
The critical role played by water in controlling Pd catalyst speciation in arylcyanation reactions†
Joshua T. W.
Bray
 a,
Mark J.
Ford
b,
Peter B.
Karadakov
a,
Adrian C.
Whitwood
a and
Ian J. S.
Fairlamb
*a
a,
Mark J.
Ford
b,
Peter B.
Karadakov
a,
Adrian C.
Whitwood
a and
Ian J. S.
Fairlamb
*a
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: ian.fairlamb@york.ac.uk
bBayer Aktiengesellschaft, Crop Science Division, Industriepark Höchst G836, 65926 Frankfurt am Main, Germany. E-mail: mark.ford@bayer.com
First published on 20th November 2018
Abstract
Pd-Catalyzed arylcyanation reactions are widely applied in academic and industrial problems. We demonstrate for the first time that water acts as a powerful trigger for heterogeneous catalysis in arylcyanation reactions. A selection of trans-Pd(OAc)2(HNR2)2 precatalysts have been synthesized, characterized and kinetically scrutinized as precatalysts for the Pd-catalyzed arylcyanation of aryl bromides. Kinetic and mechanistic studies using in operando infrared spectroscopic analysis reveal the key role played by water, specifically deriving from K4[Fe(CN)6]·3H2O, in facilitating a remarkable switch in mechanism from a homogeneous to heterogeneous catalytic manifold. Evidence for heterogeneity includes sigmoidal kinetic traces, an order in Pd of 0.26 and catalyst turnover sequestration upon addition of excess Hg. The TOF and TON values recorded using K4[Fe(CN)6] (<220 ppm H2O) were higher than when using K4[Fe(CN)6]·3H2O (>4000 ppm H2O). Catalytic cycles are configured according to the experimental evidence gained from this study. We conclude that water plays a critical role in Pd catalyst speciation in arylcyanation chemistry.
Introduction
Pd-Catalyzed cyanation of aryl/heteroaryl halides, hereafter arylcyanation, are among the most commonly used synthetic methods for the preparation of biologically-relevant aromatic nitriles,1 many of which are commercially valuable (Fig. 1).2 The reaction was first reported by Takagi and co-workers in 1973,3 using stoichiometric and toxic cyanide salts, in addition to relatively high Pd catalyst loadings (∼5 mol%). The research groups of Beller4 and Weissman5 transformed the field using K4[Fe(CN)6] as a non-toxic cyanating agent, also showing that low Pd catalyst loadings were feasible for a wide array of aryl halide coupling partners (∼0.1 to 0.01 mol%). Based on these practical advances and the reactions being run at such low catalyst loadings, it is hard to conceive anything but a homogeneous catalytic cycle being operative under working conditions. Yet the precise catalytic reaction mechanisms remain poorly understood. | ||
| Fig. 1 Biologically-relevant aryl(heteroaryl) cyanide compounds. | ||
Excess cyanide has been shown to give PdII cyanide complexes, e.g. Pd(CN)42−, as a dominant off-cycle catalytically inactive species6 and previous attempts to tackle this issue include the addition of diamines,7 Zn reductant8 or cyanide-shuttling agents.9 Further mechanistic work, using stoichiometric Pd and a bidentate phosphine ligand, has revealed the productive stage of a homogeneous catalytic cycle to be a migratory reductive elimination.10
In these reported systems, all are presumed to operate by a homogeneous catalytic cycle, i.e. involving single Pd atoms. The role of higher order Pd species (e.g. Pd clusters and nanoparticles) as catalytically competent species has received comparatively less attention, despite their confirmed role within the field of reductive Pd catalysis.11 The work of Frech et al.12 is an exception; they presented sigmoidal kinetic profiles characteristic of the presence of catalytically active Pd nanoparticles formed from a molecular PdII precursor by slow-nucleation and an autocatalytic growth mechanism.13–16
Water has previously been shown to play an integral role in Pd-catalyzed Suzuki–Miyaura reactions by aiding the leaching of Pd atoms from Pd nanoparticles (PdNPs) into the reaction mixture.17,18 It also accelerates the autocatalytic formation of Irn0 nanoparticles from polyoxoanion-supported IrI precatalysts in the hydrogenation of cyclohexene.19 Trace air plays a similar role20 and is also reported to assist in the activation of Pd(PtBu)2 to form catalytically-active PdNPs.21
In the context of arylcyanation, water aids the solubility of the cyanating agent, Zn(CN)2, changing the reaction from zeroth order in bromobenzene (anhydrous conditions) to first order in the presence of 1.7 vol% water (in DMF).22 However, despite there being a role for water, the use of either K4[Fe(CN)6] and K4[Fe(CN)6]·3H2O appears to be interchangeable throughout the literature, with little rationale for selection being made or a role for water being indicated for effective arylcyanations.
A kinetic study on the catalytic behaviour of Pd species, deriving from exemplar PdII precatalysts, in arylcyanation reactions is detailed within this paper. The use of in situ infrared spectroscopy in operando facilitated quantitative analysis of the rates of appropriate reactions, and the unprecedented, qualitative study of intermediate, solubilized K4[Fe(CN)6] concentrations throughout the course of arylcyanation reactions at Pd. The study led to the surprising finding that the presence of water triggers the reaction from being homogeneous to heterogeneous in terms of catalyst behaviour.
Results and discussion
Central to this study was the identification of a PdII precatalyst system possessing a distinct balance of stability (to enable isolation) and instability (enabling catalytically competent Pd nanoparticles to be delivered, triggered by reducing conditions). It is known that spherical particles sized 1.5–2 nm are active in many cross-couplings.23–25 One needs to avoid rapid decomposition of a PdII precatalyst, which could deliver inactive large Pd particles, i.e. Pd black >1 μm in size. The formation of catalytically competent species under reducing conditions is therefore dependent on the structural identity of the PdII complex, vide infra.We determined that trans-Pd(OAc)2(pip)2 (pip = piperidine, 1-pip) is a competent PdII precatalyst for the direct C–H bond functionalization of 2′-deoxyadenosine using aryl halides.26 PdNPs readily derive from 1-pip under reducing reaction conditions (spherical PdNPs were found to be 1.5–2 nm in size26). Both Boykin and co-workers27 and Süss-Fink and co-workers28 showed that Suzuki–Miyaura cross-couplings mediated by similar PdII precatalysts, containing amine ligands, exhibited remarkable catalyst efficacy. The kinetic studies conducted by Süss-Fink and co-workers28 supported heterogeneous Pd catalysis, also implicating PdNPs. Conversely, trans-Pd(OAc)2(HNR2)2 complexes were found to be viable catalytic intermediates in selective oxidative C–H bond activation processes, e.g. functionalization of 2,2,6,6-tetramethylpiperidine (TMP).29
For this study, five well-defined mononuclear trans-Pd(OAc)2(L)2 complexes (L = HNR2), containing different sized secondary amine ligands, were synthesized by reaction of high purity Pd3(OAc)6 material (note: Pd(OAc)2 is usually a cluster of this type)30,31 with the prerequisite secondary amine ligands (Fig. 2). It was necessary to employ two equivalents of secondary amine; any less results in the formation of a dinuclear PdII complex containing only one amine ligand at each PdII centre. The full experimental characterization for these PdII complexes can be found in the experimental section (see ESI†), but several interesting structural features are worthy of mention. Inspection of the single crystal X-ray diffraction structures of 1-pyr, 1-dmp and 1-aze (Fig. 2) indicate that there is intramolecular H-bonding between the acetate ligand and the amino proton in both 1-pyr and 1-dmp, which is inkeeping the solid-state structure of both 1-pip26 and 1-tmp.29 Boykin conjectured that this type of interaction adds to the stability of similar complexes.27 In light of recent work on concerted metalation deprotonation processes,32,33 we propose that acetate-assisted deprotonation facilitates reductive elimination of the oxidized amino ligand, with concomitant formation of Pd0 (Fig. 3). The mechanism is consistent with what has previously been reported for 1-pip.24,26 Interestingly, there is no apparent H-bond in 1-aze, presumably due to increased bulk of the azepane ring. However, upon expanding the unit cell of the X-ray data, extended intermolecular H-bonding and π–π stacking between acetate carbonyl groups is apparent (see ESI†).
