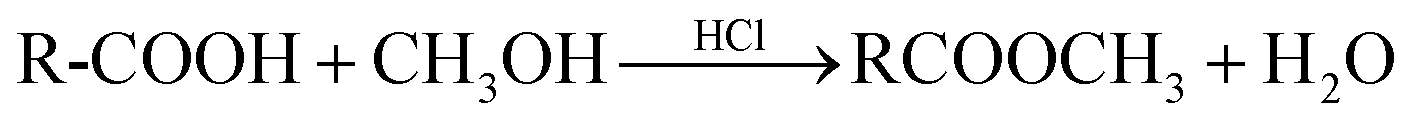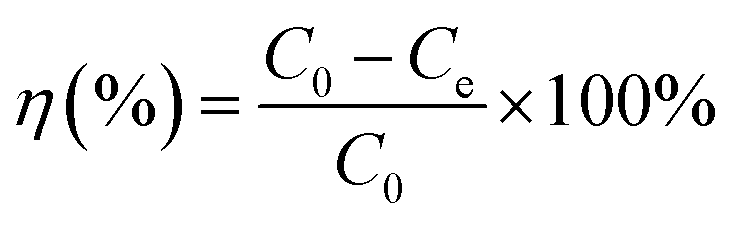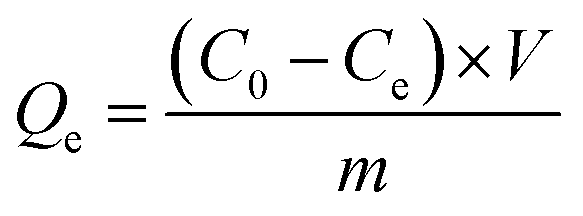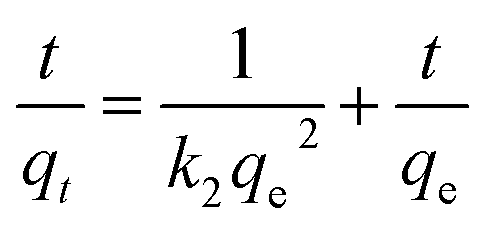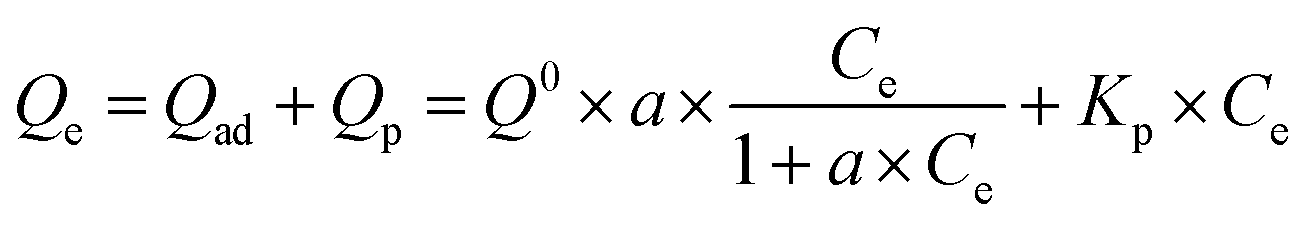DOI:
10.1039/C9RA06495H
(Paper)
RSC Adv., 2019,
9, 41745-41754
Sorption of carbendazim on activated carbons derived from rape straw and its mechanism†
Received
19th August 2019
, Accepted 25th November 2019
First published on 17th December 2019
Abstract
Due to the production and widespread application of pesticides, pesticide pollution poses a potential danger to human health and the ecosystem. Herein, activated carbons employing rape straw as a precursor were produced using H3PO4 as an activating agent at various temperatures (300–600 °C). The activated carbons differed with respect to the physicochemical properties, which were derived from elemental analysis, N2 sorption–desorption, FTIR, XPS, XRD, pHpzc, Boehm titration and blocking of the oxygen-containing groups. The oxygen-containing functional groups and the pore structure of the activated carbons obtained from the different preparation conditions were quite different. The as-prepared samples were applied as sorbents to remove carbendazim (CBD). The results indicated that the sorption of CBD was mainly dominated by partitioning at low concentrations of CBD. Meanwhile, electrostatic attractions played a more important role than hydrophobic interactions at a low initial pH; in contrast, as the initial pH increased, the hydrophobic interaction was the predominant sorption mechanism. Therefore, the results can be used to design some efficient and environmentally friendly adsorbents to reduce the risk of organic pollutants, especially organic pesticides, in aqueous solutions.
1. Introduction
Pesticides are widely used as a supplementary tool for increasing crop yields and reducing diseases and pests. However, some of these pesticides are highly toxic and easy to migrate, and their excessive use can lead to long-term accumulation in the environment, resulting in environmental pollution.1 At present, the micro-pollution of pesticides in surface water is gradually deteriorating, except that surface water, soil and groundwater sources are generally contaminated by pesticides.2–4 Organophosphorus pesticides widely exist in the soil and water of the Mekong River Delta in the Vietnam basin, and the treatment measures do not guarantee safe drinking water to the local residents.5 In addition, nearly 50% samples from the urban river sediment samples from seven metropolitan regions of the United States were contaminated by pyrethroid.6 The trace amount of pesticides frequently has high bioactivity and bioavailability and it is difficult to evaluate and reduce their environmental risks. Therefore, it is essential to eliminate pesticides.
Among the various technologies, sorption is regarded as one of the most valid methods owing to its simple operation and wide availability. The sorption of pesticides plays an important role in immobilizing organic pollutants in the environment. Through the study of the sorption mechanism, we can understand the types of pesticide sorption found in the environment. Studies on pesticide sorption have been conducted on various sorbents such as biochar,7 soil/sediments8 and mesoporous silica.9 So far, the main sorption mechanisms include pore filling, partitioning, and adsorption as well as π–π electron donor–acceptor (π–π EDA), electrostatic and hydrophobic interactions.10 Activated carbon is a kind of porous carbon material with well-developed pores, large surface area and rich surface functional groups due to the biological organic matter.11 A prominent performance of the activated carbon as a sorbent is that it possesses distinctive chemical properties and it is reusable; therefore, it has been widely used in the sorption and purification of harmful substances in the gas or liquid phase.12 In the field of sewage purification, activated carbon shows a distinguished performance in the removal of organic pollutants and inorganic heavy metal ions in water and it is one of the most widely used and most effective sorbents.13 Physical activation and chemical activation are the two main methods for the preparation of activated carbon. The process of preparing activated carbon by the chemical activation technique is comparatively convenient. As an excellent modified reagent, phosphoric acid forms a more heterogeneous pore size distribution in materials;14 thus, it is becoming more and more popular.15
Previous studies have reported that there exist several main mechanisms between the contaminants and activated carbons. Previous literature has demonstrated that electrostatic interactions are dominant for phenol sorption on activated carbons,16 while H-bonds control phenol sorption on activated carbons.17 Moreover, the π–π electron donor–acceptor (π–π EDA) interactions play an important role in nitrophenol adsorption on the activated carbons.18 These different mechanisms indicate that the complicated properties of activated carbons play an important role in the sorption process. Therefore, further studies are needed to clarify the underlying mechanisms for pesticide adsorption on activated carbons.
In this study, carbendazim (CBD; methyl-2-benzimidazolecarbamate) was chosen as a model organic pollutant because it is widely used in disease prevention and the control of crops; the chemical properties of carbendazim are very stable with a half-life of 2–14 weeks in soil and water. Remarkably, it can cause a series of environmental problems.19 The primary objectives in this work were to investigate the influential impact of the O-containing functional groups on CBD sorption onto activated carbons and then to deduce the possible mechanisms for sorption under certain conditions. This understanding will provide the detailed information required to reveal the mechanisms of CBD sorption on activated carbons and help us rationally use the activated carbons as sorbent materials to reduce the risk of CBD in our surroundings.
