have investigated Diels–Alder reactions of chiral 3-(acyloxy)acryloyl oxazolidinones with cyclopentadiene to provide direct access to optically active 3-hydroxybicyclo[2-2-1]heptane-2-carboxylate derivatives for the synthesis of a 6-5-5 tricyclic ligand alcohol for a variety of exceptionally potent HIV-1 PIs. While the acyloxy dienophiles are much less reactive than alkylacryloyl oxazolidinones, Et2AlCl-promoted reaction generally provided excellent diastereoselectivity and isolated yield. In particular, 3-(4-methoxybenzoyl)acryloyl oxazolidinone derivative 8f and cyclopentadiene afforded 98% yield of endo-diastereoselective derivative 9f. Reaction of cycloadduct in MeOH provided the corresponding methyl ester in excellent yield. The present studies provided convenient access to 3-hydroxybicyclo[2-2-1]heptane carboxylate in optically active forms using readily available commercial starting materials. Further applications of this asymmetric methodology in medicinal chemistry are in progress.Experimental section
General methods
All reactions were carried out under an atmosphere of argon in oven dried (120 °C) glassware with magnetic stirring unless otherwise noted. Solvents, reagents and chemicals were purchased from commercial suppliers. Solvents were purified as follows: CH2Cl2 was distilled from calcium hydride or purified using a solvent purification system; methanol was used without further purification; tetrahydrofuran was distilled from sodium/benzophenone; acetonitrile was purified with a solvent purification system. Purification of reaction products was carried out by flash chromatography using silica gel 230–400 mesh (60 Å pore diameter). Analytical thin layer chromatography was performed on glass-backed silica gel thin-layer chromatography plates (0.25 mm thickness, 60 Å, F-254 indicator). Optical rotations were measured by using a digital polarimeter with a sodium lamp. 1H-NMR spectra were recorded at 23 °C on a 300 and 400 MHz spectrometer and are reported in ppm relative to solvent signals (CDCl3 at δ = 7.26 ppm) as an internal standard. Data are reported as (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, ddd = doublet of doublet of doublets, dddd = doublet of doublet of doublets of doublets, td = triplet of doublets, qd = quartet of doublets, dt = doublet of triplets, dq = doublet of quartets, brs = broad singlet; coupling constant(s) in Hz; integration). Proton-decoupled 13C-NMR spectra were recorded on a 100 MHz spectrometer and are reported in ppm by using the solvent as the internal standard (CDCl3 at δ = 77.16 ppm). Low resolution mass spectra were obtained using a Quadrupole LCMS instrument under ESI+. High resolution mass spectra were obtained by the Mass Spectrometry Center at Purdue University. These experiments were performed under ESI+ and APCI+ conditions using an Orbitrap XL instrument.The enantiomer (+)-13 was prepared according to the procedure described for the compound (−)-13, using (S)-4-benzyl-2-oxazolidinone (S)-12 as chiral auxiliary.
General procedure for Diels–Alder reaction
To a stirred solution of dienophile in CH2Cl2 at specified temperature (Table 1), freshly cracked dicyclopentadiene (10 equiv.) and Lewis acid were sequentially added, and the progress of the reaction was monitored by TLC or crude NMR. The reaction was quenched at a specified temperature by the addition of a saturated aqueous NaHCO3 solution. The mixture was extracted with CH2Cl2 and the organic layer was washed with brine and dried over Na2SO4. Evaporation under reduced pressure afforded crude compound which was purified by column chromatography over silica gel (EtOAc/Hexane) to obtain cycloadduct.[α]20D = +148.3 (c 0.95, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.04–7.98 (m, 2H), 7.58–7.53 (m, 1H), 7.46–7.39 (m, 2H), 7.37–7.19 (m, 5H), 6.33 (dd, J = 5.7, 3.3 Hz, 1H), 6.12 (dd, J = 5.7, 2.8 Hz, 1H), 5.15 (ddd, J = 2.7, 1.9, 0.8 Hz, 1H), 4.76 (ddt, J = 9.9, 7.8, 3.1 Hz, 1H), 4.24 (ddd, J = 8.7, 7.9, 0.7 Hz, 1H), 4.16 (dd, J = 9.1, 2.8 Hz, 1H), 3.98 (t, J = 3.0 Hz, 1H), 3.44–3.41 (m, 1H), 3.26 (dd, J = 13.2, 3.4 Hz, 1H), 3.20 (m, 1H), 2.73 (dd, J = 13.2, 9.7 Hz, 1H), 2.09 (m, 1H), 1.82 (dq, J = 8.9, 1.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 171.9, 166.5, 153.4, 135.4, 135.2, 134.6, 132.9, 129.9, 129.5, 129.3, 128.8, 128.3, 127.3, 77.8, 66.2, 55.2, 51.6, 48.3, 48.2, 45.7, 38.0. ESI-MS (m/z): 440.1 [M + Na]+. HRMS-ESI (m/z): [M + H]+ calcd for C25H23NO5Na, 440.1468; found 440.1472.
