
Trimerization and tetramerization of ethylene in continuous gas-phase reaction using a Cr-based supported liquid phase catalyst†
Tobias
Müller
a,
John Thomas
Dixon
b,
Marco
Haumann
 *a and
Peter
Wasserscheid
a
*a and
Peter
Wasserscheid
a
aFriedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Lehrstuhl für Chemische Reaktionstechnik (CRT), Egerlandstr. 3, 91058 Erlangen, Germany. E-mail: marco.haumann@fau.de
bSasol South Africa (Pty) Ltd, 1 Klasie Havenga Street, Sasolburg, 1947, South Africa
First published on 19th November 2018
Abstract
Homogeneous Cr–PNP tri- and tetramerization catalysts were dissolved together with the co-catalyst MAO in the high-boiling, non-polar, aliphatic hydrocarbon perhydro-dibenzyltoluene (H18-DBT). The solution was then dispersed onto different high surface area supports. The resulting solid materials were tested in the gas-phase selective oligomerization of ethylene at 43 °C and pressures between 1 and 14 bar. Low activities were observed at ambient pressure. At ethylene pressures of higher than 3 bar the formed 1-hexene and 1-octene condensed inside the porous system, thereby facilitating the dissolution and redistribution of the Cr–PNP catalyst. Further studies were conducted at pressures up to 35 bar at which a change in the primary reaction selectivity from 1-hexene to 1-octene could be observed. Such a selectivity switch is in accordance with previously reported mechanistic and kinetic studies.
Introduction
Selective oligomerization of ethylene to 1-hexene and 1-octene has attracted a lot of industrial and academic attention since the discovery of selective ethylene trimerization in 1977 and the discovery of selective ethylene tetramerization in 2004.1,2 The industrial relevance of these technologies is significant given that linear α-olefins (LAOs) are important co-monomers in polyethylene production (C4–C8), and also serve as key feedstocks for the production of plasticizer alcohols (C6–C10), synthetic lubricants (C10–C12), detergent alcohols (C16–C18), and oil field chemicals (C16–C30+).3 Compared to nonselective oligomerization technologies that produce even-numbered carbon chain products in Schulz–Flory and Poisson distributions (mainly the Ziegler process of Chevron Phillips Chemical, the modified Ziegler process developed by Ethyl Corporation and currently practiced by Ineos, and the SHOP process by Shell), selective oligomerization techniques have the significant advantage in providing a more appropriate solution to the variable market growth of the independent product fractions across this broad slate. In the last decade the fastest growing demand was experienced for 1-hexene and 1-octene due to their increasing use in linear low density polyethylene (LLDPE) production.4Scheme 1 shows a simplified mechanism of selective, metallacycle-based ethylene oligomerization to 1-hexene and 1-octene via a homogeneous Cr-catalyst activated by methylaluminoxane (MAO).2,5–7 In this simplified mechanism as originally proposed in 2004, 1-hexene is formed via β-hydride elimination from the seven-membered metallacycle reaction intermediate 4, while 1-octene requires insertion of another ethylene into intermediate 4 and subsequent β-hydride elimination from the ensuing nine-membered metallacycle intermediate 5.2 More recently Britovsek et al. have shown that 1-octene can also be formed from intermediate 3via the coordination of two ethylene molecules.8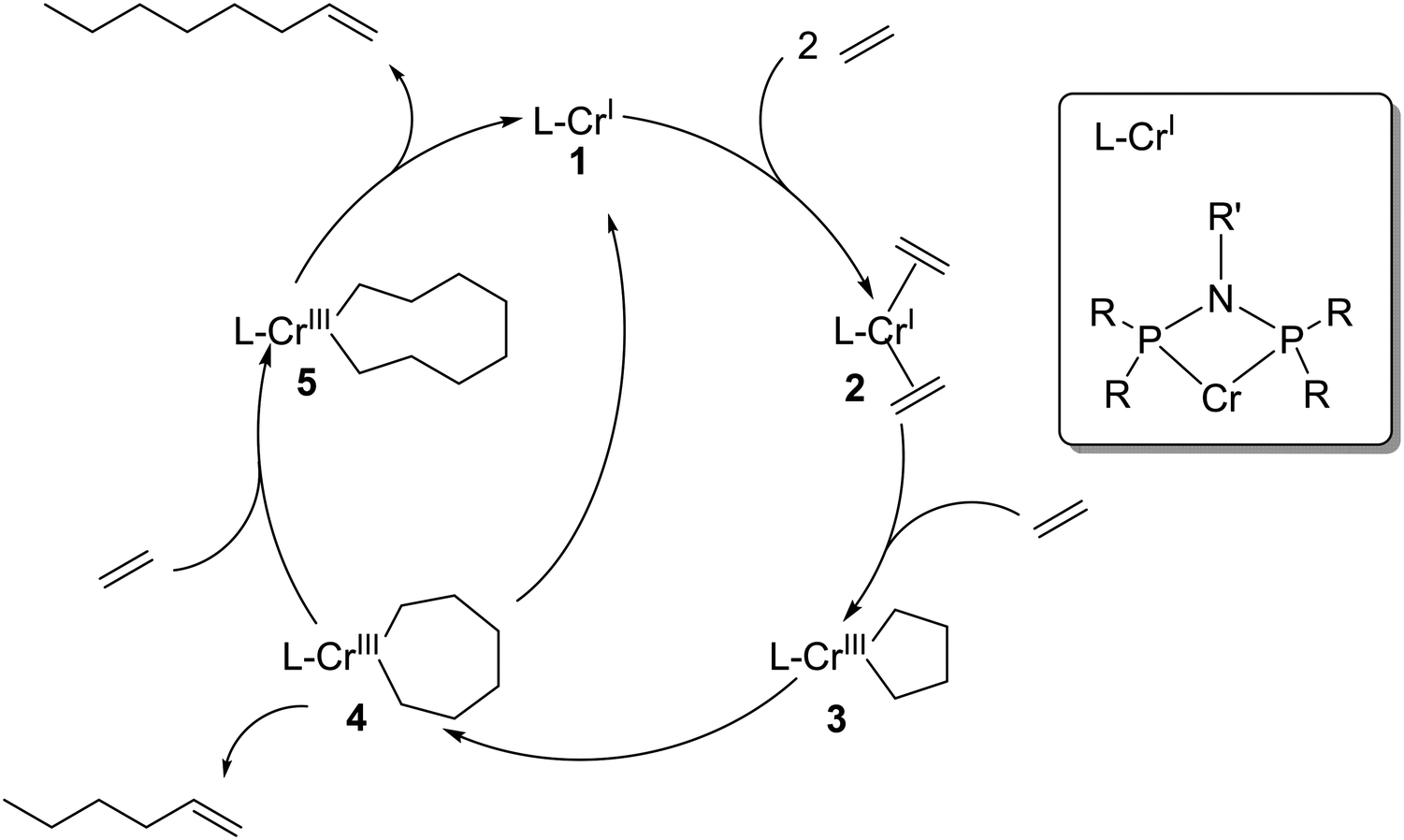 | ||
| Scheme 1 Simplified metallacycle for Cr-catalyzed ethylene tri-and tetramerization. | ||
So far, all technologies for the selective oligomerization of ethylene to α-olefins use homogeneous catalysts in solution.9 Only a few immobilization studies have been reported in literature.10–15 All of these reported concepts include covalent anchoring of the chromium species to a pre-functionalized support surface and subsequent catalyst evaluation in batch liquid-phase reactions. All these techniques require multi-step synthesis and alter the chemical nature of the molecular chromium catalyst, thereby lowering activity and selectivity. Recently, a bis-(2-dodecylsulfanyl-ethyl)-amine modified Cr (Cr-SNS) has been immobilized onto a monolayer of ionic liquid functionalized SBA-15 and again employed in batch liquid-phase ethylene trimerization reactions.16 Ionic liquids have been also applied as biphasic reaction media for N-(diphenylphosphaneyl)-N-isopropyl-1,1-diphenylphosphanamine modified Cr complexes.17 Low activity of approx. 410 gproduct gCr−1 h−1 was obtained and the activity declined even after the first recycling step, which highlights the complexities of employing ionic liquids as reaction solvent with Cr-based ethylene oligomerization catalysts. In addition, selective ethylene oligomerization catalysis in liquid-phase batch reactors require a large solvent inventory and associated purification, storage and recycling infrastructure on industrial scale. Solvents for such technologies are selected, amongst other criteria such as cost and physical properties, based on their ability to dissolve both the active catalyst and the ethylene feed. Most importantly, any applied solvent must also not compete with ethylene for the coordination sites of the highly electrophilic catalyst to prevent rate reduction by solvent coordination. Consequently, non-coordinating, extremely weakly nucleophilic solvents are typically applied in selective ethylene oligomerization.18
In this contribution, we present a novel approach to immobilize MAO-activated, homogeneous Cr–PNP catalyst complexes in the form of supported liquid phase (SLP) catalysts. This SLP catalyst allows the selective synthesis of 1-hexene and 1-octene in a continuous gas-phase reaction over extended run times of more than 200 h time on stream. Apart from thus negating most of the mentioned complexities of employing a process solvent in selective ethylene oligomerization by this technology concept, the observed catalyst life times are also significantly longer than that of the corresponding liquid-phase reactions, where rapid catalyst deactivation within 60 min is observed.19,20 Such notably longer catalyst life times reveal considerable promise for the industrial applicability of this technology concept.