2. Materials and methods
2.1 Materials
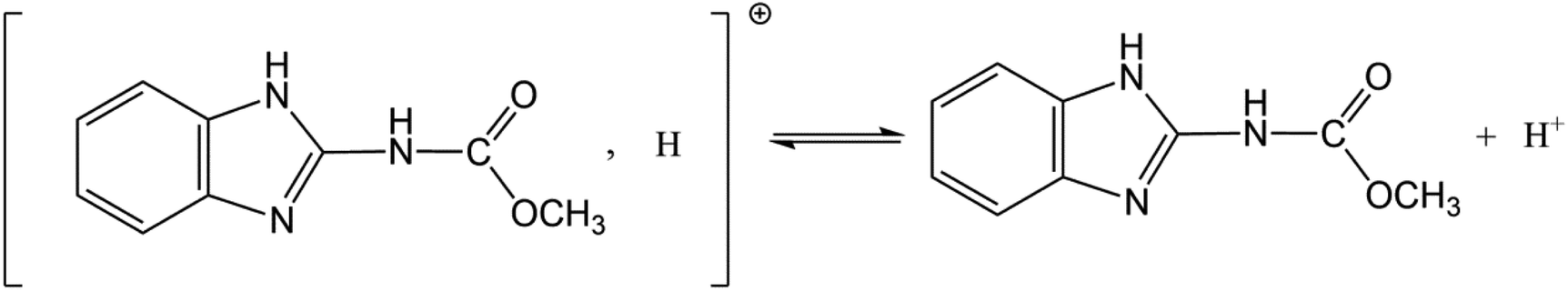
All chemical reagents used in this study were analytical grade. CBD (M = 191.2 g mol−1, purity > 97.3%) was obtained from Sword Agrochemicals (Jiangsu, China). The structure of CBD is presented in Fig. 1. It can exist as a cation (CBD+) and neutral molecule (CBD0) in water because of proton exchange ((pKa = 4.53) (Ni et al., 2002)).38 H3PO4 (85 wt%, AR), HCl (35–37 wt%, AR), and NaOH (99 wt%, AR) were purchased from Sinopharm Chemical Reagent Co. Ltd. The solutions used throughout this study were prepared with high purity water (Milli-Q Reference, Merck Millpore, USA).
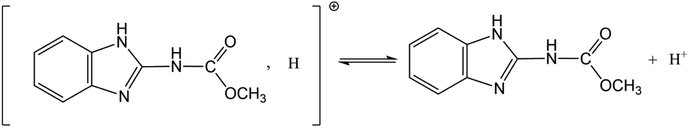 |
| | Fig. 1 Structure of carbendazim (methyl-2-benzimidazolecarbamate). | |
2.2 Preparation of activated carbon
The rape straw was collected from Huazhong Agricultural University rapeseed research field; the raw material was rinsed and dried in an oven at 105 °C for 12 h. Then, the rape straw was crushed and sieved through a 100 mesh sieve. The straw powder was impregnated in an H3PO4 solution (20 wt%) (RS/H3PO4 ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)) and set into an oven at 80 °C for 48 h. Then, the samples were pyrolysed at the desired temperatures (300 °C, 400 °C, 500 °C, and 600 °C) for 2 h in a muffle furnace under pure N2 (99.9%). The products were washed with ultra-purity water until the pH of the supernatant became stable (pH = 7.00 ± 0.10). Finally, the activated carbons were dried at 105 °C for 12 h and ground. A particle size fraction of 100 mesh was collected and labeled with ACx (x stands for the pyrolysis temperature).
:1)) and set into an oven at 80 °C for 48 h. Then, the samples were pyrolysed at the desired temperatures (300 °C, 400 °C, 500 °C, and 600 °C) for 2 h in a muffle furnace under pure N2 (99.9%). The products were washed with ultra-purity water until the pH of the supernatant became stable (pH = 7.00 ± 0.10). Finally, the activated carbons were dried at 105 °C for 12 h and ground. A particle size fraction of 100 mesh was collected and labeled with ACx (x stands for the pyrolysis temperature).
In order to further investigate the interaction between activated carbon and carbendazim, the carboxyl and phenolic hydroxyl groups on the activated carbons were masked, and the tested samples were named AC600-CGB and AC600-PGB. The reaction was as follows:20,21
| |
 | (1) |
| | |
2R-OH + HCOH → (R-O)2CH2 + H2O
| (2) |
2.3 Characterization methods
An elemental analyzer (PerkinElmer 2400, USA) was used to analyze the carbon, hydrogen, oxygen and nitrogen contents in the samples. The N2 adsorption/desorption isotherm determination of the activated carbons was carried out using ASAP-2460 at 77 K. The surface areas (SBET) and pore size distributions were determined by following the BET equation and the Density Functional Theory (DFT) method, respectively. The microspore area (Smic) and volume (Vmic) were obtained with the t-plot method. The surface functional groups of the activated carbons were analyzed with FTIR spectroscopy (VERTEX70 spectrometer, Bruker Corporation, Germany). XPS experiments were carried out with Thermo Scientific Escalab 250(USA) with an Al-Kα (1486.8 eV photons) irradiation source. All spectra were corrected using C1s (284.8 eV) absorption as a standard. The Boehm's titration method was used to quantify the acidic functional groups on the surfaces of the activated carbons.22 The determination of point of zero charge (pHpzc) was carried out following a batch method proposed in the literature.17
2.4 Sorption experiments
Parallel series of batch sorption experiments were carried out in 100 mL centrifuge tubes. About 50 mg sorbents were added into the tubes containing 50 mL solution with a specific amount of carbendazim (50–350 mg L−1). In this study, the initial pH values of the solutions were adjusted with 0.10 mol L−1 HCl or 0.10 mol L−1 NaOH solutions. Then, the centrifuge tubes were transferred to a full temperature oscillation incubator (HUAMEI-QHZ98A, Jiangsu, China) at the desired temperature and shaken at a constant speed for 6 h to ensure that the sorption process reached equilibrium. The suspensions were filtered using a 0.45 μm membrane filter, and the filtrate was checked using a UV-Vis spectrophotometer (UV-2450) at the maximum absorption wavelength of 280 nm and at the minimum absorption wavelength of 277 nm. The concentrations of the solutions were calculated by the difference between the absorbance of the peaks and troughs. All adsorption experiments were conducted in duplicate three times and averaged. The removal efficiency (η (%)) and the amounts of carbendazim adsorbed on the activated carbons, Qe (mg g−1), were calculated using the following eqn (1) and (2):| |
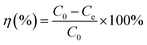 | (3) |
| |
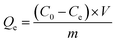 | (4) |
Here, C0 and Ce are the initial and equilibrium concentrations of the adsorbate (mg L−1), V is the volume (L) of the solutions, and m is the mass of the adsorbent (g).
2.5 Data analysis
In this work, the Freundlich model was used to describe the sorption isotherm data:| | |
logQe = logKF + NlogCe
| (5) |
Here, Qe (mg g−1) and Ce (mg L−1) are the equilibrium solid and liquid phase concentrations, respectively. KF ((mg g−1) (mg L−1)−N) is the Freundlich sorption coefficient and N is the nonlinearity index.
The equations for the pseudo-first-order kinetic, pseudo-second-order and intra particle diffusion models were applied for the experimental data of the sorption kinetics:
| | |
ln(qe − qt) = lnqe − k1t
| (6) |
| |
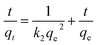 | (7) |
Here,
Qe (mg g
−1) and
Qt (mg g
−1) are the sorption capacities at equilibrium and time
t, respectively;
k1 and
k2 represent the pseudo-first- and pseudo-second-order rate constants, respectively,
Kpi (mg (g
−1 min
−1/2)) is the diffusion rate constant of the intraparticle and
Ci gives an idea about the thickness of the boundary layer.
The respective contributions of adsorption and partitioning to the total CBD sorption were calculated by the dual-mode model:23
| |
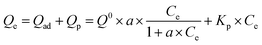 | (9) |
Here,
Qad (mg g
−1) and
Qp (mg g
−1) are the adsorption and partition fractions, respectively;
Q0 (mg g
−1) is the adsorption capacity and
a (L g
−1) and
Kp (L g
−1) are the affinity coefficient and the partitioning coefficient, respectively.