The above methyl ester (2.5 g, 8.27 mmol) was dissolved in 45 mL of MeOH and K2CO3 (1.7 g, 12.41 mmol) was added. The reaction mixture was stirred at 40 °C for 3 h and then quenched with saturated aqueous NH4Cl. The solvent was evaporated under reduced pressure and the residue was taken up with water and extracted with CH2Cl2 (3 × 10 mL). Collected organic phases were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure at 0 °C. Only a small portion of this crude compound was purified by column chromatography over silica gel (30–50% Et2O in hexane) to afford the corresponding β-hydroxy ester as a colourless oil. [α]20D = −109.90 (c 2.45, CHCl3). 1H NMR (300 MHz, CDCl3) δ 6.16 (dd, J = 5.7, 2.7 Hz, 1H), 6.08 (dd, J = 5.6, 3.3 Hz, 1H), 4.11 (s, 1H), 3.63 (d, J = 13.0 Hz, 3H), 3.09 (s, 1H), 2.78 (d, J = 0.8 Hz, 1H), 2.68–2.62 (m, 1H), 2.02 (d, J = 3.7 Hz, 1H), 1.90 (d, J = 8.8 Hz, 1H), 1.66 (dd, J = 8.8, 1.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 174.0, 137.3, 134.6, 75.7, 54.9, 51.7, 50.5, 46.3, 44.0.
The remaining crude compound was used in the next step without purification. This latter was dissolved in 100 mL of dry CH2Cl2 and the solution cooled to 0 °C. TBSOTf (2.85 mL, 12.40 mmol) and 2,6-lutidine (2.89 mL, 24.81 mmol) were sequentially added. After warming to 23 °C, the reaction was stirred for further 1 h. Then it was treated with saturated aqueous NaHCO3 and extracted with CH2Cl2 (3 × 20 mL). The organic phase was washed with saturated aqueous NaHCO3 and brine, dried (Na2SO4), filtered and concentrated under reduced pressure. The resulting residue was chromatographed on silica gel (40% CH2Cl2/hexanes) to afford 10 (495 mg, 66% over two step) as a colourless liquid. [α]20D = −69.62 (c 9.25, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.09 (dd, J = 5.7, 2.7 Hz, 1H), 6.02 (dd, J = 5.8, 3.2 Hz, 1H), 4.02 (t, J = 2.1 Hz, 1H), 3.60 (s, 3H), 3.01 (s, 1H), 2.63 (m, 1H), 2.56 (dd, J = 3.6, 2.3 Hz, 1H), 1.89 (dt, J = 8.5, 1.5 Hz, 1H), 1.59 (dq, J = 8.5, 1.8 Hz, 1H), 0.86 (s, 9H), 0.05 (d, J = 5.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 174.0, 137.2, 134.5, 75.8, 55.1, 51.3, 51.1, 46.5, 43.9, 25.7, 17.9, −4.9. ESI-MS (m/z): 283 [M + H]+. HRMS-ESI (m/z): [M + H]+ calcd for C15H26O3SiNa, 305.1543; found 305.1548.