The novelty of this approach in ethylene oligomerization is the fact that the catalyst is composed of a porous support (activated carbon or SiC) with an immobilized high-boiling organic liquid film (perhydro-dibenzyltoluene H18-DBT, Tbp > 350 °C) in which the activated Cr-catalyst is suspended (see Fig. 1). No covalent anchoring and thus modification of the Cr-catalyst or precursors thereof is required. In the past, the SLP concept has also shown promising results in other gas-phase reactions like e.g. hydrogenation and hydroformylation.20,21
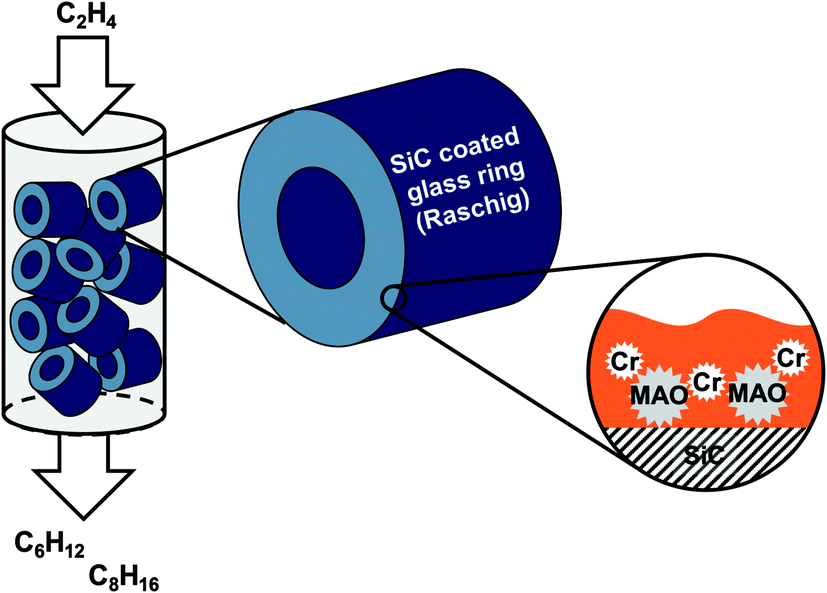 | ||
| Fig. 1 Schematic representation of the supported liquid phase (SLP) concept for Cr catalyzed selective ethylene oligomerization in gas-phase fixed bed reactors. | ||
For our study we applied Cr(acac)3 as catalyst precursor and the two PNP-ligands, N,N-bis-[bis-(o-ethylphenyl)-phosphino]-methylamine L1 (a very effective ligand for ethylene trimerization)22 and N,N-bis(diphenylphosphino)-1,2-dimethylpropylamine L2 (a very effective tetramerization ligand).23 All systems were activated by modified MAO, MMAO-3A, as detailed in the following Experimental part and in Fig. 2.
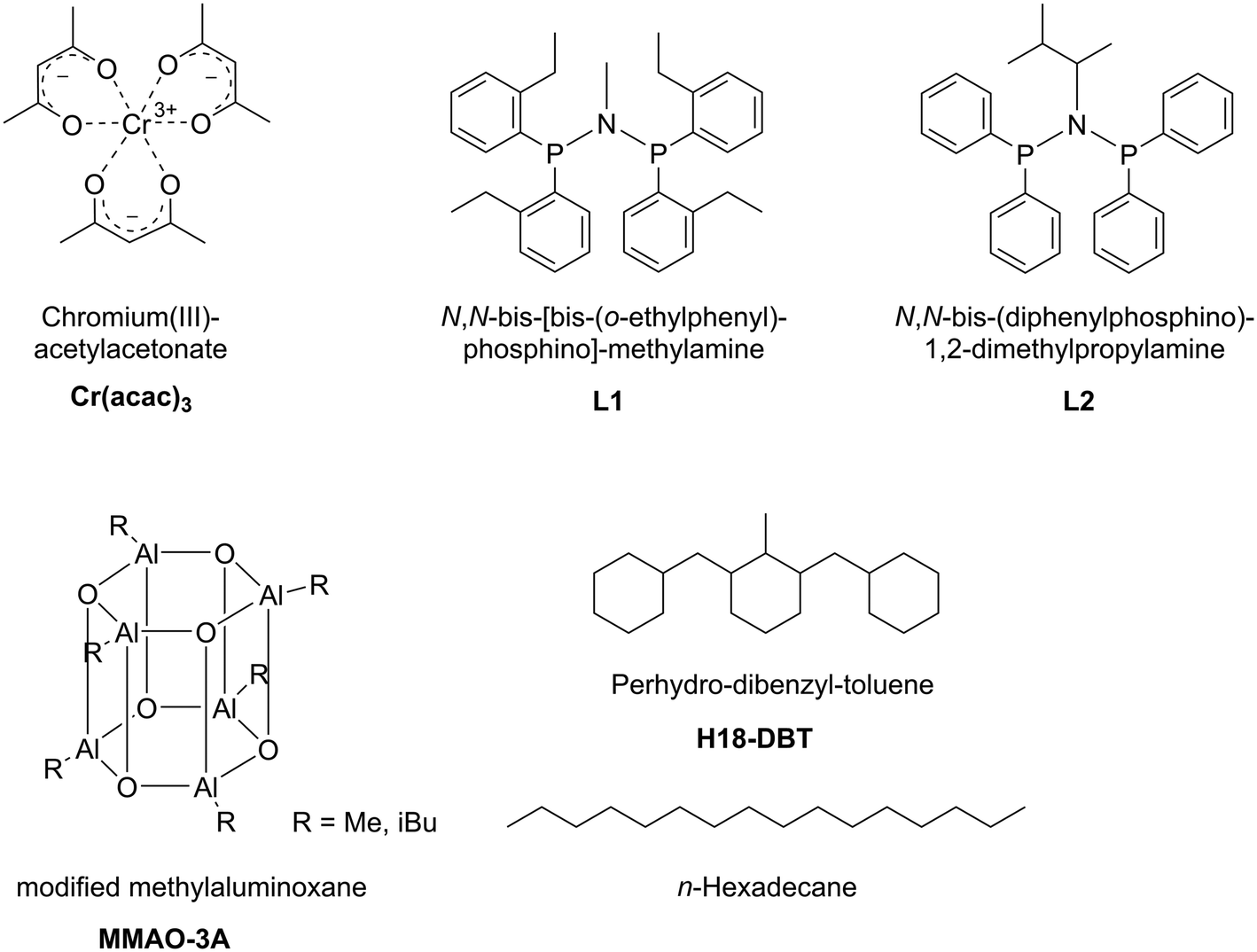 | ||
| Fig. 2 Schematic illustration of the Cr–PNP SLP components. Support materials SiC and activated carbon are not shown. Note that the structure for MMAO-3A represents one possible configuration. | ||
Experimental part
All chemicals were handled under inert conditions using Schlenk techniques or glovebox instrumentation. Polymer based spherical activated carbon beads (PBSAC-200-1, material #100050) of 200 μm particle size and with an average dpore = 2 nm, SBET = 1889 m2 g−1 have been purchased from Blücher GmbH, Erkrath, Germany. The ligands L1 and L2 have been supplied by Sasol Group Technology, a Division of Sasol Limited.23 Perhydro-dibenzyltoluene (H18-DBT) was obtained by hydrogenation of commercial dibenzyltoluene (H0-DBT, tradename Marlotherm®, Sasol Germany GmbH) using a Ru on alumina catalyst (50 bar hydrogen, 150 °C, 3 hours reaction time). The resultant liquid (Tbpca. 360 °C at ambient pressure) was purified by distillation over CaH2 prior to its use. MMAO-3A-stock solution (7 wt% Al in heptane) and n-hexadecane (anhydrous) was purchased from Akzo Nobel and Sigma-Aldrich, respectively, and were used without further purification. Cyclohexane (technical grade) was dried and purified by a continuous absorptive solvent purifications system (Pure Process Technology).Preparation of supported catalyst
A 250 mL Schlenk round bottom flask equipped with a magnetic stirring bar (cross mode for intense agitation) was transferred into the glovebox. The flask was then filled with 100 mL cyclohexane, and the required amounts of support material and liquid supporting phase (n-hexadecane or H18-DBT) as well as the volumes of Cr-precursor, ligand and MMAO-3A stock solutions added in succession. In a typical experiment, the targeted supported liquid loading αliq was 25 vol% of the total pore volume of the support. The Cr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand:MAO ratio was kept constant at 1:1:2000. Next, the tightly sealed flask was removed from the glovebox and connected to a cryo-distillation apparatus. The lower boiling solvents of the stock solutions was carefully removed by lowering the pressure under vigorous stirring. This procedure was continued until the slurry changed into a sticky, resin-like mass. Afterwards the flask was placed under an Argon atmosphere, tightly sealed, transferred back into the glovebox and stored for approx. four days prior to use (the so-called “ageing” step). The supported catalyst was then transferred into a tubular reactor whilst still inside the glovebox. Finally, after the catalyst transfer, the reactor was closed by re-assembling the ½′′ VCR fitting and sealed by closing the ball valves V-5A and V-5B (see Fig. 3 for details).