3. Results and discussion
3.1 Characteristics of ACx
The results of the elemental analysis of the activated carbons are presented in Table 1. With the increase in the pyrolysis temperatures, the content of C increased, while the contents of H and O decreased, and the N content was relatively steady. For AC600-CGB and AC600-PGB, the ratio of (O + N)/C and O/C decreased owing to the raw materials of cellulose, hemicellulose, lignin and other components with the pyrolysis processes of dehydration, decarboxylation reaction and dehydroxylation reaction.24 The H/C atomic ratio also decreased, indicating that a new unsaturated hydrocarbon or aromatic ring structure was formed during the pyrolysis process to make the activated carbon more compact.25 The ash content also increased with the increase in the pyrolysis temperature. The polarity index (O + N)/C and the O/C atomic ratio of the AC600-PGB and AC600-CGB samples exhibited a certain reduction relative to that of AC600, which showed that adding methanol and formaldehyde successfully masked the polar functional groups on the activated carbon, and the polarity index (O + N)/C of AC600-CGB was the lowest (Table 1).
Table 1 Physical and chemical properties of the samples
| Samples |
Component, wt% |
Atomic ratio |
Ash% |
| C |
H |
O |
N |
H/C |
O/C |
(O + N)/C |
| AC300 |
60.85 |
3.89 |
23.03 |
0.60 |
0.77 |
0.28 |
0.29 |
7.13 |
| AC400 |
65.74 |
3.13 |
21.40 |
0.74 |
0.57 |
0.24 |
0.25 |
10.05 |
| AC500 |
68.44 |
2.65 |
19.42 |
0.75 |
0.46 |
0.21 |
0.22 |
11.27 |
| AC600 |
71.96 |
1.88 |
17.29 |
0.81 |
0.31 |
0.18 |
0.19 |
13.06 |
| AC600-PGB |
74.23 |
1.95 |
13.72 |
0.79 |
0.29 |
0.14 |
0.15 |
13.32 |
| AC600-CGB |
76.45 |
2.01 |
12.55 |
0.75 |
0.32 |
0.12 |
0.13 |
13.74 |
The XRD patterns of the samples are shown in Fig. S1.† All samples showed two vital peaks at 25° and 42°, which were a signature of the hkl (002) and (100) planes of graphene sheets, indicating the existence of the graphitic structure on the activated carbon. However, with the increase in the pyrolysis temperature, the hkl (100) plane gradually became more prominent and narrow; it was shown that the content of graphite crystallites increased, corresponding to the increase in aromaticity (Table 1).
The FTIR spectra of the activated carbons are shown Fig. S2.† When the pyrolysis temperature increased, the wide stretching vibration peak of the hydroxyl group at approximately 3400 cm−1 was gradually weakened, and the vibration peak of aromatic C–H at approximately 882 cm−1, especially that for the high-temperature activated carbon (Fig. S2a†), became more obvious; this showed that the dehydration reaction of cellulose and lignin increased gradually,26 corresponding to the H/C atomic ratio in Table 1. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (1570–1590 cm−1) in the carboxyl and ketone groups weakened significantly after 500 °C. The intensity of the band at approximately 1704 cm−1 (ester CO) nearly disappeared at 600 °C. The peak at 1156 cm−1 (aliphatic C–O) weakened gradually with the increase in the pyrolysis temperature, corresponding to the O/C atomic ratio in Table 1. The phenolic hydroxyl stretching vibration at 1385 cm−1 gradually disappeared with the increase in the pyrolysis temperature. The peak at 1057 cm−1 may be attributed to the P+–C− bond in acid phosphate esters and to the stretching vibrations in P–O–P.27 The intensity of the AC600-CGB and AC600-PGB infrared peaks significantly weakened compared to that for AC600 (Fig. S2b†), indicating that the addition of formaldehyde and methanol successfully masked the polar functional groups on AC600. However, the intensity of the CO bond (1570–1590 cm−1) in the carboxyl groups on AC600-CGB was weaker than that for AC600-PGB; the stretching vibration intensity of the phenolic hydroxyl groups (approximately 1385 cm−1) on AC600-PGB was weak due to the masking of the carboxyl and phenolic hydroxyl groups.
O bond (1570–1590 cm−1) in the carboxyl and ketone groups weakened significantly after 500 °C. The intensity of the band at approximately 1704 cm−1 (ester CO) nearly disappeared at 600 °C. The peak at 1156 cm−1 (aliphatic C–O) weakened gradually with the increase in the pyrolysis temperature, corresponding to the O/C atomic ratio in Table 1. The phenolic hydroxyl stretching vibration at 1385 cm−1 gradually disappeared with the increase in the pyrolysis temperature. The peak at 1057 cm−1 may be attributed to the P+–C− bond in acid phosphate esters and to the stretching vibrations in P–O–P.27 The intensity of the AC600-CGB and AC600-PGB infrared peaks significantly weakened compared to that for AC600 (Fig. S2b†), indicating that the addition of formaldehyde and methanol successfully masked the polar functional groups on AC600. However, the intensity of the CO bond (1570–1590 cm−1) in the carboxyl groups on AC600-CGB was weaker than that for AC600-PGB; the stretching vibration intensity of the phenolic hydroxyl groups (approximately 1385 cm−1) on AC600-PGB was weak due to the masking of the carboxyl and phenolic hydroxyl groups.
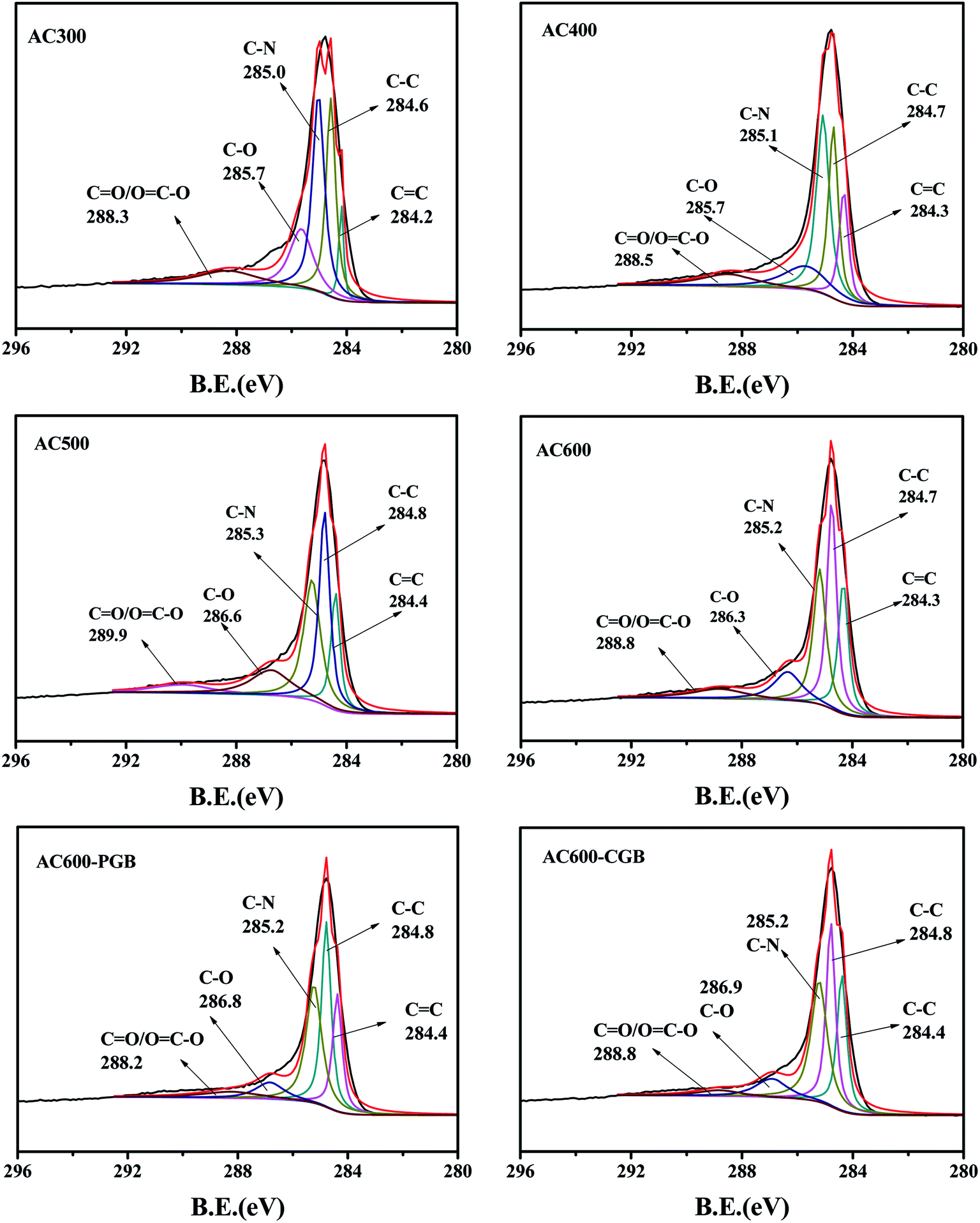
The XPS spectra of the C1s peak of the samples (a–f) are shown in Fig. 2. This shows that C1s can be fitted to five curves; they are CC/C–C (graphitized carbon, 284–285 eV), C–N (pyrrolic-N, protein-N, amine-N and pyridinic-N, 285–285.9 eV), C–O/CO ((carbon in the phenolic, alcohol, ether or carbonyl groups, 286–287 eV) (Ling et al., 2017)),28 and O–CO (carbon in the carboxyl or ester groups, 288–289 eV).28 The content of each functional group is shown in Table 2. Graphite carbon characteristic peaks existed for all samples. With the increase in the activation temperature, the contents of C–C/CC increased gradually, whereas the C–O/CO contents decreased. It was shown that with the increase in the pyrolysis temperature, carbon dioxide was released, the phosphorus compounds were reduced, and the reaction between rape straw and the carbon structure of various oxygen and nitrogen functional groups resulted in their transformation into graphite carbon, which indicated the increase in the carbonization degree on increasing the pyrolysis temperature. After being blocked by methanol and formaldehyde, the content of the acid functional groups on AC600 decreased obviously, but their change was different. Because of methanol blocking the carboxyl groups on AC600, the percent of the peak area of OC–O (carbon in the carboxyl or ester groups, 288–289 eV) for AC600-CGB was less than that for AC600-PGB. At the same time, due to formaldehyde blocking the phenolic hydroxyl groups on AC600, the percent of the peak area of C–O (carbon in the phenolic, alcohol or ether groups, 286–287 eV) for AC600-PGB was less than that for AC600-CGB. The result of XPS is consistent with that of FTIR.