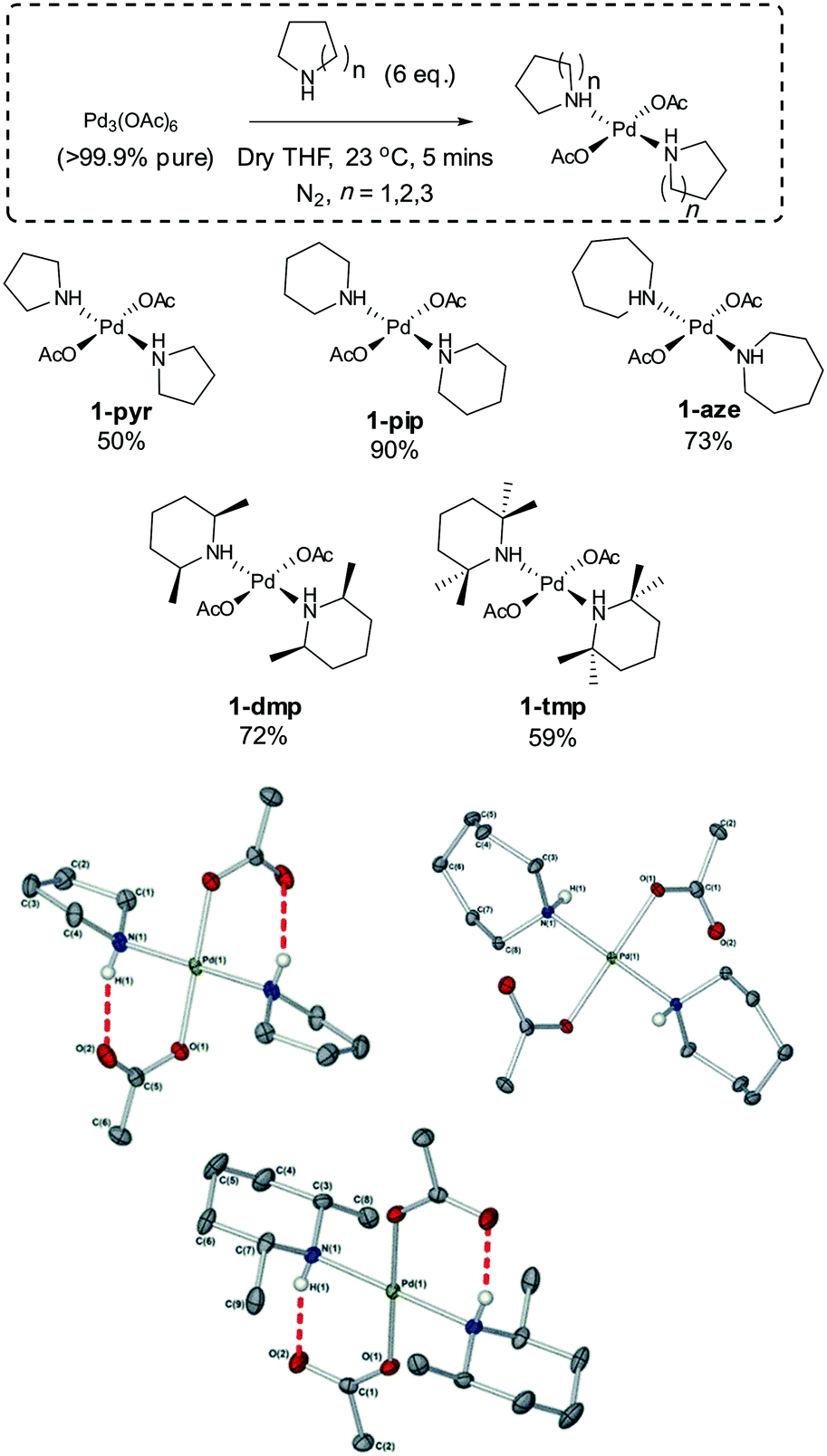 | ||
| Fig. 2 trans-Pd(OAc)2(HNR2)2 precatalysts. (top) The synthesis of a series of trans-Pd(OAc)2(HNR2)2 complexes. (bottom) Structures of 1-pyr, 1-aze and 1-dmp respectively, determined by X-ray diffraction; arbitrary atom numbering used. Selected hydrogen atoms removed for clarity. Thermal ellipsoids shown with a probability of 50%. Selected bond lengths (Å): 1-pyr N(1)–Pd(1): 2.043(3), O(1)–Pd(1): 2.015(2), C(1)–N(1): 1.484(4), C(5)–O(1): 1.282(4), C(5)–O(2): 1.243(4); 1-aze N(1)–Pd(1): 2.0638(14), O(1)–Pd(1): 2.0050(12), C(1)–O(1): 1.286(2), C(1)–O(2): 1.236(2); 1-dmp N(1)–Pd(1): 2.0696(18), O(1)–Pd(1): 2.0191(16), C(1)–O(1): 1.266(3), C(1)–O(2): 1.229(3), C(3)–N(1): 1.481(3). | ||
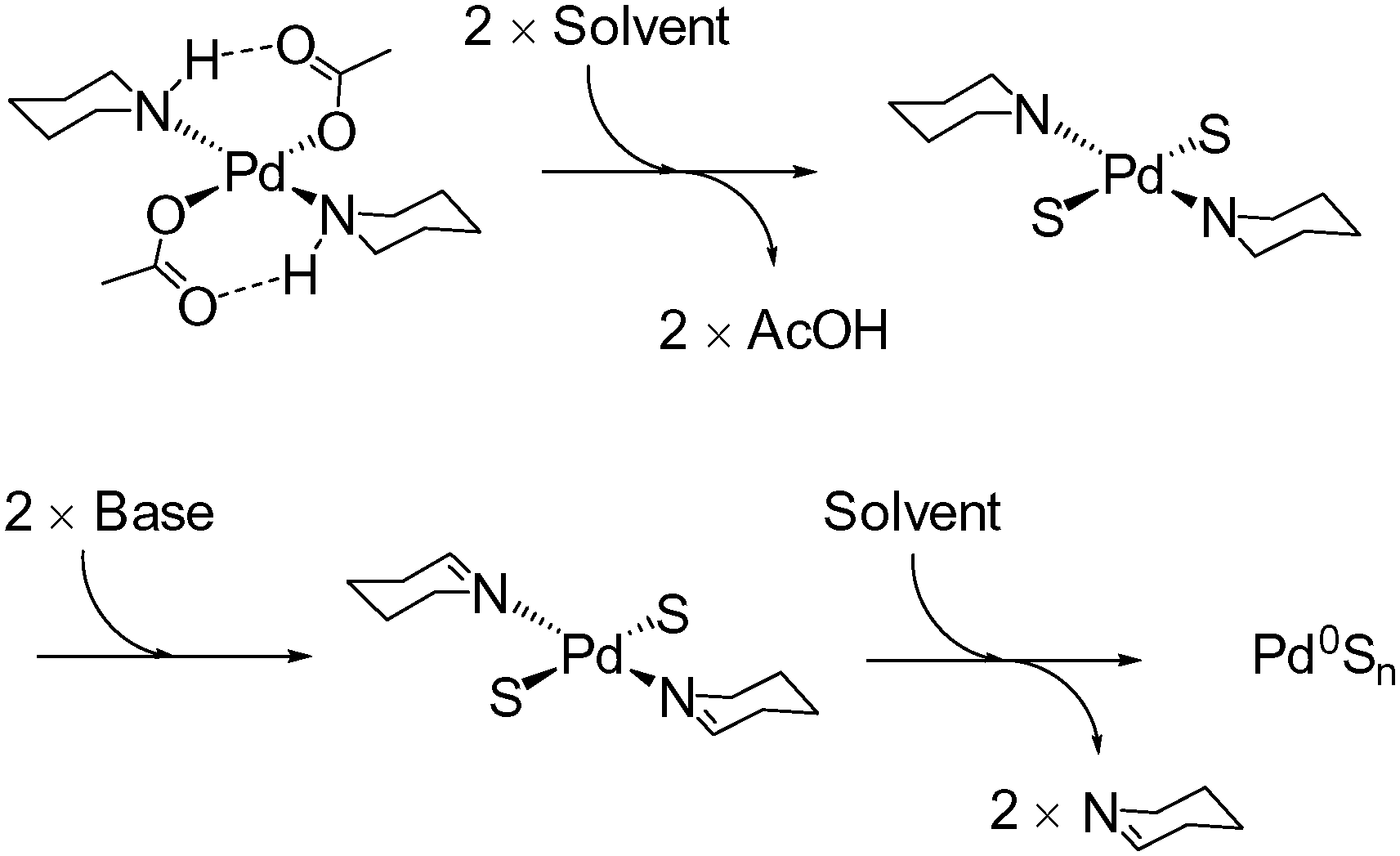 | ||
| Fig. 3 Postulated mechanism for activation of 1-pip; we propose that the related trans-Pd(OAc)2(L)2 are activated in forming Pd0 species by a similar mechanism. | ||
Kinetic examination of the behavior of trans-Pd(OAc)2(L)2 (L = secondary amine) precatalysts in arylcyanation reactions
To examine the catalytic behavior of the Pd(OAc)2(L)2 precatalysts the identification of a suitable benchmark organohalide substrate was required to: (a) be suitably challenging, i.e. containing a sensitive ‘palladaphilic’ amide functionality; (b) provide a spectroscopic handle to monitor consumption by in operando real-time analysis; (c) be electron-deficient, providing a test for the reductive elimination step.10 In terms of application, a chemical structure comparable to commercial targets such as olprinone and milrinone were taken into consideration (Fig. 1).2,34N-Benzyl-4-bromo-6-methyl-2-pyridone, 2, was chosen as a benchmark substrate (Fig. 4). Confirmation of its aromatic character, by comparison with N-methyl-4-bromo-6-methyl-2-pyridone, benzene, pyridine and bromobenzene, was gained by computational (DFT) studies – see ESI.†
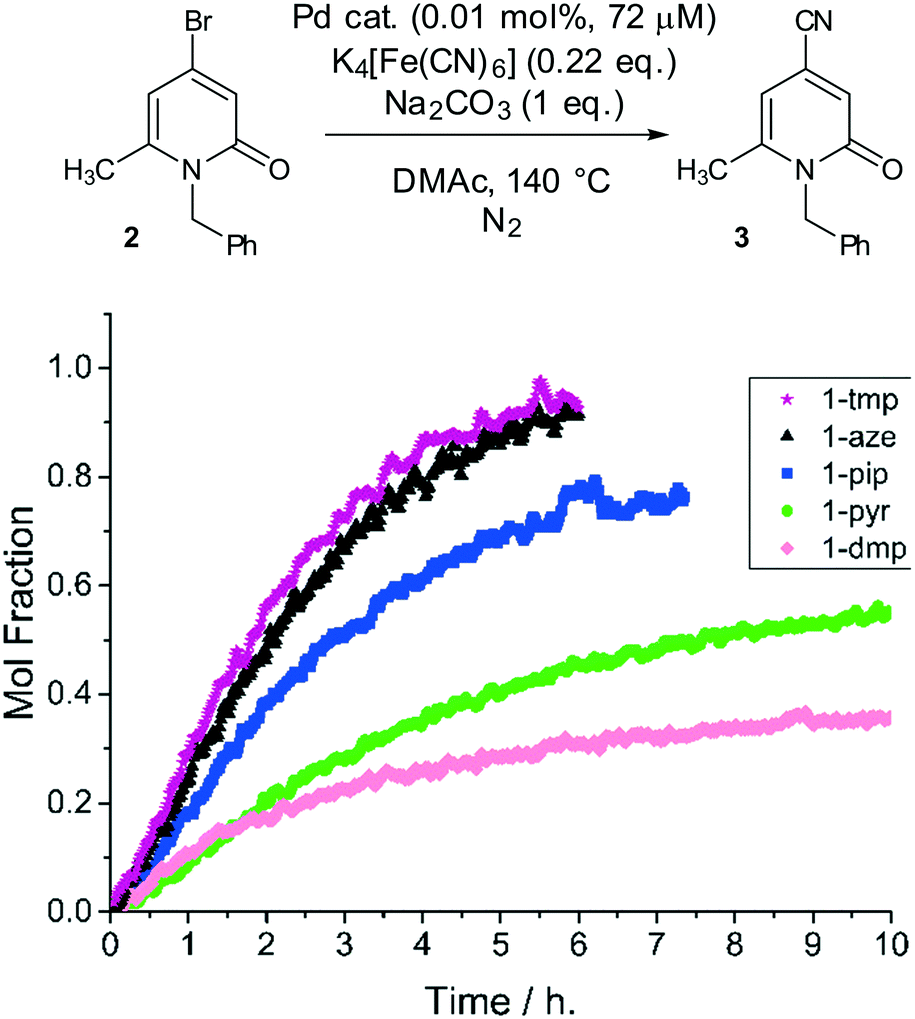 | ||
| Fig. 4 Precatalyst activity screen. Comparison of precatalyst activity in the cyanation of N-benzyl-4-bromo-6-methyl-2-pyridinone, 2 (see chemical scheme). Reaction conditions: [2]0: 0.72 M, catalyst (0.01 mol%, 72 μM Pd), K4[Fe(CN)6] (0.22 eq.), Na2CO3 (1 eq.), DMAc, N2, 140 °C. Data cut off at 10 h. The Pd precatalyst structures are given in Fig. 2. | ||
The initial catalyst screening studies involved reaction of 2 mediated by anhydrous K4[Fe(CN)6]35 and confirmed the reaction as a binary system – no homocoupling or dehalogenation was observed under any conditions tested. Displacement of bromide by cyanide requires a Pd catalyst species. The catalytic activity of the Pd precatalyst series was examined using low loadings of Pd (0.01 mol%, 72 μM Pd in the system).
The kinetic profiles showing the formation of product 3 over time are presented in Fig. 4. The best precatalyst under the reaction conditions examined was found to be 1-aze, containing azepane ligands, or 1-tmp containing 2,2,6,6-tetramethylpiperidine ligands. The piperidine-derived precatalyst, 1-pip, exhibited comparable activity up to 50% conversion to product 3 while the pyrrolidine-derived precatalyst 1-pyr was considerably less active than either 1-aze or 1-pip. In all reactions, a first order consumption with respect to 2 was determined. By comparison with 1-pip, the cis-2,6-dimethylpiperidine-derived precatalyst 1-dmp, was poor, suffering from significant catalyst deactivation after 2 h (ca. 15% conversion).