:ligand:MAO ratio was kept constant at 1:1:2000. Next, the tightly sealed flask was removed from the glovebox and connected to a cryo-distillation apparatus. The lower boiling solvents of the stock solutions was carefully removed by lowering the pressure under vigorous stirring. This procedure was continued until the slurry changed into a sticky, resin-like mass. Afterwards the flask was placed under an Argon atmosphere, tightly sealed, transferred back into the glovebox and stored for approx. four days prior to use (the so-called “ageing” step). The supported catalyst was then transferred into a tubular reactor whilst still inside the glovebox. Finally, after the catalyst transfer, the reactor was closed by re-assembling the ½′′ VCR fitting and sealed by closing the ball valves V-5A and V-5B (see Fig. 3 for details).
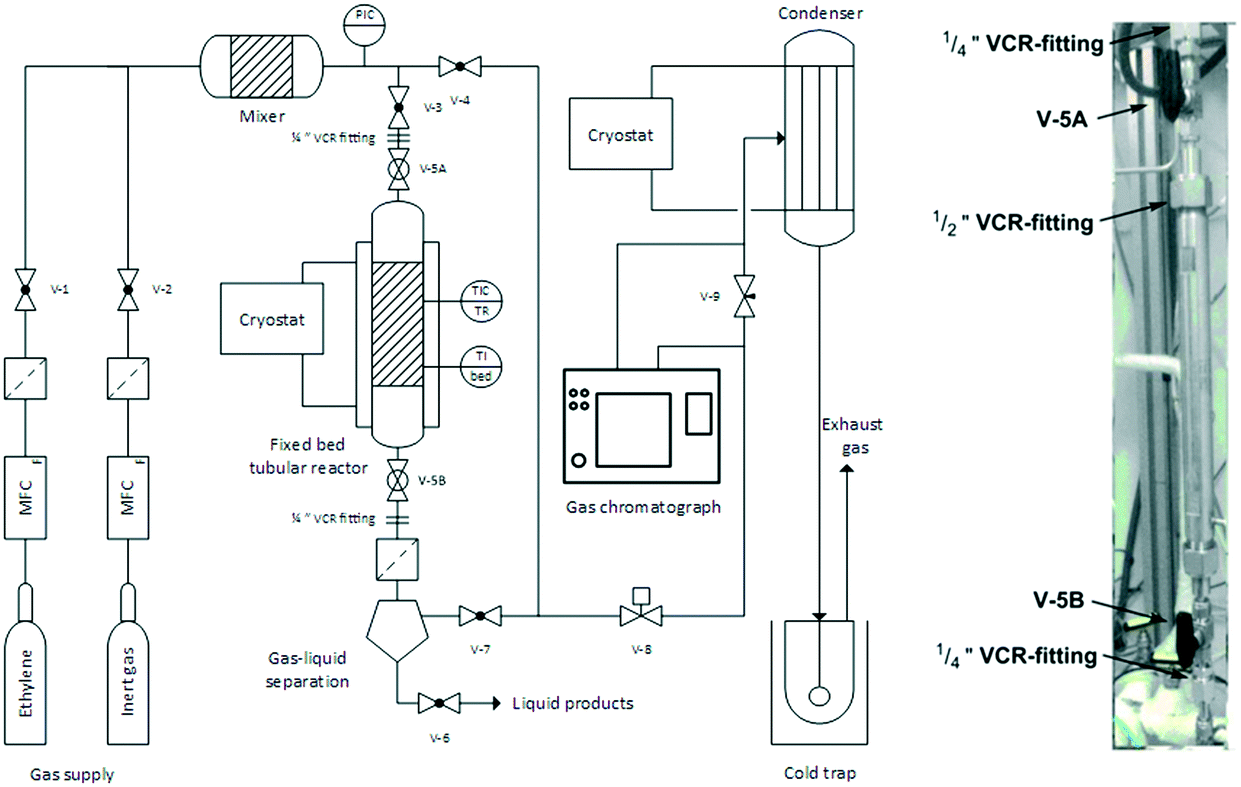 | ||
| Fig. 3 Flowsheet of the reactor for continuous gas-phase oligomerization (left) and detailed photograph of the removable tubular reactor (right). | ||
Gas-phase reactor setup
A reactor set-up previously described by Scholz et al. was used for the selective gas-phase oligomerization.24 A simplified flow sheet of the reactor configuration (i.e. the “rig”) is shown in Fig. 3. The whole rig comprised V4A stainless steel (1.4571) components and experiments were performed in an in-house designed fixed-bed tubular reactor with the following dimensions: length = 285 mm, inner diameter = 10 mm, wall thickness = 6.5 mm.The catalyst bed was held in place by a metal frit at the bottom. The reactor temperature was controlled by a cryostat for heat removal from the exothermic reaction. Additionally, the tubular reactor could be heated by a heating jacked and the catalyst bed temperature was continuously logged via software. The gaseous feed components (reactant and inertization gas) were pre-heated by a heated feed line. Gas supply was controlled by electronic mass flow meter and controllers (= MFC) (Bronkhorst GmbH and Brooks Instruments) and different feed compositions could be adjusted by blending ethylene 3.5 with inert gas (helium 4.6 or argon 6.0). In order to exclude any exposure of the extremely oxygen- and moisture-sensitive catalyst to air during the reactor filling procedure, the whole tubular reactor could be removed from the rig using the ¼′′ VCR fittings. A gas–liquid separator was installed below the reactor to retain condensed products and to protect the downstream part of the rig from contamination in case of catalyst leaching. The reaction pressure ptotal was monitored by an electronic pressure indicator and controller (= PIC) (Ashcroft Technologies) and could be regulated by a pneumatic needle valve V-8 (SAMSON AG). Downstream of V-8, the process stream was at ambient pressure and its composition determined by an online gas chromatograph (Agilent 7820A with fused silica capillary column CP7531, length l = 50 m, film thickness = 0.5 μm, inner diameter di = 0.21 mm). For collection of the oligomeric products the process stream was passed through a cooled condenser (Tcryostat = −30 °C) and finally through a cold trap (i-PrOH/CO2, Tcoldtrap = −78 °C).