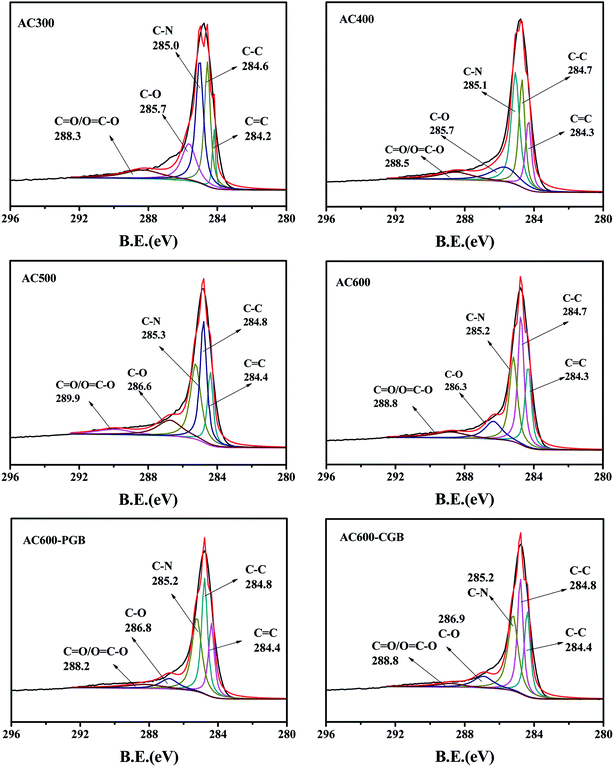 |
| | Fig. 2 XPS analysis of the samples. | |
Table 2 The percent of the peak area determined by XPS
| Samples |
CC/C–C |
C–O |
CO/OC–O |
| AC300 |
33.74 |
19.84 |
12.09 |
| AC400 |
39.89 |
16.17 |
10.17 |
| AC500 |
46.86 |
15.56 |
7.35 |
| AC600 |
51.91 |
12.73 |
8.21 |
| AC600-PGB |
51.30 |
8.76 |
6.90 |
| AC600-CGB |
51.64 |
10.32 |
4.40 |
In order to understand the chemical properties of the surfaces of different samples, the quantities of the surface functional groups and pHpzc were measured. The results are shown in Table 3. The change in the Boehm titration results indicated that the surface functional groups on the samples decreased with the increase in the temperature, which coincided with the FTIR spectra and the XPS survey. Similarly, the numbers of carboxyl groups on AC600-CGB and phenolic hydroxyl groups on AC600-PGB were greatly reduced compared to that for AC600 due to group masking. The pHpzc value of the activated carbon was consistent with the surface functional groups. When the surface functional group content was low, the pHpzc value increased. Therefore, both the elevated temperature and blocking experiments reduced the content of surface functional groups on activated carbon and pHpzc increased.
Table 3 Surface characteristics of the samples
| Samples |
pHpzc |
Carboxyl (mmol g−1) |
Lactone (mmol g−1) |
Phenolic (mmol g−1) |
Total acidic (mmol g−1) |
| AC300 |
3.68 |
0.78 |
0.46 |
1.21 |
2.45 ± 0.12 |
| AC400 |
5.02 |
0.69 |
0.35 |
0.82 |
1.86 ± 0.14 |
| AC500 |
5.61 |
0.46 |
0.45 |
0.67 |
1.58 ± 0.08 |
| AC600 |
5.96 |
0.41 |
0.24 |
0.51 |
1.16 ± 0.11 |
| AC600-PGB |
6.39 |
0.32 |
0.14 |
0.21 |
0.67 ± 0.09 |
| AC600-CGB |
6.55 |
0.10 |
0.11 |
0.32 |
0.53 ± 0.07 |
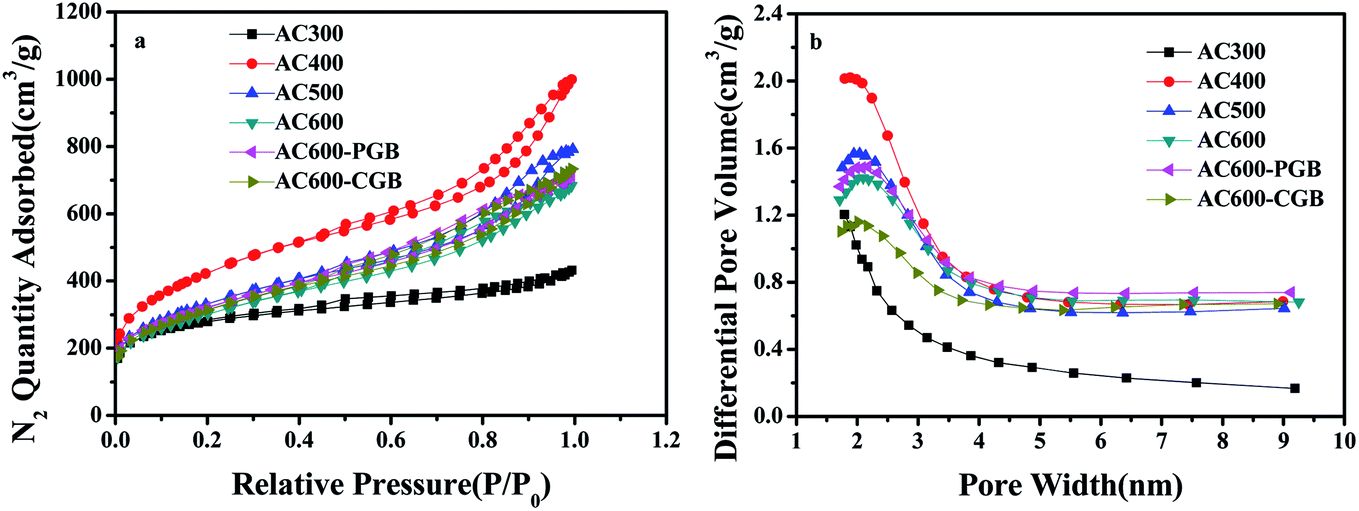
According to the IUPAC classification,29 six kinds of N2 sorption/desorption isotherms showed a mixture of type I and type IV isotherms and obvious hysteresis loops (Fig. 3a), which usually appear for a mixed microporous and mesoporous sorbent material. It could be seen that some of them also had a part of the microporous structure in the six kinds of activated carbons (Fig. 3b). The pyrolysis temperature exhibited a significant influence on the textural properties, as shown in Table S1.† The BET surface area and the total pore volume gradually increased with the increase in the temperature from 300 °C to 400 °C; however, they gradually decreased from 400 °C to 600 °C. At a lower temperature (300 °C), the pore structure was still not fully open. The BET surface area and pore volume of air reach the maximum values at 400 °C: 1490 m2 g−1 and 1.40 cm3 g−1, respectively. At a higher temperature, the violent gasification reaction led to a severe hole collapse in the structure or them being cross-linked together, leading to decrease in the BET surface area and pore volume.30 After group masking, the structure and properties were basically the same as those of AC600, which showed that the reaction did not destroy the surface structure of the activated carbon.