In continuation of the kinetic studies, efforts were focused on 1-pip. As a precatalyst 1-pip: (a) exhibits very good activity for the reaction of 2 → 3; (b) is the most straight-forward to prepare within the series; (c) the post-catalysis PdNPs derived from it after the direct arylation of 2′-deoxyadenosine have been characterized by TEM as spherical PdNPs.24,26
The catalyst efficiency of 1-pip was optimum at increased concentration of Pd while maintaining constant catalyst loadings (at 0.01 and 0.005 mol%). At a Pd concentration of 72 μM (0.01 mol%) the reaction proceeds to 77% conversion to 3, after 6 hours at 140 °C (Fig. 5). This compares with 40% conversion at [Pd] = 36 μM (0.01 mol%). Similar catalyst turnover numbers (TONs)‡ were recorded for 0.01 mol% (72 μM) and 0.005 mol% (72 μM) catalyst loadings (7700 and 7600 respectively). Comparison of the catalyst turnover frequencies (TOFs)‡ revealed that catalyst efficiency was highest when the reaction was charged with 0.01 mol% 1-pip at [Pd] = 72 μM (Fig. 5) and a substrate (2) concentration of 0.72 M. This provided a TOF of 0.527 s−1 and TON of 7700.36 It was these optimum conditions under which the precatalyst series were screened (see Fig. 4).
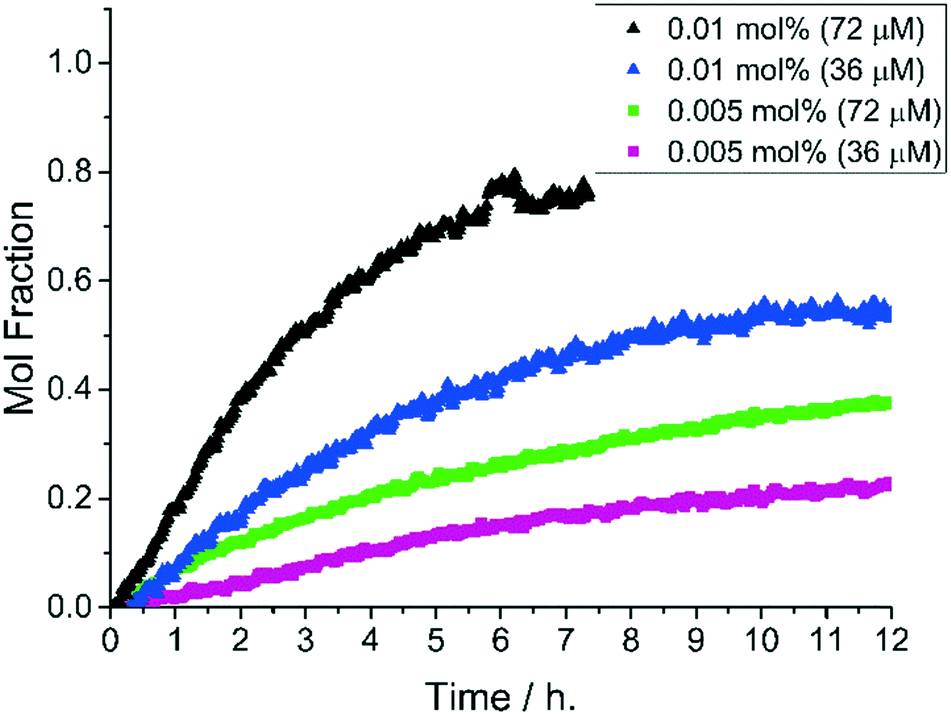 | ||
| Fig. 5 Reaction profiles for formation of 3 from 2 with increased [Pd] and reaction concentrations. Constant catalyst loadings were maintained over two different [Pd] under pseudo-anhydrous conditions (data points cut-off at 12 h). Reaction conditions: 1-pip (0.005–0.01 mol%), K4[Fe(CN)6] (0.22 eq.), Na2CO3 (1 eq.), DMAc, N2, 140 °C. Data cut off at 12 h. Black triangles [2]0: 0.72 M, blue triangles [2]0: 0.36 M; green squares [2]0: 1.44 M; purple squares [2]0: 0.72 M. Reaction conditions: [2]0: 0.36 M, 1-pip (0.05 mol%, 180 μM Pd), hexacyanoferrate salt (0.22 eq.), Na2CO3 (1 eq.), DMAc, N2, 120 °C. | ||
The order with respect to Pd under pseudo-anhydrous conditions was determined using the Burés normalized time scale method (Fig. 6, top).37 The rate of reaction shows a second-order dependence upon Pd catalyst concentration, indicating a homogeneous reaction mechanism and rate-determining state involving two discrete catalyst molecules38,39 or one involving a dominant dinuclear [Pd2] complex.40 Given this, the mechanism must go via a rate determining step involving either bi-metallic Pd-to-Pd transmetallation (i.e. independent of [2]) or oxidative addition involving two atoms of Pd (i.e. dependent on [2]). The reaction was determined to be first order with respect to the concentration of 2 (Fig. 6, bottom), again using the Burés time normalized scale method.41 Therefore, due to the limited solubility of K4[Fe(CN)6] in the reaction solvent a rate-determining oxidative addition step involving two atoms of Pd is thus confirmed.