To a stirred solution of difluoro isostere 4 (14 mg, 0.025 mmol) in acetonitrile (1 mL) were added above carbonate 18 (7 mg, 0.023 mmol) and N,N-diisopropylethylamine (20 μL, 0.117 mmol). The reaction was stirred at 23 °C for 18 h. After this period, the reaction mixture was concentrated under reduced pressure to afford a crude residue which was purified by column chromatography (2% MeOH/CHCl3) to give inhibitor 2 (16 mg, 69% over two steps) as an amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.10–8.07 (m, 1H), 7.69 (dd, J = 8.6, 1.9 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 6.93 (brs, 1H), 6.82–6.73 (m, 2H), 6.66 (tt, J = 9.0, 2.4 Hz, 1H), 5.43 (d, J = 6.7 Hz, 1H), 5.29 (d, J = 9.0 Hz, 1H), 4.84 (dd, J = 9.1, 5.7 Hz, 1H), 3.96–3.80 (m, 5H), 3.62 (ddd, J = 19.1, 10.3, 7.0 Hz, 2H), 3.15 (dd, J = 15.1, 8.3 Hz, 1H), 3.09–2.96 (m, 3H), 2.90–2.81 (m, 2H), 2.79–2.70 (m, 2H), 2.69–2.63 (m, 1H), 2.37–2.30 (m, 1H), 1.90–1.78 (m, 2H), 1.45 (dt, J = 12.2, 4.3 Hz, 1H), 0.98–0.91 (m, 5H), 0.89 (d, J = 6.6 Hz, 3H), 0.82–0.77 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 172.9, 164.2 (d, J = 13.2 Hz), 161.7 (d, J = 13.5 Hz), 155.7 (d, J = 13.5 Hz), 141.9 (t, J = 9.3 Hz), 131.4, 130.2, 125.3, 120.9, 118.7, 112.3–112.1 (m), 104.3, 102.1 (t, J = 25.3 Hz), 75.1, 72.8, 68.3, 59.8, 59.0, 54.8, 53.7, 44.8, 42.0, 37.4, 35.0, 27.4, 26.7, 23.5, 20.1, 19.9, 8.0. ESI-MS (m/z): 707.2 [M + H]+. HRMS-ESI (m/z): [M + H]+ calcd for C33H41F2N4O7S2, 707.2385; found 707.2378.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work was provided by the National Institutes of Health (AI150466). NMR and Mass Spectrometry were all performed using shared resources which are partially supported by the Purdue Center for Cancer Research through NIH grant (P30CA023168).Notes and references
- H. Zheng, J. Hou, M. D. Zimmerman, A. Wlodawar and W. Minor, Expert Opin. Drug Discovery, 2014, 9, 125 CrossRef CAS PubMed.
- A. K. Ghosh and S. Gemma, Structure-Based Design of Drugs and Other Bioactive Molecules: Tools and Strategies, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- A. Wlodawer and J. Vondrasek, Annu. Rev. Biophys. Biomol. Struct., 1998, 27, 249 CrossRef CAS PubMed.
- A. K. Ghosh, H. L. Osswald and G. Prato, J. Med. Chem., 2016, 59, 5172 CrossRef CAS PubMed.
- A. S. Fauci and H. D. Marston, N. Engl. J. Med., 2015, 373, 2197 CrossRef PubMed.
- J. A. Esté and T. C. Cihlar, Antiviral Res., 2010, 85, 25 CrossRef PubMed.
- A. K. Ghosh, D. D. Anderson, I. T. Weber and H. Mitsuya, Angew. Chem., Int. Ed., 2012, 51, 1778 CrossRef CAS PubMed.
- A. K. Ghosh, B. Chapsal, I. Weber and H. Mitsuya, Acc. Chem. Res., 2008, 41, 78 CrossRef CAS PubMed.
- A. K. Ghosh, Z. L. Dawson and H. Mitsuya, Bioorg. Med. Chem. Lett., 2007, 15, 7576 CrossRef CAS PubMed.
- M. P. de Béthune, V. Sekar, S. Spinosa-Guzman, M. Vanstockem, S. De Meyer, P. Wigerinck and E. Lefebvre, Darunavir (Prezista, TMC114): From Bench to Clinic, Improving Treatment Options for HIV-Infected Patients in Antiviral Drugs: From Basic Discovery Through Clinical Trials, John Wiley & Sons, Inc., New Jersey, 2011, pp. 31–45 Search PubMed.
- H. Hayashi, N. Takamune, T. Nirasawa, M. Aoki, Y. Morishita, D. Das, Y. Koh, A. K. Ghosh, S. Misumi and H. Mitsuya, Proc. Natl. Acad. Sci. U. S. A., 2014, 79, 5697 Search PubMed.