Cr–PNP catalyzed gas-phase oligomerization
Prior to an oligomerization experiment, the feed line and downstream line had to be heated to the desired set points and the whole rig was flushed with inert gas for approximately 30 minutes. When the reactor was removed (i.e. for catalyst transfer), the ¼′′ VCR fittings were sealed by isolation plugs. In order to fit the reactor, V-3 and V-4 were first opened and the ½′′ VCR isolation plugs then removed. Next, the reactor was connected to the rig under a constant flow of inert gas until the connections were tight. V5-A and V5-B were then opened simultaneously and the bypass valve V-4 closed. Following this, the desired feed composition was obtained by adjusting the MFCs and the reaction pressure and temperature were adjusted by the PIC and the TIC (temperature indicator and controller, WIKA), respectively. Samples of the process stream downstream of the reactor were taken automatically at regular intervals (typically 52 minutes). When the cold trap was employed, the frigorific mixture (solid CO2/i-PrOH) had to be refilled regularly to prevent the loss of volatile products.Termination of a run was achieved by setting the ethylene flow to 0 mLN min−1, opening the bypass valve V-4 and releasing the excess reaction pressure through V-8 by setting ptotal to 1 bar. If the cold trap was used, the collected liquids were removed immediately to prevent loss caused by the enhanced gas flow during depressurization. Afterwards the whole rig was purged with inert gas for 30 min and finally all the pressure was released by opening V-6. The reactor could then be removed from the rig and opened to recover the used catalyst. The reactor was cleaned mechanically by a steel brush and subsequently chemically with acetone. Accumulated high boiling oligomers, waxes and polyethylene (PE) were removed from the metal frit by pyrolysis (Bunsen burner) before cleaning in an ultrasonic bath. Additionally, the condenser and V-6 were thoroughly flushed with acetone. All removed and cleaned parts were dried in the oven before reuse.
Calculation of Cr–PNP catalyst performance
Evaluation of the Cr–PNP in the gas-phase oligomerization performance was based on both the quantities of detectable compounds ṁproducts,GP in the online GC and the collected liquids mliq-products that were analyzed by offline GC. The conversion of ethylene XC2,GP was based solely on the composition of the gaseous effluent stream (online GC) and was calculated according to eqn (1).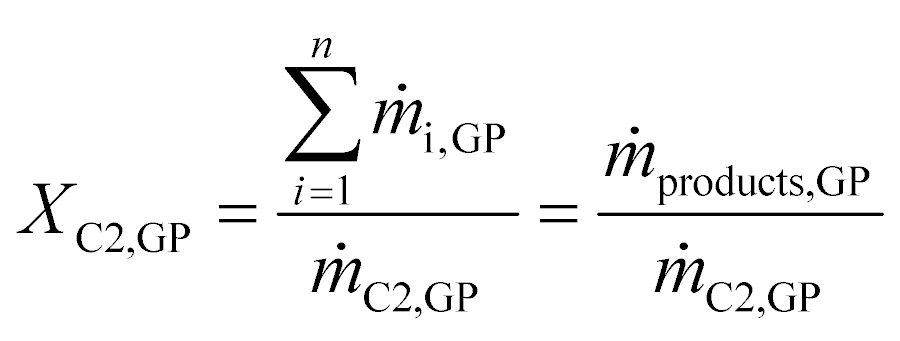 | (1) |
The selectivity of any individual compound Si,GP in the gas-phase was calculated according to eqn (2).
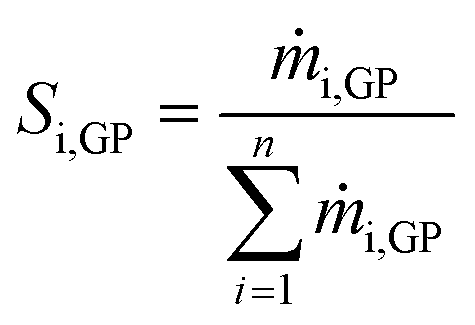 | (2) |
The activity of the Cr–PNP catalyst (in gproduct gCr−1 h−1) in the gas-phase reaction is related to the mass of Cr in the system according to eqn (3).
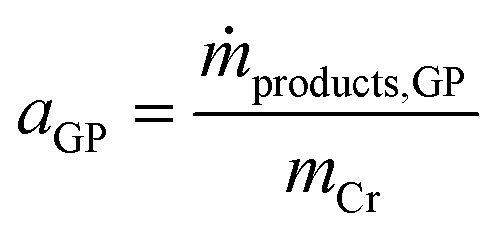 | (3) |
The productivity of the Cr–PNP catalysts (in gproduct gCr−1) is calculated by multiplication of the activity with the time on stream according to eqn (4).