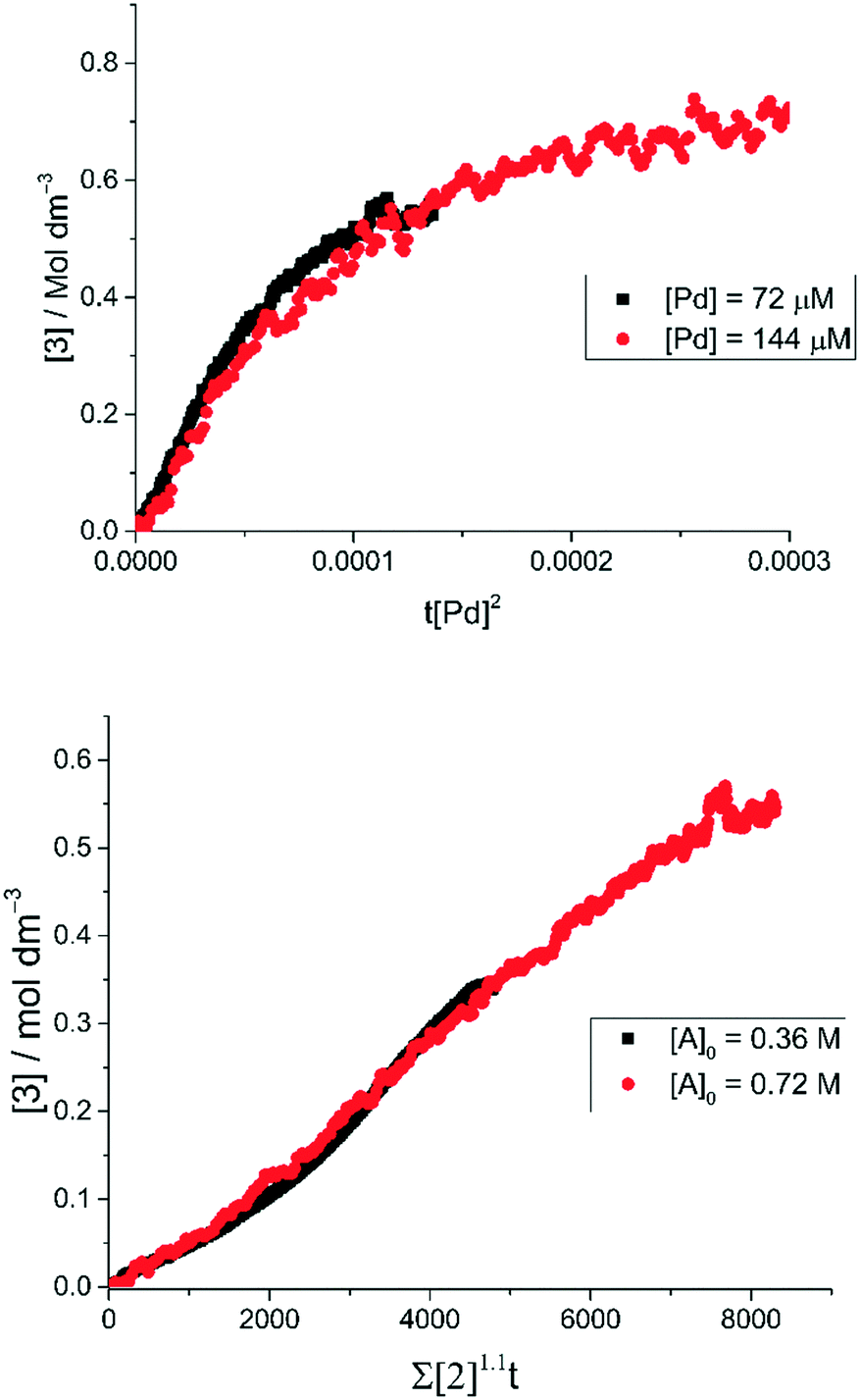 | ||
Fig. 6 Normalized time scale reaction profiles. (top) Analysis of two different concentrations of Pd using the Burés37 normalized time scale method, shows that under ‘low water’ conditions the arylcyanation reaction is second order in 1-pip. Equation used: [3] = t[Pd]n where n is the order with respect to [Pd] when the traces overlay. (bottom) By performing the cyanation reaction at two different concentrations of 2 under ‘low water’ conditions, the Burés normalized time scale method shows that the order of reaction is first order in 2.41 Equation used: 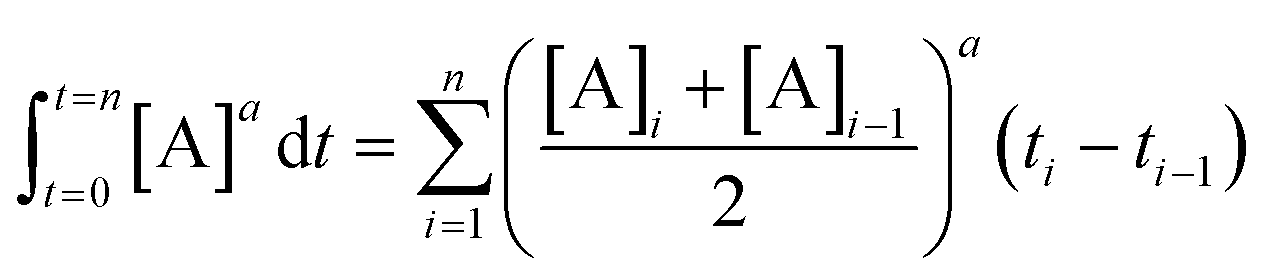 , where a is the order with respect to [A] when the traces overlay. , where a is the order with respect to [A] when the traces overlay. | ||
Effect of ‘excess water’
Upon changing the cyanide source to K4[Fe(CN)6]·3H2O, i.e. water present albeit not fully solubilized, a characteristic sigmoidal kinetic curve became apparent (Fig. 7, top). When the 1-pip catalyst loading was reduced to 0.05 mol% the sigmoidal reaction profile exhibited a pronounced induction period of up to 1.5 h. This outcome stands in stark contrast to the reactions conducted using anhydrous K4[Fe(CN)6]. The direct addition of degassed water (0.66 eq., 4290 ppm) to an equivalent reaction performed with anhydrous K4[Fe(CN)6], resulted in a similar, sigmoidal kinetic profile (Fig. 7, bottom kinetic profiles).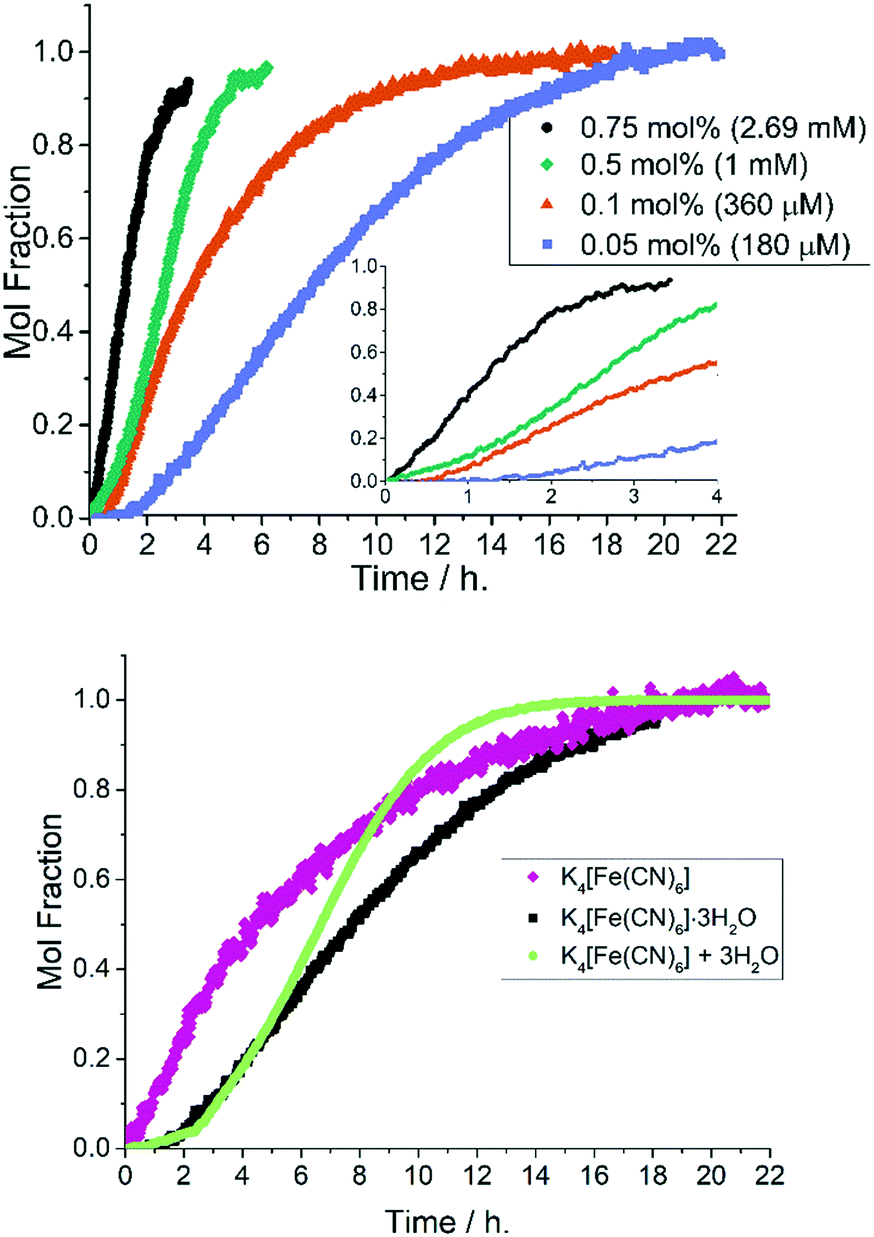 | ||
| Fig. 7 Sigmoidal reaction profiles under hydrous conditions. (Top) Formation of 3 from 2 with varying Pd catalyst loading results in an induction period at low catalyst loadings when using K4[Fe(CN)6]·3H2O. The inset graph shows kinetic profiles to 4 h. Reaction conditions: [2]0: 0.36 M, 1-pip (0.05–0.75 mol%, 180–2700 μM Pd), K4[Fe(CN)6]·3H2O (0.22 eq.), Na2CO3 (1 eq.), DMAc, N2, 120 °C. (bottom) The addition of degassed water (0.66 eq.) to a reaction performed under otherwise ‘low water’ conditions results in a sigmoidal reaction profile. | ||
The non-identical curves for the reactions conducted in the presence of either K4[Fe(CN)6]·3H2O (black squares) and K4[Fe(CN)6] with additional water (3 equivalents, light green circles) is to be expected as added water is fully soluble (free to influence catalysis), whereas the water derived from K4[Fe(CN)6]·3H2O is not.