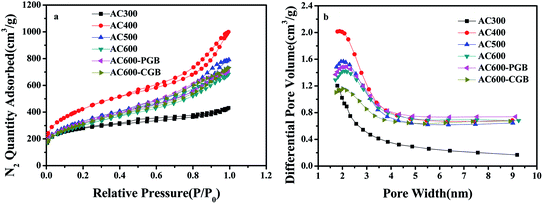 |
| | Fig. 3 (a) N2 sorption and desorption isotherms and (b) pore size distributions of the samples. | |
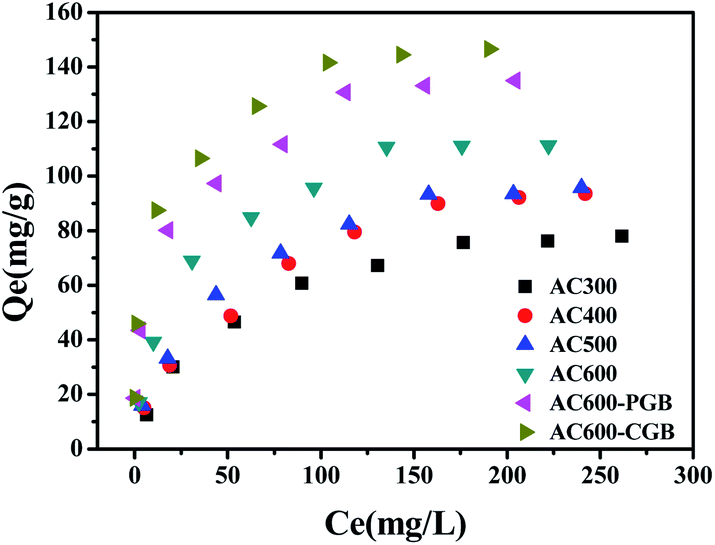
3.2 Sorption isotherms of carbendazim onto ACx
The sorption isotherms of CBD on ACx are presented in Fig. 4 and the parameters of the Freundlich model are listed in Table 4. The Freundlich model was used to describe the isotherms well (R2 > 0.946). Usually, the nonlinear index, N, is an important parameter to analyze the sorption theory. The energy distribution of the ACx sorption sites and the heterogeneity of sorption sites were reflected. With the reduction in the O-containing functional groups of ACx, the N values decreased from 0.507 to 0.273, which indicated a more heterogeneous glassy, hard or condensed sorption domain in ACx and a wider sorption site energy distribution.31 As can be seen from Table 4, KF increases with the decrease in the O-containing functional groups on ACx; however, it cannot reasonably respond to the sorption capacity of ACx. Therefore, the sorption coefficient, Kd, values (Kd = Qe/Ce, L g−1) were determined by the equilibrium concentration, which enabled a reasonable comparison of the sorption capacity of ACx. The Kd values of all ACx samples decreased significantly with the increase in the concentration of CBD. The most likely reason for this is that the CBD sorption on ACx was nonlinear and the sorption sites of high energy on ACx were occupied by CBD.32 At a low CBD concentration range, the Kd value increased with the decrease in the O-containing functional groups. However, this was not the case at high concentrations of CBD (for example, 1000 mg L−1). The results stated that the sorption capacity of CBD on ACx was dependent on the CBD concentration. However, the Kd values were significantly negatively correlated with the N values for the whole concentration range in this study (0–350 mg L−1). On the one hand, the O-containing functional groups of ACx could be combined with organic compounds through a special interaction. Previous studies indicated that the Kd value of the sorption of sulfamethoxazole biochar at 450 °C was higher than that for biochar at 600 °C, which was because the functional groups of biochar obtained by the low pyrolysis temperature were more abundant, and the sorption of the surface functional groups was more important than the sorption of the surface area.33 On the other hand, it showed that O-containing functional groups could also be combined with water molecules, which resulted in competition for the sorption of target compounds. In this experiment, there was a significant negative correlation between the polarity index (O + N)/C atomic ratios and the Kd values at a low concentration (Fig. S3†), indicating that more surface functional groups on ACx limited the sorption of CBD on its surface, and the hydrophobic reaction played an important role in the sorption of CBD on ACx. However, there was not a correlation between the polarity index (O + N)/C atomic ratios and the Kd values at a high concentration, which implied that adsorption was controlled by other mechanisms.34 The negative correlation between the Kd values and the content of O-containing functional groups measured by XPS matched the number of surface functional groups obtained with the Bohem titration method.
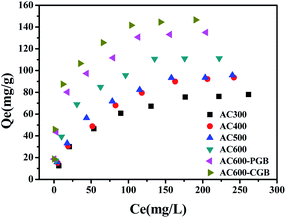 |
| | Fig. 4 Sorption isotherms of CBD by the samples (temperature = 20 ± 0.5 °C; initial pH = 7.00 ± 0.10; time = 360 ± 5 min). | |
Table 4 Isotherm fitting results of CBD sorption on the samples
| Samples |
FMa |
Kdc (L g−1) |
| KFb |
N |
R2 |
10 (mg L−1) |
300 (mg L−1) |
1000 (mg L−1) |
| FM: Freundlich model. KF: [(mg g−1) (mg L−1)−N]. The Kd values (L g−1) were calculated from FM at different equilibrium concentrations of CBD (10, 300 and 1000 mg L−1). |
| AC300 |
5.458 ± 1.227 |
0.507 ± 0.046 |
0.946 |
1.754 |
0.333 |
0.181 |
| AC400 |
7.211 ± 1.096 |
0.487 ± 0.021 |
0.987 |
2.213 |
0.386 |
0.208 |
| AC500 |
9.120 ± 1.117 |
0.452 ± 0.025 |
0.978 |
2.582 |
0.401 |
0.207 |
| AC600 |
14.158 ± 1.132 |
0.412 ± 0.030 |
0.963 |
3.656 |
0.495 |
0.244 |
| AC600-PGB |
31.696 ± 1.308 |
0.290 ± 0.009 |
0.993 |
6.180 |
0.552 |
0.235 |
| AC600-CGB |
39.446 ± 1.062 |
0.273 ± 0.016 |
0.977 |
7.379 |
0.624 |
0.260 |
Similar results were reported that the pHpzc of wheat biochar was 4.2, while the pHpzc of activated carbon was 7.8, and the content of the O-containing functional groups of biochar was 5.4 times that of activated carbon.35 The more the content of O on the surface of biochar, the larger and denser the water molecules formed around the O-containing functional groups, resulting in intense competition between the water molecules and CBD to adsorb on the biochar surface. Therefore, the biochar adsorption of pesticides was about 30–50% of that for activated carbon. As shown in Fig. 4, the equilibrium adsorption capacity of AC600 is 111.31 mg g−1, while the equilibrium sorption capacities of AC600-PGB and AC600-CGB increase by 21.31 and 31.67%, respectively. This also stated that the hydrophobic interaction could play an important role in the sorption process.