| PGP = aGP·t | (4) |
Results and discussion
In order to probe if an immobilized MAO activated Cr-based oligomerization catalyst would exhibit activity without any further solvent, the prepared Cr-SLP catalyst was tested initially in a standard semi-batch reactor (i.e. autoclave with ethylene fed on demand, see ESI† for details). The sticky Cr-L1-SLP immobilized on microporous activated carbon (PBSAC) converted ethylene at 45 °C and 45 bar into predominantly 1-hexene (90% selectivity) with a total productivity of approx. 2000 gproduct gCr−1 over 6 h run time. This value is three orders of magnitude lower than the productivities obtained with the corresponding homogeneous Cr catalyst under identical conditions.25 The lower activity can be attributed to mass transport limitations, most probably extrinsic in nature i.e. slow diffusion of ethylene through the liquid supporting phase. Nevertheless, since this test confirmed the activity of the supported Cr catalyst, a first gas-phase experiment was conducted at 43 °C and an ethylene partial pressure ranging between 1 and 14 bar as shown in Fig. 4. No ethylene conversion was detectable at 1 bar during the first 8 hours of the experiment. Raising the partial pressure to 3 bar resulted in traces of products being formed with a catalyst activity of 25 gproduct gCr−1 h−1. At this very low activity level, the reaction was maintained for 10 hours. A further increase of partial pressure to 5 bar resulted in an increase of catalyst activity to 100 gproduct gCr−1 h−1. However, the catalyst performance at this production level was not stable and the catalyst activity gradually declined during the following hours to reach the initial level of 25 gproduct gCr−1 h−1 after a total of 96 hours time-on-stream. The catalyst activity could be partially regained by increasing the ethylene partial pressure in a stepwise manner to 7.5 bar, 8.75 bar and 10 bar, respectively, but in each instance this was again followed by periods of a gradual decrease in activity.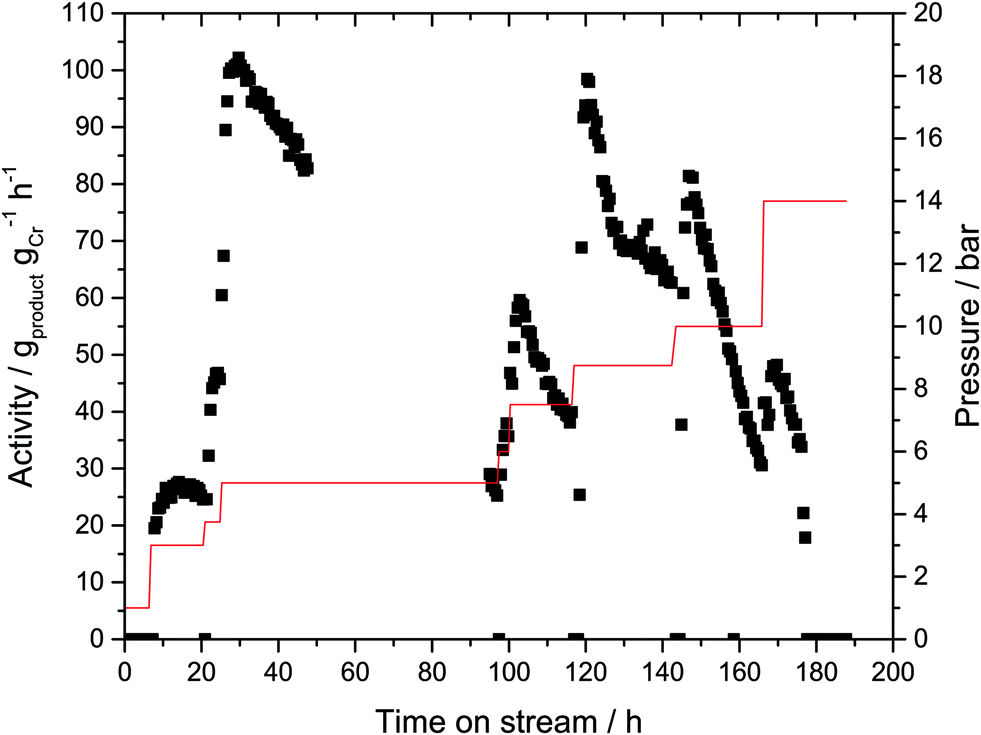 | ||
| Fig. 4 Gas-phase ethylene trimerization using a Cr-L1-SLP catalyst with H18-DBT dispersed onto microporous activated carbon (AC). T = 43 °C, ethylene partial pressure as indicated by red line, total volume flow = 140 mLN min−1, Cr-loading = 0.013 wt%, 2000 eq. MAO, αliq = 25 vol%, 1 g of AC, solvent = H18-DBT. No data acquisition between 40 and 100 h due to GC shutdown. 0–6.5 h: volume flow ethylene FC2 = 14 mLN min−1, volume flow helium FHe = 126 mLN min−1, ptotal = 10 bar. 6.5–25 h: FC2 = 21 mLN min−1, FHe = 119 mLN min−1, ptotal = 10 bar. 25–100 h: FC2 = 28 mLN min−1, FHe = 112 mLN min−1, ptotal = 30 bar. 100–116 h: FC2 = 35 mLN min−1, FHe = 105 mLN min−1, ptotal = 30 bar. 116–143 h: FC2 = 35 mLN min−1, FHe = 105 mLN min−1, ptotal = 35 bar. 143–166 h: FC2 = 35 mLN min−1, FHe = 105 mLN min−1, ptotal = 40 bar. 166–187 h: FC2 = 49 mLN min−1, FHe = 91 mLN min−1, ptotal = 40 bar. | ||
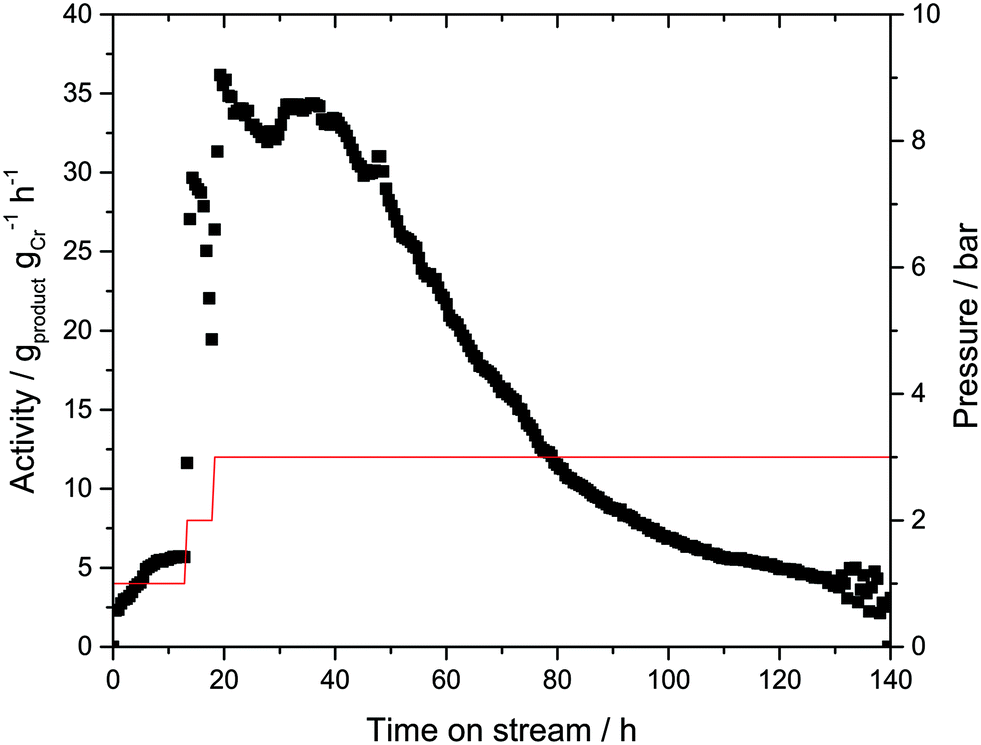 | ||
| Fig. 5 Gas-phase ethylene trimerization using a Cr-L1-SLP catalyst with H18-DBT dispersed onto microporous activated carbon (AC). T = 43 °C, ethylene partial pressure as indicated by red line, total volume flow = 70 mLN min−1, Cr-loading = 0.013 wt%, 2000 eq. MAO, αliq = 50 vol%, 1 g of AC, solvent = H18-DBT. 0–13 h: volume flow ethylene FC2 = 7 mLN min−1, volume flow argon FAr = 63 mLN min−1, ptotal = 10 bar. 13–18 h: FC2 = 14 mLN min−1, FAr = 56 mLN min−1, ptotal = 10 bar. 18–140 h: FC2 = 21 mLN min−1, FAr = 49 mLN min−1, ptotal = 10 bar. | ||
The observed catalytic behavior indicates that the active Cr–PNP-based catalytic species is still present in the SLP material, but is most probably becoming increasingly inaccessible due to a growing/thickening polyethylene coating around the activated carbon particles (see Fig. S6 in the ESI†). It should be noted that the activated carbon support used in this instance was a highly microporous material (SBET = 1889 m2 g−1, dP = 1.5 nm).26 It is therefore also unlikely that the macromolecular MAO would have been ample to penetrate the entire pore system. Thus, a further contributing factor to the low activity observed in the gas-phase experiments may be the comparatively small amount of active Cr–PNP species in the SLP phase on the external surface of the carbon support. The reaction selectivity toward 1-hexene was high at >99.5% throughout the entire experiment. In retrospect this was to be expected to some extent given that gas-phase catalysis would in principle limit the presence of 1-hexene near the active catalyst species (a prerequisite for the formation of C10 secondary reaction product).6
The gas-phase experiment was reproduced using a second portion of the same batch of the Cr-SLP catalyst that had been stored for 8 days inside the glovebox. It was noticed that during the storage, the general appearance of the catalyst changed from a sticky/gluey to a hard/solid material. With this “aged” catalyst, no product could be detected at 1, 2, 4 or even 6 bar ethylene pressure. After 20 h time-on-stream the partial pressure was increased further to 10 bar and this led to an increase in activity to 100 gproduct gCr−1 h−1. As in the first run, this activity level was not stable and declined constantly. After 140 hours time-on-stream, the reaction was stopped, with the spent catalyst again showing significant PE-coating. Our interpretation of the differences in catalyst activity of the freshly prepared and the “aged” catalyst at low ethylene pressures is as follows: during the aging period in the glove box, the small quantity of residual solvent evaporates which results in the exposure of MMAO-compounds on the outer surface which in turns forms a solid coating that has a low penetrability for ethylene gas at low pressures (see ESI† on the color change of the SLP from grey to white). Therefore, no product formation is seen at low ethylene pressures. With higher ethylene pressure, ethylene solubility and diffusion rates increase and, this together with the first formed 1-hexene molecules, “swells” the catalyst particulates and finally re-established the organic active coating. At this point the activity increases drastically. At this stage, 1-hexene is released into the gas phase and detected by GC. The fact that the catalyst activity at this point is similar to that of the freshly prepared catalyst under the same operating conditions shows that catalyst decomposition during the aging period is unlikely.