The observation of a sigmoidal kinetic curve for a metal-catalyzed reaction under reducing conditions is powerful evidence for the in situ formation of a heterogeneous catalyst.14 Extended induction periods indicate that mononuclear Pd (as 1-pip) is catalytically incompetent under the conditions used, requiring activation before productive catalysis can occur. The Finke–Watzky 2-step mechanism13 was developed precisely to elucidate the rate of the pseudoelementary steps involved in the formation of catalytically active metal nanoclusters from a catalytically inactive precursor by slow continuous nucleation (k1) and fast autocatalytic surface growth (k2).
Using the Finke–Watzky 2-step mechanism, it was possible to use a non-linear least squares fit to support our hypothesis of heterogeneous catalyst behaviour when using 1-pip together with K4[Fe(CN)6]·3H2O. The values of k1 and k2 for each catalyst loading could be measured by this approach (Fig. 8, left). Analysis of all the 1-pip catalyst loadings showed that a good fit was only obtained with a 1-pip catalyst loading below and including 0.5 mol% ([Pd] = 1.8 mM), indicating that precatalyst activation was occurring via the Finke–Watzky 2-step mechanism at lower catalyst loadings. Further information regarding the goodness of fit and subsequent error analysis using Fisher's F distribution is included in the ESI.†42 Furthermore when a large excess of metallic Hg was added to a reaction performed with 1-pip (0.5 mol%, 1.8 mM Pd) at 93 min, no further product 3 was formed (Fig. 8, right). The Hg drop test provides a means of showing that a metallic surface is playing some role in terms of productive catalysis;14 importantly, this test is only supportive evidence for the presence of heterogeneous catalyst species, and cannot be interpreted as conclusive proof for catalyst heterogeneity. Other powerful poisoning tests, such as the quantitative addition of 1,10-phenanthroline,43,44 were considered but deemed to be chemically incompatible with the precatalyst (1,10-phenantroline would outcompete piperidine as a ligand at PdII, resulting in a chelated N,N-Pd(OAc)2 species), inhibiting the catalyst activation mechanism proposed vide supra.
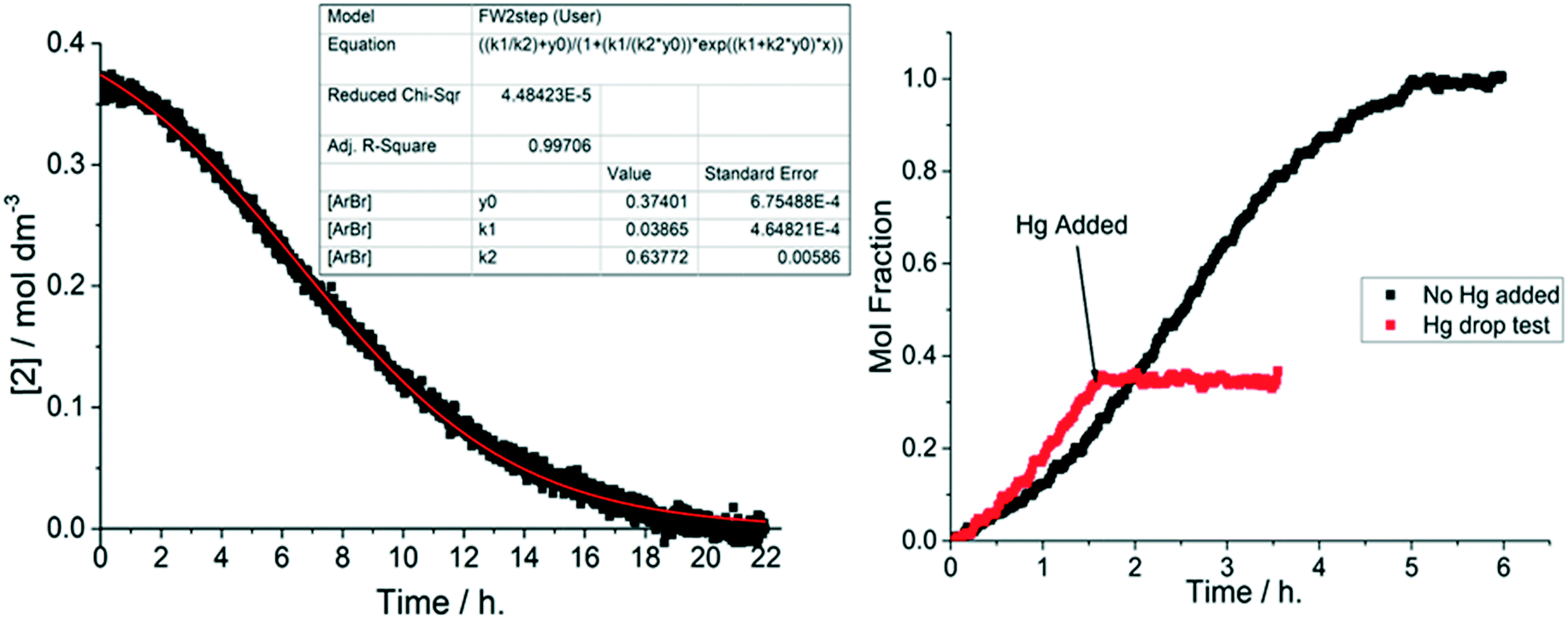 | ||
Fig. 8 A good fit to the Finke–Watzky 2-step mechanism and a positive Hg drop test are indicators for heterogeneous catalysis. (left) The 2-step, Finke–Watzky nucleation and autocatalytic growth mechanism* gives a good fit for the data at low catalyst loadings. Reaction conditions: [2]0: 0.36 M, 1-pip (0.05 mol%, 180 μM Pd), K4[Fe(CN)6]·3H2O (0.22 eq.), Na2CO3 (1 eq.), N2, 120 °C, DMAc. (right) Mercury drop test experiment. Reaction conditions: [2]0: 0.36 M, 1-pip (0.5 mol%, 1.8 mM Pd), K4[Fe(CN)6]·3H2O (0.22 eq.), Na2CO3 (1 eq.), N2, 120 °C, DMAc. Hg (200 eq. with respect to Pd) added with vigorous stirring after 93 min. *Equation used: 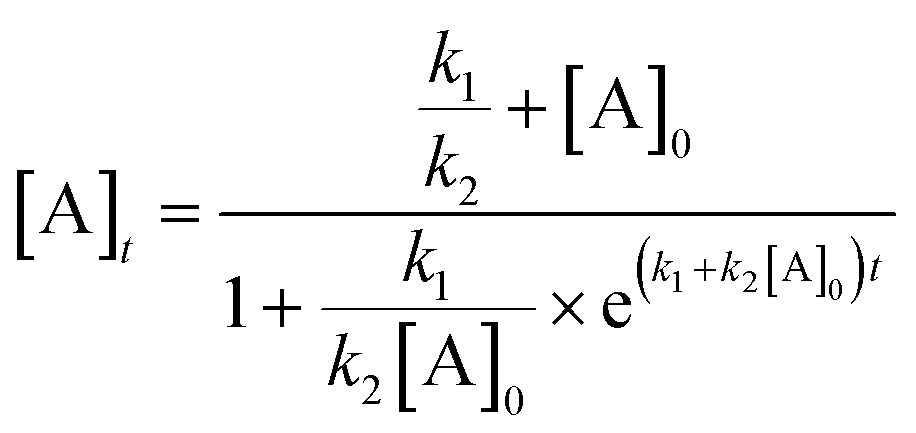 . . | ||
Plotting TOF vs. catalyst45 loading indicates a counter-intuitive, negative effect upon catalyst efficiency caused by increased catalyst loadings (Fig. 9). At low Pd catalyst loading and concentration (0.05 mol%, 180 μM), the catalyst turned-over steadily for ca. 12 hours until reaction completion, however when the Pd catalyst loading was increased to 0.1 mol% a higher TOF was achieved (Fig. 9, left). This indicates that in the presence of water the catalyst turns over most efficiently at a [Pd] of ca. 360 μM and Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate ratio of 1:1000. However, upon increasing the Pd catalyst loading and concentration further to 0.5 mol% (1.8 mM Pd) and above, the catalyst TOF suffers a significant decrease. The lower TOF resulting from the increased [Pd] can be attributed to fewer catalytically active sites per mole of Pd, indicating the presence and role for larger Pd clusters at higher [Pd]. Increased TON and TOF upon decreasing catalyst loading has also been attributed to catalyst degradation by aggregation in Sonogashira reactions: Fairlamb et al. report higher TOFs at 0.001 mol% than at 0.01 mol% (ref. 46) catalyst loadings and a significant decrease in catalyst activity above that catalyst concentration. A similar outcome noted by De Vries et al. where higher yields were obtained in a Heck47,48 reaction at 0.02 and 0.08 mol% Pd catalyst loadings than at 0.00125 and 1.28 mol%. When stabilized PdNPs of a range of sizes (1.8–4 nm) were utilized in a Suzuki reaction it was reported that TOF decreased upon increased NP size.49 An inverse TOF/[Pd] relationship therefore indicates that heterogeneous species are likely involved in the reaction. Extraction of the observed rate constants from peak rates of the sigmoidal reaction profiles allows for the calculation of the order with respect to Pd via the differential method. This revealed a reaction order of 0.257 (±0.02) (Fig. 9, right) and shows that 1 in every 3.89 Pd atoms are catalytically competent; providing further evidence for the involvement of small Pd clusters when K4[Fe(CN)6]·3H2O is used as the cyanating agent, i.e. under ‘excess water’ conditions.