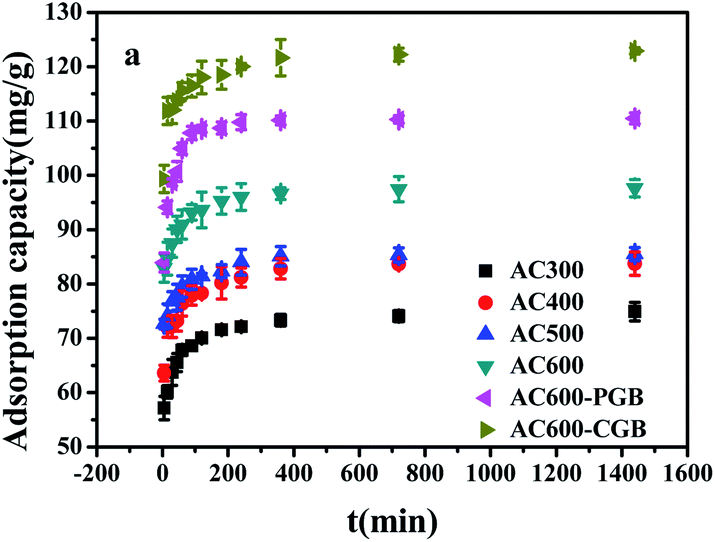
3.3 The sorption kinetics of CBD onto ACx
The CBD sorption kinetics on ACx are shown in Fig. 5. It can be seen from the figure that the sorption amount of CBD on ACx and the amount of CBD increased rapidly in the first 5 minutes of contact with CBD, reaching about 80% of the total sorption capacity. This was because the higher driving force in the initial stage caused carbendazim to transfer rapidly to the surface sites of the adsorbent particles. After 5 minutes, the sorption rate increased slowly until an equilibrium was reached. This was mainly caused by the reduction in the vacant sites on the adsorbents and the repulsion between the solute molecules on the solid surfaces and the solute molecules in the bulk phase.36 Based on the tested material prepared in this experiment, it was worth noting that the sorption capacity in the first 5 minutes could reach 80% of the total sorption capacity, which indicated that ACx is an ideal adsorbent. In addition, AC600-CGB had the best sorption performance among all the selected samples for CBD sorption.
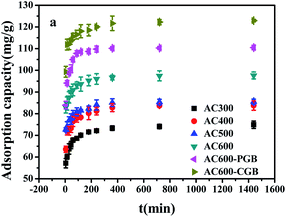 |
| | Fig. 5 The effect of contact time for CBD sorption on the samples (temperature = 20 ± 0.5 °C; initial pH = 7.00 ± 0.10; time = 360 ± 5 min). | |
Sorption kinetics analysis is an important parameter in the process of sorption and the potential mechanism of sorption (Fig. S4†). The correlation coefficient values (R2) calculated from the pseudo-second-order kinetic model were above 0.999, and they were higher than those of the pseudo-first-order model. In addition, the calculated qe values were in good agreement with the experimental values, thus showing quite good linearity (Table S2†). This suggested that the sorption process was affected by chemical interactions.36
The sorption process of CBD on the tested sample was described by an intra-particle diffusion model (Table S2†). The multi-linear relationship between Qt and t1/2 indicated that in this study, the intra-particle diffusion model of CBD absorbed on the activated carbon sample was multi-linear and multi-stage. The first stage represented the instantaneous sorption of CBD from the solution to the outer surface of the tested sample, which was mainly the sorption on the outer surface. The second stage demonstrated that the CBD molecules passed through the liquid membrane into the solid surface, as described by the CBD gradual sorption on the activated carbon samples. This suggested that at this stage in the activated carbon particles in the samples, diffusion dominated and the surface was saturated by the sorption of the first-order phase; when the surface sorption increased gradually, the CBD of the diffusion resistance increased and therefore, this stage of Kpi was less than the first stage. In the third stage, the adsorption reached an equilibrium state. In this stage, the CBD molecules interacted with the sample surface and reacted with the sample through chemical bonds. In the three stages, the boundary thickness of the AC600-CGB sample was the largest compared with that of the other five samples, indicating that the boundary layer of AC600-CGB had the greatest influence on the sorption of CBD. This was mainly attributed to the maximum hydrophobicity shown by the sample. In addition, the figure showed that the linear adsorption relationship of all samples with CBD did not pass through the origin, which indicated that the diffusion film model was not the only speed control step in the sorption process and there were other sorption mechanisms in existence.
3.4 The sorption thermodynamics of CBD onto ACx
The thermodynamic parameters (ΔS0, ΔG0, and ΔH0) derived from the CBD adsorption on the as-prepared samples were calculated from the temperature-dependent adsorption isotherms (Text S1, ESI†), and the results are presented in Table S3.† As shown in Table S3,† the ΔG0 values of the as-prepared samples are negative; therefore, the adsorption process was spontaneous. However, the increase in ΔG0 indicated that the as-prepared samples had the most efficient adsorption for CBD at a higher temperature. The negative value of ΔH0 for CBD adsorption showed that the CBD adsorption was an exothermic process. The negative value of ΔS0 for CBD demonstrated the increase in the degree of disorder at the solid–liquid interface.
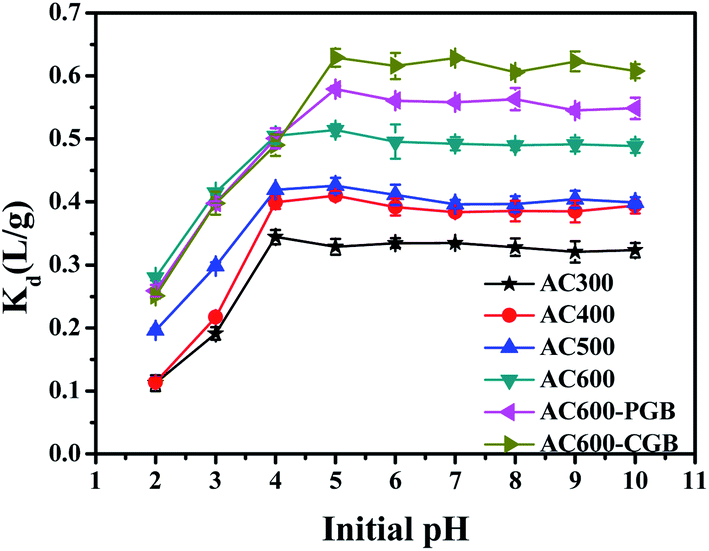
3.5 Effect of initial pH on CBD sorption
The sorption of ionic compounds on carbonaceous materials depends on the solution pH; the system pH can not only affect the existence of ionic organic compounds, but also change the surface charge of the activated carbons, which affects the adsorption of the ionic organic compounds on activated carbons.37 CBD is a weak basic compound and its pKa value is 4.53;38 when pH is greater than pKa, it exists as a neutral molecule in water. However, when pH is less than pKa, it exists in water in the form of a cation (Fig. S5†). The effect of the initial pH on the adsorption of CBD on ACx is presented in Fig. 6. With the increase in the initial pH, all samples except AC300 showed the same sorption trend; the Kd values increased with the increase in the initial pH from 2.00 to 5.00 and then, they remained steady. Since the pHpzc value of AC300 was 3.68, when the pH value of the solution was 4, AC300 had a negative charge, while CBD had a positive charge. Therefore, there was electrostatic attraction between them, which made the adsorption amount of CBD at this pH value of AC300 maximum. The pHpzc values of the other five samples were greater than 5; therefore, when the pH value of the solution was 4, there was electrostatic repulsion between the positive charge and CBD with the same positive charge. Therefore, when the pH value of the solution was 5, the adsorption amount of CBD for the other five samples was the largest. The sorption of ionic compounds was largely influenced by the electrostatic interactions between the sorbate and the sorbent.39 At initial pH < 3.00, CBD+ was dominant and the ACx surface was positively charged; therefore, electrostatic repulsion occurred between CBD and ACx, resulting in decrease in the CBD adsorption on ACx. However, the positively charged activated carbons still had a certain sorption capacity for CBD, indicating that a major driving force may be the interaction between the aromatic ring of ACx and CBD; it was a π–π EDA bond instead of ordinary electrostatic cation exchange.40
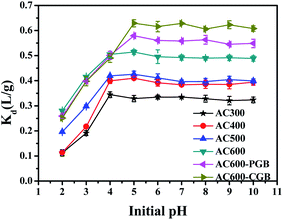 |
| | Fig. 6 CBD adsorption capacities of the samples in different initial pH ranges (temperature = 20 ± 0.5 °C; initial pH = 7.00 ± 0.10; time = 360 ± 5 min). | |
When the initial pH is above 4.53, CBD exists as a neutral molecule in solutions; the electrostatic interaction is not the main mechanism of sorption, and the main sorption mechanism is very likely discussed above: π–π EDA, hydrophobic interaction and pore-filling. At the same time, when the pH was between 2.00 and 4.00, the Kd values of the three samples of AC600, AC600-PGB and AC600-CGB were basically the same and the pore structure of the three samples was basically the same. This showed that the electrostatic interaction played a far more important role than the hydrophobic interaction and π–π EDA in this pH range. The Kd values of AC600-CGB and AC600-PGB compared to the Kd value of AC600 increased to 22.37% and 12.68%, respectively. In addition, combined with the initial pH increasing from 4.00 to 5.00, the Kd value of AC600 increased only to 1.8%. As explained above, hydrophobic interactions played a far more important role than π–π EDA.