In order to verify this hypothesis, a new batch of Cr-L1-SLP catalyst was synthesized. Twice the amount of liquid phase (αliq = 50 vol%) was used during catalyst preparation in this instance in an attempt to prevent “solidification” of the outer surface of the supported liquid layer. In addition, during the continuous catalytic experiment, the ethylene flow was lowered from 140 to 70 mLN min−1 to limit any “stripping” of the immobilizing liquid from the SLP catalyst (Fig. 5).
Finally, argon 6.0 (99.99990 vol%) was used as inertization gas instead of helium 4.5 (99.995 vol%) to minimize the addition of possible catalyst poisons (most notable oxygen and moisture). In contrast to the previous experiments, traces of 1-hexene were now detectable even at a low ethylene pressure of 1 bar. Increasing the pressure to 3 bar led to an increase in catalyst activity to 35 gproduct gCr−1 h−1, a marginally higher value to that obtained in the previous experiments under comparable conditions. This improved activity can be attributed to the use of argon 6.0.
The activity level of 35 gproduct gCr−1 h−1 was maintained for almost 30 h, after which a gradual decline in activity was observed. Similar to the previous experiment, the 1-hexene selectivity remained unchanged at >99.5% over the complete run-time. After 140 h, the catalyst was removed from the reactor, again showing signs of PE coating on the outer surface. The extent of PE formation was determined by a post-run TGA analysis on the spent Cr-L1-SLP catalysts (see ESI† for details). In this analysis, a mass loss around 7 wt% was detected in the temperature range between 25 and 250 °C, which can be attributed to the loss of light hydrocarbons (mainly 1-hexene) from the SLP material. A further increase in temperature to 650 °C resulted in an additional mass loss of 65 wt%. This high value clearly indicates that the Cr-SLP catalyst gained mass during the gas-phase experiments by accumulation of PE polymer (and some oligomers). One reason for the high PE formation levels could be the poor heat management inside the catalyst bed. It is known that temperatures above 70 °C would turn the applied Cr-complex into a very effective wax/PE forming catalyst.27 Thus, hot spots that may form in the catalyst bed could be a reason for the observed PE formation.
In a next experiment, the SLP catalyst bed was mixed with inert graphite material with the intention to lower the average ethylene conversion rate per reactor volume unit, and hence improve the heat removal per reactor volume unit. Similar to the previous runs, no activity was observed at 1 bar ethylene pressure even after a runtime of 20 hours. The first product could be detected after 24 hours, when the pressure had been raised to 4 bar (see ESI† for details). As this stage, the ethylene pressure was increased significantly to 15 bar to determine whether the targeted improved heat removal in the diluted bed would also allow more drastic reaction conditions without excessive PE formation in the catalyst bed. As was hoped for, at 15 bar ethylene pressure, a drastic increase of activity to a level of 1400 gproduct gCr−1 h−1 was obtained which corresponds to an ethylene conversion XC2,GP = 3.8% in our set-up. This value could be kept constant over a period of 5 hours. Similar to the previous observations, the catalyst activity subsequently declined slowly over the next 20 hours and the spent Cr-L1-SLP catalyst recovered from the reactor after 100 hour run time regrettably again showed clear signs of PE coating. It should be noted that no catalyst leaching was detected during the experiment; the full amount of chromium was still present on the catalyst after 100 h time-on-stream as confirmed by ICP-OES. In this run, a cumulative catalyst productivity of 32.000 gproduct gCr−1 was achieved with the 1-hexene selectivity exceeding >99.5% over the complete run-time.
From these gas-phase experiments using porous carbon (AC) as support we concluded that coating of the SLP particle with PE is the major reason for catalyst deactivation. Therefore, we prepared the next set of immobilized catalysts on meso-porous SiC powder that was fixed to the macro-porous structure of glass Raschig rings (ring opening: 6 mm). Again, H18-DBT was used as the immobilizing organic solvent. In addition, in the following experiments the tetramerization ligand N,N-bis(diphenylphosphino)-1,2-dimethylpropylamine L2 was applied to demonstrate that our novel selective continuous gas-phase ethylene oligomerization methodology is also able to produce 1-octene selectively.
The run was started with an ethylene feed flow of FC2 = 65 mLN min−1 at 5 bar. Immediately, 1-alkene products were detected in the effluent gas stream (molar ratio 1-C6:1-C8 = 3:1). The gas-phase activity was 70 gproduct gCr−1 h−1 under these conditions. These values remained constant for 2 hours, after which the pressure was increased to 15 bar in an attempt to push the overall productivity upwards. Following an equilibration phase, the catalyst activity stabilized at 250 gproduct gCr−1 h−1 under these conditions (Fig. 6).
Under these conditions, the product selectivity changed in favor of 1-C8 production with selectivities of SC6,GP of 55% and SC8,GP = 45% being obtained. This was expected for higher ethylene pressure because formation of 1-octene was found to be approximately second order with respect to ethylene, while 1-hexene formation was attributed to be nearly first order dependency in respect to ethylene concentration during previous kinetic studies in gas–liquid phase catalysis.8,20,25,28 The activity and selectivity levels remained constant for 20 hours time-on-stream and no deactivation was observed under these conditions.