:substrate ratio of 1:1000. However, upon increasing the Pd catalyst loading and concentration further to 0.5 mol% (1.8 mM Pd) and above, the catalyst TOF suffers a significant decrease. The lower TOF resulting from the increased [Pd] can be attributed to fewer catalytically active sites per mole of Pd, indicating the presence and role for larger Pd clusters at higher [Pd]. Increased TON and TOF upon decreasing catalyst loading has also been attributed to catalyst degradation by aggregation in Sonogashira reactions: Fairlamb et al. report higher TOFs at 0.001 mol% than at 0.01 mol% (ref. 46) catalyst loadings and a significant decrease in catalyst activity above that catalyst concentration. A similar outcome noted by De Vries et al. where higher yields were obtained in a Heck47,48 reaction at 0.02 and 0.08 mol% Pd catalyst loadings than at 0.00125 and 1.28 mol%. When stabilized PdNPs of a range of sizes (1.8–4 nm) were utilized in a Suzuki reaction it was reported that TOF decreased upon increased NP size.49 An inverse TOF/[Pd] relationship therefore indicates that heterogeneous species are likely involved in the reaction. Extraction of the observed rate constants from peak rates of the sigmoidal reaction profiles allows for the calculation of the order with respect to Pd via the differential method. This revealed a reaction order of 0.257 (±0.02) (Fig. 9, right) and shows that 1 in every 3.89 Pd atoms are catalytically competent; providing further evidence for the involvement of small Pd clusters when K4[Fe(CN)6]·3H2O is used as the cyanating agent, i.e. under ‘excess water’ conditions.
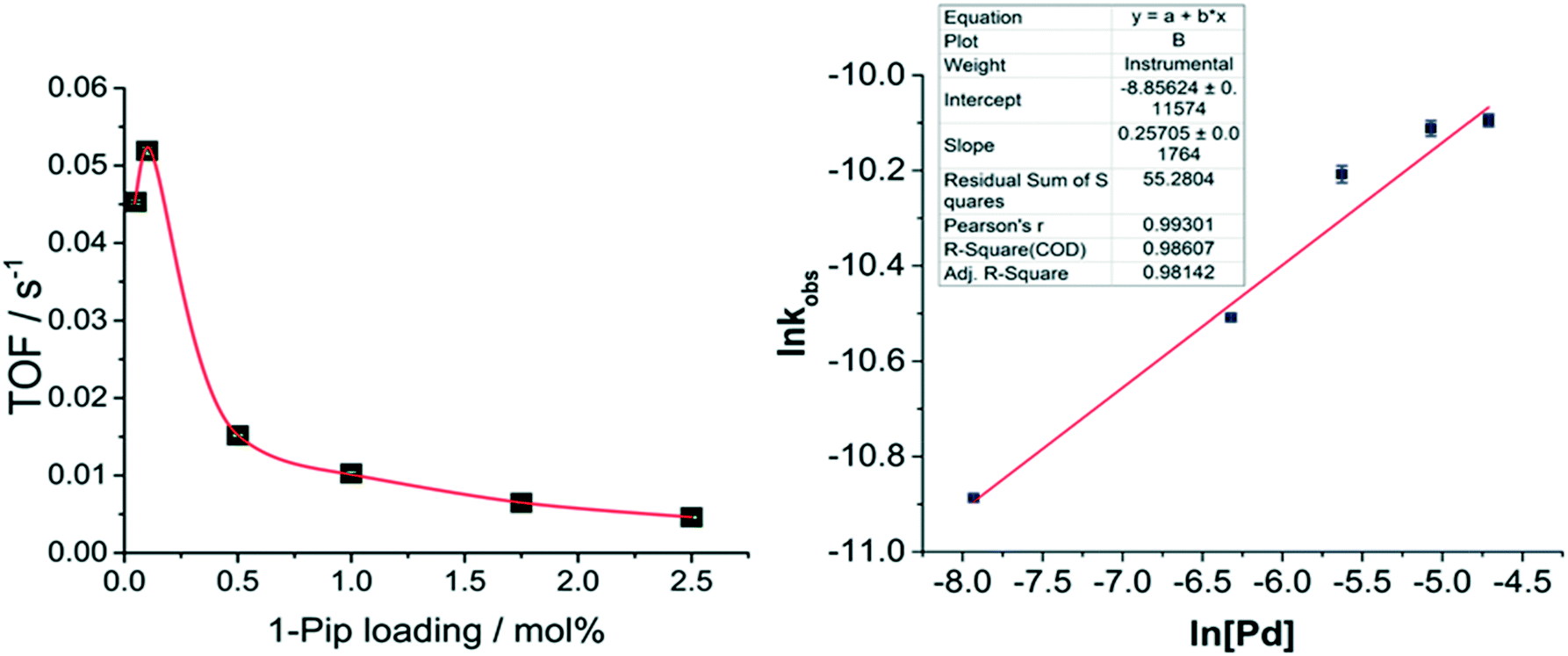 | ||
| Fig. 9 TOF and Kobs data support heterogeneous-type catalysis under hydrous conditions. (left) A graph to show TOF vs. Pd precatalyst (1-pip) loading, revealing an inverse correlation of catalytic activity with Pd catalyst loading. The TOFs of each of the different precatalyst loadings were measured as a gradient of TON s−1 for the peak rates of the kinetic profiles. Error bars are shown in green. Reaction conditions: [2]0: 0.36 M, 1-pip (0.05–2.5 mol%, 180 μM–9 mM Pd), K4[Fe(CN)6]·3H2O (0.22 eq.), Na2CO3 (1 eq.), DMAc, N2, 120 °C. (right) The order with respect to Pd as determined by the differential method provides a value of 0.257 (±0.02). Error bars are shown in blue. | ||
It is worthy of note that the hexacyanoferrate and additive counterions were also found to have a significant effect on the efficiency of catalysis. Using kinetic methods, this has been extensively studied by following the characteristic [Fe(CN)6]4− infrared absorbance (seen at 2045 cm−1) in correlation with reaction progression. The additive, Na2CO3, has been shown to assist salt metathesis with K4[Fe(CN)6] which tensions dissociative ligand exchange reactivity and complex solubility. The use of alternative carbonate additives (K2CO3 and Cs2CO3) results in no catalytic turnover; this has been attributed to a differing hexacyanoferrate solubilities following partial counterion exchange. The direct addition of Na4[Fe(CN)6] as a cyanating agent resulted in inhibited catalytic turnover caused by its increased solubility in hot DMAc (shown by in situ IR spectroscopy). These findings are summarized in the context of a complete mechanism in Fig. 10, however further discussion, including kinetic traces, is included in the ESI.†
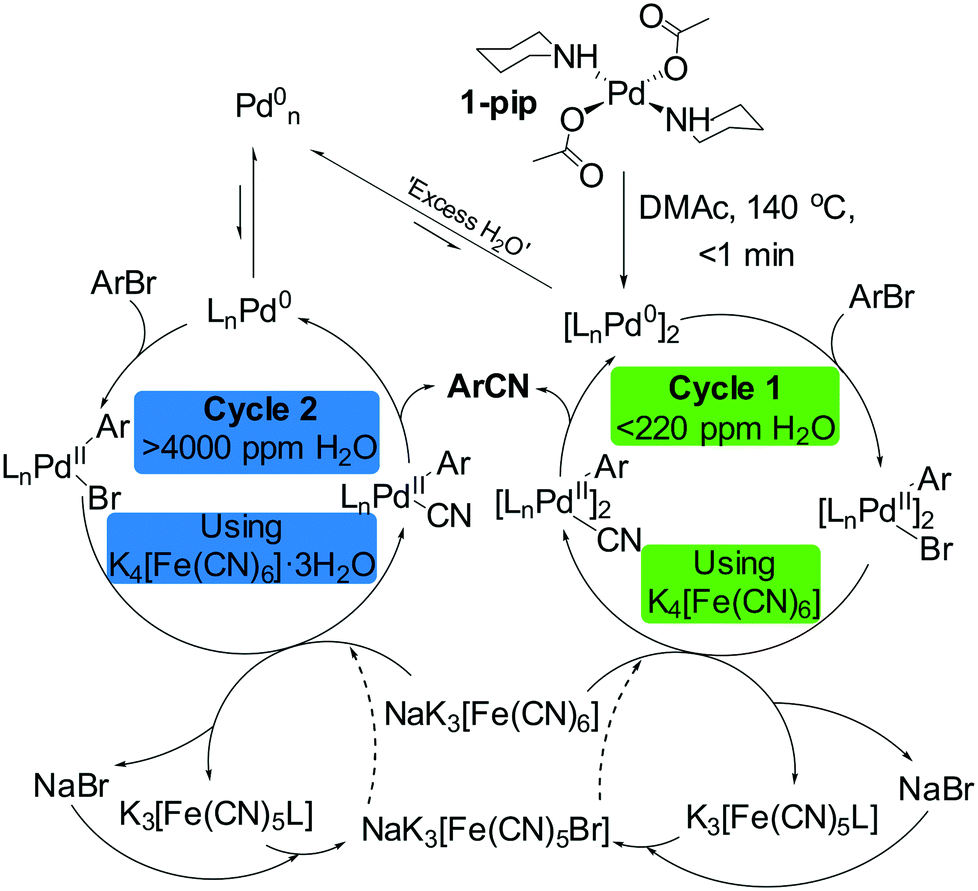 | ||
| Fig. 10 Postulated mechanism for the Pd catalyzed arylcyanation of 2. The presence or absence of excess water dictates which mechanism the reaction proceeds by while Na2CO3 assists dissociative ligand exchange of −CN from FeII to PdII. A simplified cluster is depicted as suggested by order in Pd obtained experimentally. The water content of anhydrous DMAc obtained using Karl-Fisher titration. NaK3[Fe(CN)6] formed following counterion metathesis of K4[Fe(CN)6] with Na2CO3. X is acetate or bromide. The dotted lines describe the second dissociative ligand exchange step following complete consumption of NaK3[Fe(CN)6]. | ||
Implications for the delineating the mechanistic complexity of the arylcyanation reaction manifold
This study has allowed us to fully assess the mechanistic complexity of catalytic arylcyanation reaction at Pd. When a PdII precatalyst is used in a ‘reductive’ cross-coupling-type reaction, the involvement of a multi-ensemble of Pd catalyst species should be questioned. It is not the case that aggregated Pd species, particularly nanoparticles, are moribund forms of the catalyst – they can be catalytically competent, exhibiting different catalytic behavior. Examination of five trans-Pd(OAc)2(HNR2)2 complexes, all of which are excellent precursors to Pd0, has revealed a profound effect of catalyst structure with catalyst performance. Given the number of potential reaction variables for catalytic arylcyanation reactions, the study has enabled us to define measurable conclusions, with implications for whether catalysis is occurring by a homogenous or heterogeneous process.For example, when water is introduced into the arylcyanation reaction in the form of K4[Fe(CN)6]·3H2O, it triggers the reaction to proceed via heterogeneous catalytic manifold (H2OTOTAL > 4000 ppm, as measured using Karl-Fisher titration) while inhibiting homogeneous catalysis. The proposal is supported by an inverse TOF/catalysis loading relationship, a partial order in Pd (0.26), sigmoidal kinetics fitted to the Finke–Watzky mechanism, and a positive Hg drop test (testing for surface catalysis). The order in Pd suggests that 1 in 3.8 Pd atoms are catalytically active, which taken alongside the positive Hg drop test and sigmoidal kinetic behaviour, confirms the involvement of higher order Pd species in the catalysis. Water facilitates the leaching of atomic Pd from PdNPs.17 In our study, ‘excess water’ is the trigger for a catalytic cycle involving heterogeneous Pd species.