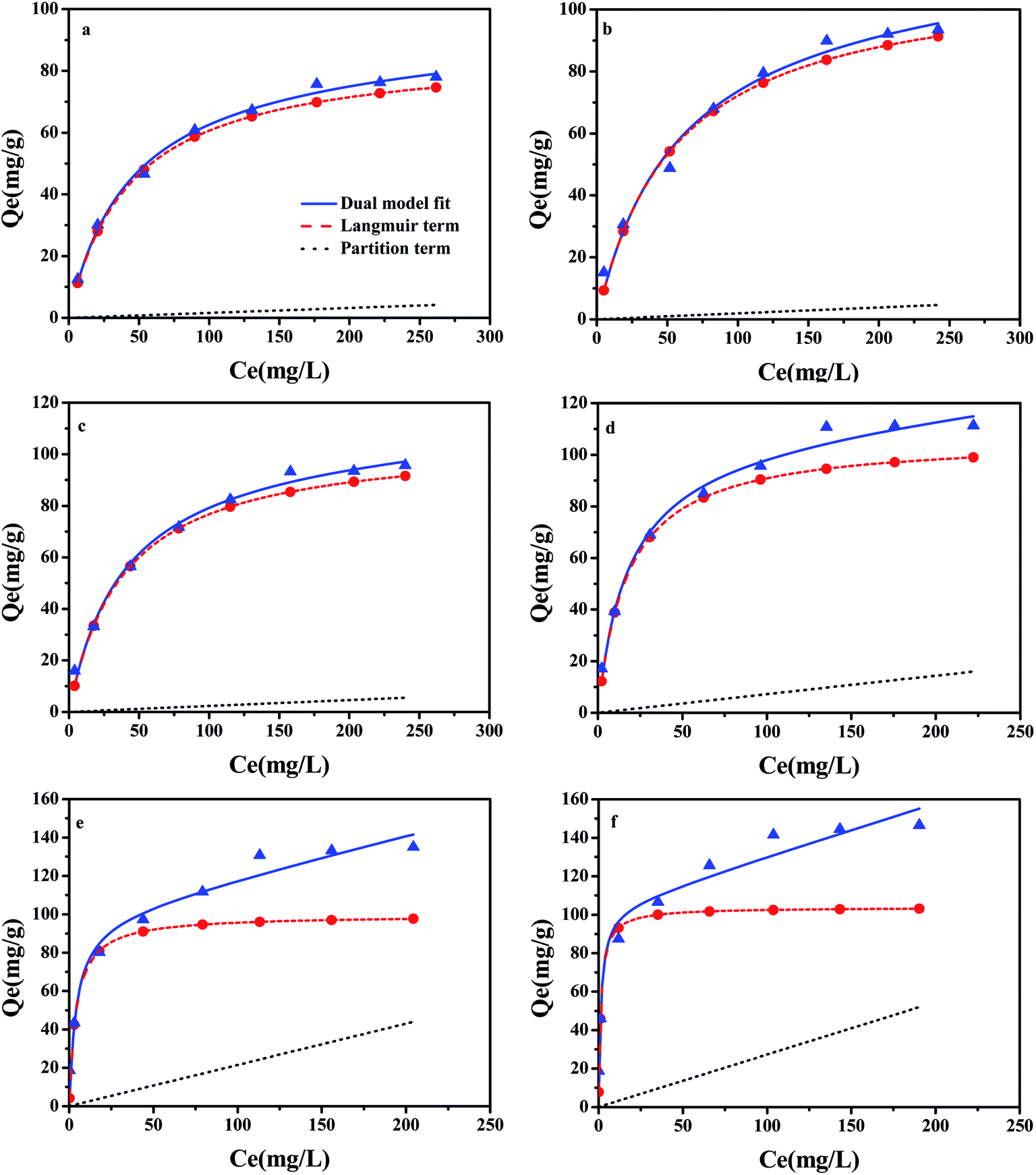
3.6 The mechanisms of CBD adsorption on ACx samples
A large number of studies have shown that the adsorption of organic pollutants on a water medium includes the role of partition and adsorption.41 In order to evaluate the contribution of adsorption in the carbonized phase and partitioning in the non-carbonized phase in the sorption of compounds on carbonaceous materials, the dual-mode model was successfully referenced in this area of research.23 The partition and adsorption of CBD on ACx at different concentrations were calculated by the dual-mode model. In this experiment, the dual-model provided a good fit to the isotherm for all the samples (Fig. 7) and the results are shown in the Table S4.† The adsorption of CBD on all activated carbons was more than that for partitioning for all of the CBD concentrations in this study. However, with the increase in the CBD concentration, the contribution of partition (Qp) and adsorption (Qad) to the overall sorption was also different. In the low concentration range, adsorption was greater than partitioning on ACx and the Qad value contribution was over 97% of the overall sorption. However, in the high concentration range, the contribution of CBD to the partition and adsorption of different samples in the overall sorption was different. When the concentration of CBD reached 300 mg L−1, on AC300, AC400 and AC500, the contribution of adsorption to the overall sorption was still far greater than that of partitioning. While the content of the O-containing functional groups in ACx decreased gradually, the contribution ratio of partitioning to the overall sorption increased gradually, especially for AC600-PGB and AC600-CGB; their Qp values reached 39.4% and 44.13%, respectively. The main reason might be that the reduction of the O-containing functional groups on the surface of the samples led to a better exposure to the surface of the samples and an increase in the CBD concentration, resulting in saturation of the sorption process. Therefore, CBD could be assigned to the non-carbonized phase.42
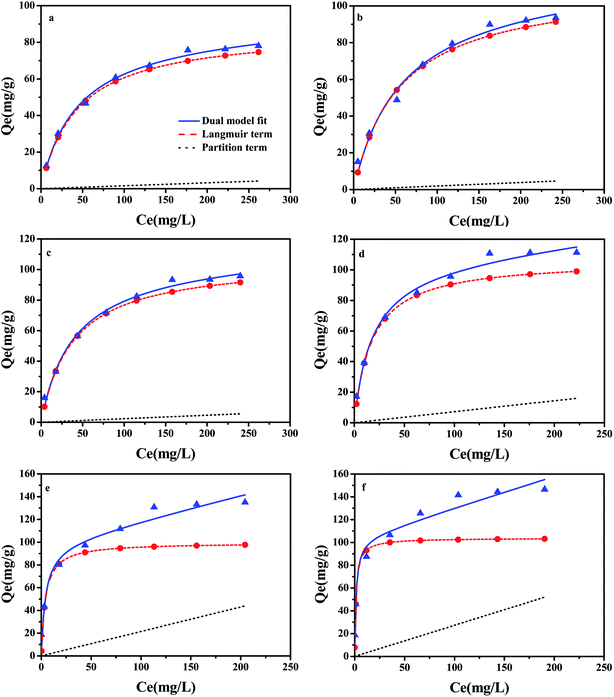 |
| | Fig. 7 CBD isotherms of the samples fitted by the dual-model sorption model. (a) AC300, (b) AC400, (c) AC500, (d) AC600, (e) AC600-PGB, and (f) AC600-CGB; dashed line is the adsorption contribution and dotted line is the partition contribution (temperature = 20 ± 0.5 °C; initial pH = 7.00 ± 0.10; time = 360 ± 5 min). | |
4. Conclusions
This study revealed that the main contribution of the CBD adsorption to ACx was a partitioning mechanism at the selected CBD concentrations. In addition, the adsorption of CBD was greater than partitioning for ACx at a low concentration range; in contrast, at the high concentration range, when the O-containing functional groups on ACx were significantly reduced, the effect of partitioning on the overall sorption increased gradually. The results demonstrated that there were different adsorption mechanisms at different initial pH values and various CBD concentrations. At a low initial pH, the electrostatic interaction played a more important role than the hydrophobic interaction. However, as the initial pH increased, the hydrophobic interaction took up a large proportion in the adsorption of CBD. Therefore, the results indicated that farm residues can be used to produce efficient and environmentally friendly adsorbents to reduce the risk of organic matter, especially organic pesticides, in an aqueous environment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the National Key R&D Program of China (2018YFD1000900) and the Fundamental Research Funds for the Central Universities (2662017JC023).