To determine whether the reaction selectivity can be further pushed towards higher 1-octene, the ethylene pressure was further increased to 30 bar after 22 hours time-on-stream. This led to another large increase of activity during the equilibration period with a maximum activity of 3700 gproduct gCr−1 h−1. However, within the next 25 hours the activity strongly decreased before stabilizing at a level of approximately 1100 gproduct gCr−1 h−1. Under these conditions, 1-octene formation was indeed predominant, with a SC6,GP = 35% and a SC8,GP = 58%, respectively (plus minor amounts of C10+ oligomers). For a period of 20 hours the activity and selectivity did not change, indicative of an improved heat management as anticipated by the use of SiC. After 75 h a slow decrease in catalyst activity was observed, presumable due to continuous/further deposition of by-product PE on the catalyst surface. After 150 h the ethylene pressure was set to 35 bar which resulted in a minor enhancement of activity from 570 gproduct gCr−1 h−1 to 630 gproduct gCr−1 h−1, but this production level was maintained for a further 70 h without significant deactivation. Remarkably, the SC8,GP increased from 59% to 67% over this time period. After the run, the cold trap was emptied and 8.6 g of liquid products was isolated from the cold trap. The mixture was analyzed by offline GC, using n-hexane as internal standard. The two main products were 1-hexene (32.3%) and 1-octene (57%), respectively. TGA analysis of the spent Cr-L2-SLP catalyst revealed that the masses of higher oligomers and PE were moligomers = 0.817 g and mPE = 2.83 g, respectively. The full product distributions obtained using the L2-modified Cr-system running both in semi-batch mode and in continuous fixed bed mode using SLP are summarized in Table 1.
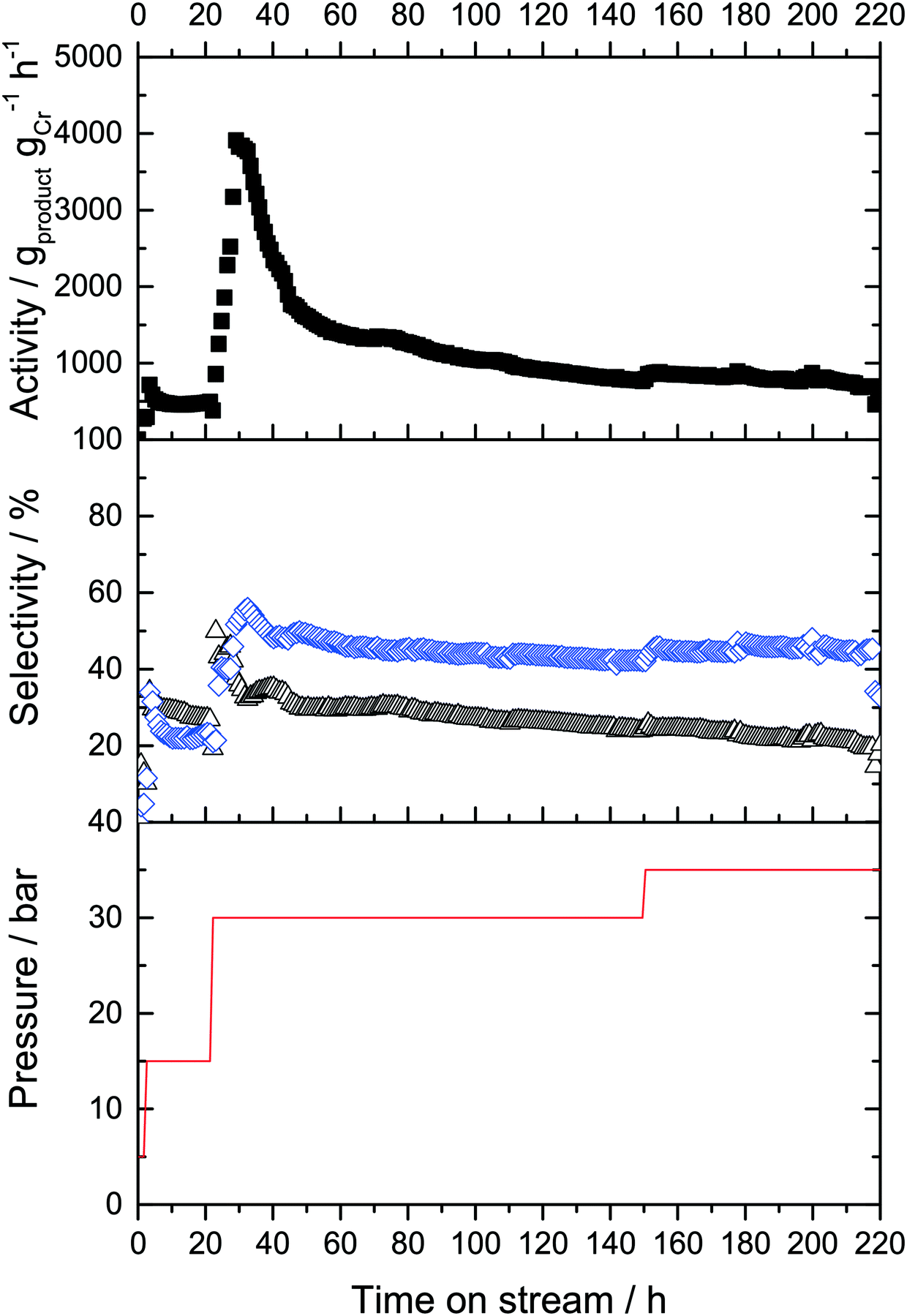 | ||
| Fig. 6 Gas-phase ethylene oligomerization using a Cr-L2-SLP catalyst dissolved in H18-DBT. The catalytic liquid was coated onto SiC/Raschig rings. T = 43 °C, ethylene partial pressure as indicated by red line, total volume flow Ftotal = 65 mLN min−1, Cr-loading = 0.013 wt%, 2000 eq. MAO, αliq not assigned, 1 g of SiC, solvent = H18-DBT. Selectivity toward 1-hexene (black triangles), selectivity toward 1-octene (blue diamonds). | ||
| System | S 1-C6 /% | S 1-C8 /% | S C6-cyclics /% | S >C10 /% | S solids /% |
|---|---|---|---|---|---|
|
a 1-Hexene selectivity.
b 1-Octene selectivity.
c Cyclic hexenes selectivity.
d Higher olefin selectivity.
e Polymer selectivity.
f Conditions for semi-batch mode: 0.8 μmol Cr(acac)3, Cr:L:Al = 1:1:2000, pC2 = 45 bar, T = 50 °C.
g Calculated with mass of product mixture from condenser.
h Calculated with mass of product mixture from the gas-phase by online GC.
|
|||||
| Semi-batchf | 13.5 | 70.9 | 3.5 | 7.4 | 4.0 |
| SLP condensedg | 22.2 | 39.3 | 1.4 | 5.1 | 28.0 |
| SLP gas phaseh | 25.2 | 44.6 | 1.6 | 5.8 | 18.3 |
It is notable that the 1-octene:1-hexene ratio of the product formed in the continuous gas-phase experiment is significantly lower than that in the semi-batch experiment. The reason for the lower C8 yield is the lower average ethylene pressure that was applied throughout the gas-phase run (there is also a pressure limit of 35 bar due to technical/safety limitations). Both the condensed phase and gas-phase SLP data sets indicate significantly larger PE yields compared to the corresponding gas–liquid-phase catalysis in a batch reactor, or even the semi-batch catalysis case in Table 1. This can be explained in terms of the comparative poor heat removal within the fixed-bed during gas-phase catalysis when compared to the much more efficient heat removal during gas–liquid-phase catalysis. Still, as can be seen from Table 2, significant improvements have been obtained already by selecting the correct support material (SiC instead of AC), diluting the catalyst material as well as controlling the ethylene low through the reactor. Arguably further such improvements could be achieved by additional optimization studies.
| Run | Catalyst | Support | p total/bar | Activity/gproduct gCr h−1 | Stability/h | PE formation |
|---|---|---|---|---|---|---|
| H18-DBT was used as solvent in all experiments. Reactor temperature was set to 43 °C in all experiments. a He (99.995 vol%) carrier gas, 140 mLN min−1 ethylene, αliq = 25 vol% H18-DBT loading. b Ar (99.99990 vol%) carrier gas, 70 mLN min−1 ethylene, 50 vol% H18-DBT loading. | ||||||
| 1a | Cr-L1 | AC | 3 | 28 | 2 | High |
| 1 | 10 | 85 | <1 | |||
| 2b | Cr-L1 | AC | 3 | 33 | 25 | Moderate |
| 3 | Cr-L1 | AC/SiC | 3 | 300 | <1 h | Moderate |
| 3 | 15 | 1400 | 5 h | |||
| 4 | Cr-L2 | SiC/Raschig | 15 | 500 | 20 h | Moderate |
| 4 | 30 | 1000 | 100 h | |||
The selection of support, supporting liquid phase, catalyst, activation methodology and process parameters are far from being optimized and the right combination of these parameters holds significant promise for further optimization.