Under ‘low water’ conditions, using anhydrous K4[Fe(CN)6] as a cyanide source, kinetic behaviour representative of a homogeneous catalytic cycle has been evidenced (H2OTOTAL < 220 ppm). Under these pseudo-anhydrous conditions the rate of reaction shows a second order dependence upon 1-pip concentration, indicating that two Pd atoms, i.e. a dinuclear Pd2 species, is involved in the rate-determining transition state.40 Under pseudo-anhydrous conditions it is possible to achieve lower catalyst loadings (down to 0.005 mol%) and optimum conditions were found to be 0.01 mol% with a Pd concentration of 72 μM. Under such conditions 1-pip achieved a TOF of 0.527 s−1. The relative activity of Pd(OAc)2(HNR2)2 precatalysts was found to be in order: 1-tmp > 1-aze > 1-pip > 1-pyr > 1-dmp.
Lastly, further experimental evidence confirmed that sodiated additives assist the solubility of K4[Fe(CN)6] by partial counterion swapping to NaxKy[Fe(CN)6], where x + y = 4. This process facilitates the formation of NaBr (lattice enthalpy: 747 kJ mol−1) upon dissociative ligand exchange with Pd, rather than KBr (lattice enthalpy: 682 kJ mol−1). Other carbonate salts do not facilitate productive catalytic turnover, but significantly influence the concentration of [Fe(CN)6]4− observed in solution (see ESI†). Using Na4[Fe(CN)6] results in a significantly reduced reaction efficiency, i.e. low product conversions and slow reaction, due to increased solubility.
A proposed mechanistic rationale from the observations deduced from this study is depicted in Fig. 10 for 1-pip. Reduction of 1-pip occurs almost instantaneously (typically within seconds), delivering a catalytically active, bimetallic [LnPd0]2 species to the reaction system. In the presence of ‘low water’ (<220 ppm), [LnPd0]2 is then able to enter homogeneous catalytic cycle 1. In both cycles, the oxidative addition product then undergoes dissociative ligand exchange with NaK3[Fe(CN)6], which is formed through counterion exchange with Na2CO3 (not displayed in Fig. 10). Reductive elimination from LnPdII(Ar)(CN) yields the ArCN product and regenerates the active catalyst species. Under any of the reaction conditions tested, LnPd0 is in equilibrium with aggregated Pd particles, either as a reservoir, moribund form or catalytically-competent form. The ‘excess H2O’ shifts the position of the equilibrium from LnPd0 to Pdn0 (representative of the total active aggregated Pd), which allows cycle 2 to become operative, with concomitant shutdown of cycle 1. The dissociative ligand exchange step is undoubtedly complicated, as there are several species that can act as the cyanide source. The steps proposed within the catalytic manifold involve salt metathesis and eventually lead to NaK3[Fe(CN)5X], the fate of which is likely halogenated Fe species.
Conclusions
The findings from this study are important for Pd catalyst efficacy of arylcyanation reactions. More broadly, there is significant message concerning the role for water in related reductive Pd-catalyzed cross-coupling-type reactions employing PdII precatalysts, which will be useful for those working in applied chemical synthesis, especially scale-up processes.Key experimental details
Method for the kinetic analysis of all arylcyanations of 2 using in situ infrared spectroscopy
An oven dried, 100 mL three necked round bottomed flask equipped with a small stirrer bar was attached to the React-IR, then backfilled with nitrogen three times. At all points of the experiment hereafter, the reaction vessel was open to a steady flow of N2. In parallel, a small oven-dried Schlenk tube was charged with catalyst (10 mg,) and backfilled with nitrogen three times.After backfilling the round bottomed flask with dry N2, a background spectrum was collected, and then dry, degassed DMAc added (6 mL). At this point a reference spectrum was recorded with stirring. After sequence initiation, the reaction was stirred at room temperature for 5 minutes, before being heated to reaction temperature (as monitored by the internal thermocouple) by being placed in a silicon oil bath set to an appropriate level above the desired reaction temperature. After the reaction mixture reached the correct temperature (determined using an external, digital thermometer), the solvent was stirred for a further five minutes and a spectrum used as a reference. To the hot solvent was then added the Na2CO3 (0.25–1 eq., freshly ground in a pestle and mortar) and was stirred for a further 5 minutes. After the system reached equilibrium, the cyanating agent (K4[Fe(CN)6]·3H2O or K4[Fe(CN)6], 0.167–0.5 eq., freshly ground in a pestle and mortar) was added and stirred for 5 minutes. A spectrum was then selected as a second reference. To the mixture was then added N-benzyl-4-bromo-6-methylpyridin-2-one (2, 1 eq.), and the sides of the flask rinsed with dry, degassed DMAc (0.5–1 mL, adding the appropriate amount to make the final total reaction volume up to 7 mL) via a septum. The reaction mixture was then stirred for 5 minutes, to allow for temperature equilibration back to the desired reaction temperature and dissolution of the substrate. During this time the appropriate amount of dry, degassed DMAc was added to the small Schlenk containing the precatalyst to make a stock solution. All catalyst stock solutions were made to a concentration of 25 mmol dm−3 prior to their addition to reaction mixtures. After the reaction mixture had achieved a constant temperature and immediately after complete dissolution of the catalyst, the reaction was initiated by addition of an appropriate quantity of catalyst stock solution using a syringe to the round bottomed flask via a septum.