- Y. Koh, S. Matsumi, D. Das, M. Amano, D. A. Davis, J. Li, S. Leschenko, A. Baldridge, T. Shioda, R. Yarchoan, A. K. Ghosh and H. Mitsuya, J. Biol. Chem., 2007, 282, 28709 CrossRef CAS PubMed.
- S. De Meyer, H. Azijn, D. Surleraux, D. Jochmans, A. Tahri, R. Pauwels, P. Wigerinck and M. P. de Béthune, Antimicrob. Agents Chemother., 2005, 49, 2314 CrossRef CAS PubMed.
- Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents, https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf, accessed on March 31, 2018 Search PubMed.
- G. Sterrantino, M. Zaccarelli, G. Colao, F. Baldanti, S. Di Giambenedetto, T. Carli, F. Maggiolo and M. Zazzi, Infection, 2012, 40, 311 CrossRef CAS PubMed.
- S. De Meyer, E. Lathouwers, I. Dierynck, E. De Paepe, B. Van Baelen, T. Vangeneugden, S. Spinosa-Guzman, E. Lefebvre, G. Picchio and M.-P. de Béthune, AIDS, 2009, 23, 1829 CrossRef PubMed.
- A. K. Ghosh, K. V. Rao, P. R. Nyalapatla, S. Kovela, M. Brindisi, H. L. Osswald, B. S. Reddy, J. Agniswamy, Y.-F. Wang, M. Aoki, S.-i. Hattori, I. T. Weber and H. Mitsuya, ChemMedChem, 2018, 13, 803 CrossRef CAS PubMed.
- M. Aoki, H. Hayashi, K. V. Rao, D. Das, N. Higashi-Kuwata, H. Bulut, H. Aoki-Ogata, Y. Takamatsu, R. S. Yedidi, D. A. Davis, S.-i. Hattori, N. Nishida, K. Hasegawa, N. Takamune, P. R. Nyalapatla, H. L. Osswald, H. Jono, H. Saito, R. Yarchoan, S. Misumi, A. K. Ghosh and H. Mitsuya, eLife, 2017, 6, e28020 CrossRef PubMed.
- A. K. Ghosh, K. V. Rao, P. R. Nyalapatla, H. L. Osswald, C. D. Martyr, M. Aoki, H. Hayashi, J. Agniswamy, Y.-F. Wang, H. Bulut, D. Das, I. T. Weber and H. Mitsuya, J. Med. Chem., 2017, 60, 4267 CrossRef CAS PubMed.
- S. Mukherjee and E. J. Corey, Org. Lett., 2010, 12, 1024 CrossRef CAS PubMed.
- K. M. Reddy, E. Bhimireddy, B. Thirupathi, S. Breitler, S. Yu and E. J. Corey, J. Am. Chem. Soc., 2016, 138, 2443 CrossRef PubMed.
- J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325 CrossRef CAS PubMed.
- K. Narasaka, N. Iwasawa, M. Inoue, T. Yamada, M. Nakashima and J. Sugimori, J. Am. Chem. Soc., 1989, 111, 5340 CrossRef CAS.
- K. Narasaka and I. Yamamoto, Tetrahedron, 1992, 48, 5743 CrossRef CAS.
- M. P. Sibi and H. Matsunaga, Tetrahedron Lett., 2004, 45, 5925 CrossRef CAS.
- D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1984, 106, 4261 CrossRef CAS.
- D. A. Evans, K. T. Chapman, D. T. Hung and A. T. Kawaguchi, Angew. Chem., Int. Ed., 1987, 26, 1184 CrossRef.
- D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1988, 110, 1238 CrossRef CAS.
- M. Zeller, Single-Crystal X-Ray analysis was performed in-house, X-Ray Crystallography laboratory, Department of Chemistry, Purdue University, West Lafayette, IN 47907 Search PubMed.
- CCDC 1955735 contains the supplementary crystallographic data for compound 17.†.
- A. K. Ghosh, T. T. Doung, S. P. McKee and W. J. Thompson, Tetrahedron Lett., 1992, 33, 2781 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1955735. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra10178k |
| ‡ Current address: Department of Excellence of Pharmacy, University of Naples Federico II, Naples, Italy. |
| This journal is © The Royal Society of Chemistry 2019 |