References
- A. Alvarez-Martin, M. S. Rodriguez-Cruz, M. S. Andrades and M. J. Sanchez-Martin, Environ. Sci. Pollut. Res. Int., 2016, 23, 9192–9203 CrossRef CAS PubMed.
- I. K. Konstantinou, D. G. Hela and T. A. Albanis, Environ. Pollut., 2006, 141, 555–570 CrossRef CAS PubMed.
- M. Arias-Estevez, E. Lopez-Periago, E. Martinez-Carballo, J. Simal-Gandara, J. C. Mejuto and L. Garcia-Rio, Agric., Ecosyst. Environ., 2008, 123, 247–260 CrossRef CAS.
- F. Worrall and T. Besien, J. Hydrol., 2005, 303, 92–107 CrossRef CAS.
- P. V. Toan, Z. Sebesvari, M. Blasing, I. Rosendahl and F. G. Renaud, Sci. Total Environ., 2013, 452, 28–39 CrossRef PubMed.
- K. M. Kuivila, M. L. Hladik, C. G. Ingersoll, N. E. Kemble, P. W. Moran, D. L. Calhoun, L. H. Nowell and R. J. Gilliom, Environ. Sci. Technol., 2012, 46, 4297–4303 CrossRef CAS PubMed.
- B. W. Zhao, H. Xu, F. F. Ma, T. Zhang and X. J. Nan, RSC Adv., 2019, 9, 5218–5223 RSC.
- K. Sun, J. Jin, M. J. Kang, Z. Y. Zhang, Z. Z. Pan, Z. Y. Wang, F. C. Wu and B. S. Xing, Environ. Sci. Technol., 2013, 47, 5138–5145 CrossRef CAS PubMed.
- R. Otero, D. Esquivel, M. A. Ulibarri, C. Jimenez-Sanchidrian, F. J. Romero-Salguero and J. M. Fernandez, Chem. Eng. J., 2013, 228, 205–213 CrossRef CAS.
- L. Zhu, N. Zhao, L. H. Tong and Y. Z. Lv, RSC Adv., 2018, 8, 21012–21019 RSC.
- S. Ismadji, Y. Sudaryanto, S. B. Hartono, L. E. K. Setiawan and A. Ayucitra, Bioresour. Technol., 2005, 96, 1364–1369 CrossRef CAS PubMed.
- I. Martin-Gullon, J. P. Marco-Lozar, D. Cazorla-Amoros and A. Linares-Solano, Carbon, 2004, 42, 1339–1343 CrossRef CAS.
- L. K. Zhang, J. Y. Guo, X. M. Huang, W. D. Wang, P. Sun, Y. M. Li and J. H. Han, RSC Adv., 2019, 9, 365–376 RSC.
- Q. Y. Zhou, X. Jiang, X. Li, C. Q. Jia and W. J. Jiang, RSC Adv., 2018, 8, 30171–30179 RSC.
- M. Al Bahri, L. Calvo, M. A. Gilarranz and J. J. Rodriguez, Chem. Eng. J., 2012, 203, 348–356 CrossRef CAS.
- K. Laszlo and A. Szucs, Carbon, 2001, 39, 1945–1953 CrossRef CAS.
- M. Franz, H. A. Arafat and N. G. Pinto, Carbon, 2000, 38, 1807–1819 CrossRef CAS.
- J. S. Mattson, H. B. Mark, M. D. Malbin, W. J. Weber and J. c. Crittend, J. Colloid Interface Sci., 1969, 31, 116–130 CrossRef CAS.
- J. G. Cuppen, P. J. Van den Brink, E. Camps, K. F. Uil and T. C. Brock, Aquat. Toxicol., 2000, 48, 233–250 CrossRef CAS PubMed.
- J. P. Chen and L. Yang, Langmuir, 2006, 22, 8906–8914 CrossRef CAS PubMed.
- J. L. Gardeatorresdey, M. K. Beckerhapak, J. M. Hosea and D. W. Darnall, Environ. Sci. Technol., 1990, 24, 1372–1378 CrossRef CAS.
- H. P. Boehm, Carbon, 1994, 32, 759–769 CrossRef CAS.
- B. S. Xing and J. J. Pignatello, Environ. Sci. Technol., 1997, 31, 792–799 CrossRef CAS.
- B. B. Sun, F. Lian, Q. L. Bao, Z. Q. Liu, Z. G. Song and L. Y. Zhu, Environ. Pollut., 2016, 214, 142–148 CrossRef CAS PubMed.
- H. Zheng, Z. Y. Wang, J. Zhao, S. Herbert and B. S. Xing, Environ. Pollut., 2013, 181, 60–67 CrossRef CAS PubMed.
- M. Keiluweit, P. S. Nico, M. G. Johnson and M. Kleber, Environ. Sci. Technol., 2010, 44, 1247–1253 CrossRef CAS PubMed.
- Z. F. Wang, E. Nie, J. H. Li, Y. J. Zhao, X. Z. Luo and Z. Zheng, J. Hazard. Mater., 2011, 188, 29–36 CrossRef CAS PubMed.
- L. L. Ling, W. J. Liu, S. Zhang and H. Jiang, Environ. Sci. Technol., 2017, 51, 10081–10089 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- Y. Chen, S. R. Zhai, N. Liu, Y. Song, Q. D. An and X. W. Song, Bioresour. Technol., 2013, 144, 401–409 CrossRef CAS PubMed.
- B. S. Xing, J. J. Pignatello and B. Gigliotti, Environ. Sci. Technol., 1996, 30, 2432–2440 CrossRef CAS.
- K. Sun, B. Gao, K. S. Ro, J. M. Novak, Z. Y. Wang, S. Herbert and B. S. Xing, Environ. Pollut., 2012, 163, 167–173 CrossRef CAS PubMed.
- Y. Yao, B. Gao, H. Chen, L. J. Jiang, M. Inyang, A. R. Zimmerman, X. D. Cao, L. Y. Yang, Y. W. Xue and H. Li, J. Hazard. Mater., 2012, 209, 408–413 CrossRef PubMed.
- J. Z. Ni, J. J. Pignatello and B. S. Xing, Environ. Sci. Technol., 2011, 45, 9240–9248 CrossRef CAS PubMed.
- X. B. Yang, G. G. Ying, P. A. Peng, L. Wang, J. L. Zhao, L. J. Zhang, P. Yuan and H. P. He, J. Agric. Food Chem., 2010, 58, 7915–7921 CrossRef CAS PubMed.
- Y. Y. Sun, Q. Y. Yue, B. Y. Gao, L. H. Huang, X. Xu and Q. Li, Chem. Eng. J., 2012, 181, 790–797 CrossRef.
- B. L. Chen and Z. M. Chen, Chemosphere, 2009, 76, 127–133 CrossRef CAS PubMed.
- N. Ni, T. Sanglivi and S. H. Yalkowsky, Int. J. Pharm., 2002, 244, 99–104 CrossRef CAS PubMed.
- M. Teixido, J. J. Pignatello, J. L. Beltran, M. Granados and J. Peccia, Environ. Sci. Technol., 2011, 45, 10020–10027 CrossRef CAS PubMed.
- J. Jin, K. Sun, F. C. Wu, B. Gao, Z. Y. Wang, M. J. Kang, Y. C. Bai, Y. Zhao, X. T. Liu and B. S. Xing, Sci. Total Environ., 2014, 473, 308–316 CrossRef PubMed.
- F. Lian, F. Huang, W. Chen, B. S. Xing and L. Y. Zhu, Environ. Pollut., 2011, 159, 850–857 CrossRef CAS PubMed.
- C. T. Chiou and D. E. Kile, Environ. Sci. Technol., 1998, 32, 338–343 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06495h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Shuijiao Liaoa,
Xiali Yuea and
Guangsheng Zhouc
*a,
Shuijiao Liaoa,
Xiali Yuea and
Guangsheng Zhouc