The reaction progress was monitored by measuring the increase in absorbance at 2238 cm−1, corresponding to the product's nitrile stretch (in 3). After the absorbance at 2238 cm−1 had reached a constant value (indicating reaction completion) the experiment was stopped, and the reaction mixture allowed to cool to room temperature. Once the mixture had cooled, the final conversion of each reaction was measured by direct 1H NMR spectroscopic analysis of the crude reaction mixture. An aliquot of the reaction mixture was filtered through a small Celite/cotton wool plug to remove any organic-insoluble salts and diluted with CDCl3 before running the NMR experiment. The final conversion was calculated by measuring the ratio of the diagnostic peaks at 6.80 ppm for N-benzyl-4-bromo-6-methylpyridin-2-one (2) and 6.85 ppm for N-benzyl-4-cyano-6-methylpyridin-2-one (3).
All peak absorption data for 2238 cm−1, 2045 cm−1 and any other peaks of interest were exported into Excel in three forms: i) with no reference subtraction; ii) after subtraction of a reference spectrum of DMAc at reaction temperature; iii) after subtraction of a reference spectrum of the reaction mixture before substrate addition, at reaction temperature °C. All subsequent analysis was carried out on dataset iii). All kinetic traces (conversions) were normalised using the final reaction conversion (as calculated vide supra) and the absorption value of an appropriate data point at 2238 cm−1 (mid data scatter) after reaction completion. All data was smoothed with a moving four-point average. An annotated figure detailing the experimental set up is included in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The results presented within this paper were made possible by funding from Bayer AG (PhD studentship to J. T. W. B.). The equipment and facilities used in this study have been supported by the University of York and EPSRC (grant number EP/K031589/1).Notes and references
- P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049–5067 RSC.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed.
- K. Takagi, T. Okamoto, Y. Sakakibara and S. Oka, Chem. Lett., 1973, 2, 471–474 CrossRef.
- T. Schareina, A. Zapf and M. Beller, J. Organomet. Chem., 2004, 689, 4576–4583 CrossRef CAS.
- S. A. Weissman, D. Zewge and C. Chen, J. Org. Chem., 2005, 70, 1508–1510 CrossRef CAS PubMed.
- K. D. Dobbs, W. J. Marshall and V. V. Grushin, J. Am. Chem. Soc., 2007, 129, 30–31 CrossRef CAS PubMed.
- M. Sundermeier, A. Zapf, S. Mutyala, W. Baumann, J. Sans, S. Weiss and M. Beller, Chem. – Eur. J., 2003, 9, 1828–1836 CrossRef CAS PubMed.
- R. Chidambaram, Tetrahedron Lett., 2004, 45, 1441–1444 CrossRef CAS.
- C. Yang and J. M. Williams, Org. Lett., 2004, 6, 2837–2840 CrossRef CAS PubMed.
- J. L. Klinkenberg and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5758–5761 CrossRef CAS PubMed.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- R. Gerber, M. Oberholzer and C. W. Frech, Chem. – Eur. J., 2012, 18, 2978–2986 CrossRef CAS PubMed.
- M. A. Watzky and R. G. Finke, J. Am. Chem. Soc., 1997, 119, 10382–10400 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- E. E. Finney and R. G. Finke, J. Colloid Interface Sci., 2008, 317, 351–374 CrossRef CAS PubMed.
- S. Özkar and R. G. Finke, Langmuir, 2016, 32, 3699–3716 CrossRef.
- P.-P. Fang, A. Jutand, Z.-Q. Tian and C. Amatore, Angew. Chem., Int. Ed., 2011, 50, 12184–12188 CrossRef CAS PubMed.
- J.-S. Chen, A. N. Vasiliev, A. P. Panarello and J. G. Khinast, Appl. Catal., A, 2007, 325, 76–86 CrossRef CAS.
- Y. Lin and R. G. Finke, Inorg. Chem., 1994, 33, 4891–4910 CrossRef CAS.
- K. Köhler, R. G. Heidenreich, J. G. E. Krauter and J. Pietsch, Chem. – Eur. J., 2002, 8, 622–631 CrossRef.
- D. Heijnen, F. Tosi, C. Vila, M. C. A. Stuart, P. H. Elsinga, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 3354–3359 CrossRef CAS PubMed.
- F. G. Buono, R. Chidambaram, R. H. Mueller and R. E. Waltermire, Org. Lett., 2008, 10, 5325–5328 CrossRef CAS PubMed.
- M. Hyotanishi, Y. Isomura, H. Yamamoto, H. Kawasaki and Y. Obora, Chem. Commun., 2011, 47, 5750–5752 RSC.
- C. G. Baumann, S. De Ornellas, J. P. Reeds, T. E. Storr, T. J. Williams and I. J. S. Fairlamb, Tetrahedron, 2014, 70, 6174–6187 CrossRef CAS.
- A. F. Lee, P. J. Ellis, I. J. S. Fairlamb and K. Wilson, Dalton Trans., 2010, 39, 10473–10482 RSC.
- T. E. Storr, C. G. Baumann, R. J. Thatcher, S. De Ornellas, A. C. Whitwood and I. J. S. Fairlamb, J. Org. Chem., 2009, 74, 5810–5821 CrossRef CAS PubMed.
- B. Tao and D. W. Boykin, J. Org. Chem., 2004, 69, 4330–4335 CrossRef CAS PubMed.
- L. Chahen, B. Therrien and G. Süss-Fink, Eur. J. Inorg. Chem., 2007, 5045–5051 CrossRef CAS.
- A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS PubMed.
- S. E. Bajwa, T. E. Storr, L. E. Hatcher, T. J. Williams, C. G. Baumann, A. C. Whitwood, D. R. Allan, S. J. Teat, P. R. Raithby and I. J. S. Fairlamb, Chem. Sci., 2012, 3, 1656–1661 RSC.
- W. A. Carole and T. J. Colacot, Chem. – Eur. J., 2016, 22, 7686–7695 CrossRef CAS PubMed.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef.
- D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed.
- K. Mizushige, T. Ueda, K. Yukiiri and H. Suzuki, Cardiovasc. Drug Rev., 2002, 20, 163–174 CrossRef CAS PubMed.
- To obtain anhydrous potassium hexacyanoferrate, the hydrate salt was heated to 80 °C under high vacuum for 3 h (0.1 mmHg), i.e. until the vacuum remained stable, this was then confirmed as anhydrous by complete mass loss of 3H2O and stored under argon.
- S. Kozuch and J. M. L. Martin, ACS Catal., 2012, 2, 2787–2794 CrossRef CAS.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed.
- M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936–938 CrossRef CAS PubMed.
- L. P. C. Nielsen, C. P. Stevenson, D. G. Blackmond and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 1360–1362 CrossRef CAS PubMed.
- E. H. P. Tan, G. C. Lloyd-Jones, J. N. Harvey, A. J. J. Lennox and B. M. Mills, Angew. Chem., Int. Ed., 2011, 50, 9602–9606 CrossRef CAS PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef PubMed.
- G. Kemmer and S. Keller, Nat. Protoc., 2010, 5, 267–281 CrossRef CAS PubMed.
- E. Bayram, J. C. Linehan, J. L. Fulton, J. A. S. Roberts, N. K. Szymczak, T. D. Smurthwaite, S. Özkar, M. Balasubramanian and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 18889–18902 CrossRef CAS PubMed.
- E. Bayram and R. G. Finke, ACS Catal., 2012, 2, 1967–1975 CrossRef CAS.
- T. Rosner, J. Le Bars, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 1848–1855 CrossRef CAS PubMed.
- I. J. S. Fairlamb, A. R. Kapdi, A. F. Lee, G. Sánchez, G. López, J. L. Serrano, L. García, J. Pérez and E. Pérez, Dalton Trans., 2004, 3970–3981 RSC.
- A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS PubMed.
- J. G. A. de Vries, Dalton Trans., 2006, 421–429 RSC.
- P. J. Ellis, I. J. S. Fairlamb, S. F. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820–1824 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, X-ray details, results of all kinetic studies and characterization data accompanies this paper. See DOI: 10.1039/c8re00178b |
| ‡ The methods used for determining turnover numbers (TONs)/turnover frequencies (TOFs) are detailed in the viewpoint paper reported by Kozuch and Martin.36 The TOF errors were calculated using the gradient of TON vs. time during the period where the reactions were at their optimum rates. |
| This journal is © The Royal Society of Chemistry 2019 |