Conclusion
In this work the immobilization of Cr–PNP complexes in a supported liquid phase (SLP) mode is demonstrated for the selective oligomerization of ethylene. For this purpose, established trimerization and tetramerization catalysts were dissolved in the high-boiling hydrocarbon H18-DBT (Tbp > 350 °C) and this catalytic solution was dispersed as thin film onto activated carbon and porous SiC supports. The resulting solid Cr–PNP-SLP catalysts were placed in a fixed-bed reactor and exposed to various partial pressures of ethylene. At pressures of 30 bar activities of up to 3700 gproduct gCr h−1 could be realized. This was accompanied by the formation of a polyethylene layer which led to continuous catalyst deactivation. Using a support with improved pore structure and better heat transfer properties (porous SiC on glass Raschig rings) a stable oligomerization activity of 1000 gproduct gCr h−1 could be maintained over 150 h time-on-stream. The variation in ethylene pressure resulted in the expected change in selectivity, with more 1-octene being formed at higher pressures due to its second order dependency on ethylene partial pressure. Thus, the similar kinetic trends were found for the continuous gas-phase experiments as for the liquid-phase experiment. This indicates that the active Cr-system in solution and immobilized on the novel SLP system here are similar in their chemical nature and the observed significant difference in productivity is due to mass and heat transfer limitations in the immobilized system. Therefore, we anticipate that by better choice of the materials (support, immobilizing liquid, Cr-complex), of the reactor (dimensions, flow regime) and of the process conditions (pressure, flow rate, temperature), these limitations could be leveraged and significantly more productive continuous gas-phase systems could be obtained. These experiments will be in the focus of future studies in this ongoing process development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from Sasol Technology (Pty) Limited, South Africa, for this project and thank for the allowance to publish these results. Stephen Evans from Sasol Technology (Pty) Limited is gratefully acknowledged for fruitful discussions.References
- R. M. Manyik, W. E. Walker and T. P. Wilson, J. Catal., 1977, 47, 197–209 CrossRef CAS
.
- A. Bollmann, K. Blann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. S. McGuinness, D. H. Morgan, A. Neveling, S. Otto, M. Overett, A. M. Z. Slawin, P. Wasserscheid and S. Kuhlmann, J. Am. Chem. Soc., 2004, 126, 14712–14713 CrossRef CAS PubMed
- D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS PubMed
-
A. Jess and P. Wasserscheid, Chemical Technology: An integral textbook, Wiley-VCH, Weinheim, 2013, pp. 749 and 803 Search PubMed
- W. J. van Rendsburg, J.-A. van den Berg and P. J. Steynberg, Organometallics, 2007, 26, 1000 CrossRef
- M. J. Overett, K. Blann, A. Bollmann, J. T. Dixon, D. Haasbroek, E. Killian, H. Maumela, D. S. McGuiness and D. H. Morgan, J. Am. Chem. Soc., 2005, 127, 10723 CrossRef CAS PubMed
- A. Brückner, J. K. Jabor, A. E. C. McConnell and P. B. Webb, Organometallics, 2008, 27, 3849 CrossRef
- G. J. P. Britovsek, D. S. McGuiness, T. S. Wierenga and C. T. Young, ACS Catal., 2015, 5, 4152 CrossRef CAS
- K. A. Alferov, G. P. Belov and Y. Meng, Appl. Catal., A, 2017, 542 Search PubMed
- T. Monoi and Y. Sasaki, J. Mol. Catal. A: Chem., 2002, 187, 135–141 CrossRef CAS
-
(a) C. N. Nenu and B. M. Weckhuysen, Chem. Commun., 2005, 1865–1867 RSC
- N. Peulecke, B. H. Müller, S. Peitz, B. R. Aluri, U. Rosenthal, A. Wöhl, W. Müller, M. H. Al-Hazmi and F. M. Mosa, ChemCatChem, 2010, 2, 1079–1081 CrossRef CAS
- S. Liu, Y. Zhang, Y. Han, G. Feng, F. Gao, H. Wang and P. Qiu, Organometallics, 2017, 36, 632 CrossRef CAS
- M. L. Shozi and H. B. Friedrich, S. Afr. J. Chem., 2012, 65, 214 CAS
- M. J. Lamb, D. C. Apperly, M. J. Watson and P. W. Dyer, Top. Catal., 2018, 61, 213–224 CrossRef CAS
- M. Fallahi, E. Ahmadi, A. Ramazani and Z. Mohamadnia, J. Organomet. Chem., 2017, 848, 149–158 CrossRef CAS
- X. Gao, M. Wang, Y. Chen, Y. Sun, B. Dong and T. Jiang, React. Kinet., Mech. Catal., 2014, 113, 159–167 CrossRef CAS
- K. Blann, A. Bollmann, H. de Bod, J. T. Dixon, E. Killian, P. Nongodlwana, M. C. Maumela, H. Maumela, A. E. McConnell, D. H. Morgan, M. J. Overett, M. Pretorius, S. Kuhlmann and P. Wasserscheid, J. Catal., 2007, 249, 244–249 CrossRef CAS
- R. Walsh, D. H. Morgan, A. Bollmann and J. T. Dixon, Appl. Catal., A, 2006, 306, 184 CrossRef CAS
-
(a)
S. Kuhlmann, Selective Tri- and Tetramerization of Ethylene: from Ligand Design to Mini-Plant Operation, Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), 2006 Search PubMed
-
(a) J. M. Herman, P. J. van den Berg and J. J. F. Scholten, Chem. Eng. J., 1986, 32, 101 CrossRef CAS
-
(a) K. Blann, A. Bollmann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. H. Morgan, A. Neveling, S. Otto and M. J. Overett, Chem. Commun., 2005, 620–621 RSC
- M. C. Maumela, K. Blann, H. de Bod, J. T. Dixon, W. F. Gabrielli and D. B. G. Williams, Synthesis, 2007, 24, 3863–3867 CrossRef
- J. Scholz, V. Hager, X. Wang, F. T. U. Kohler, M. Sternberg, M. Haumann, N. Szesni, K. Meyer and P. Wasserscheid, ChemCatChem, 2014, 6, 162–169 CrossRef CAS
-
C. Paetz, Towards a technical oligomerisation process: Hin zu einem technischen Oligomerisierungs-Prozess, Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), 2009 Search PubMed
- K. Schute, Y. Louven, C. Detoni and M. Rose, Chem. Ing. Tech., 2016, 88, 355–362 CrossRef CAS
-
K. Blann, D. de Wet-Roos, D. J. Joubert, E. Lillian, J. T. Dixon, N. J. Phelembe and A. Du Toit, US 2006/0020091 A1, 2006
- S. Kuhlmann, J. T. Dixon, M. Haumann, D. H. Morgan, J. Ofili, O. Spuhl, N. Taccardi and P. Wasserscheid, Adv. Synth. Catal., 2006, 348, 1200–1206 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8re00179k |
| This journal is © The Royal Society of Chemistry 2019 |