
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible homolytic activation of water via metal–ligand cooperativity in a T-shaped Ni(II) complex†
Mu-Chieh
Chang‡
,
Kate A.
Jesse
,
Alexander S.
Filatov
and
John S.
Anderson
 *
*
Department of Chemistry, The University of Chicago, Chicago, Illinois 60637, USA. E-mail: jsanderson@uchicago.edu
First published on 6th November 2018
Abstract
A T-shaped Ni(II) complex [Tol,PhDHPy]Ni has been prepared and characterized. EPR spectra and DFT calculations of this complex suggest that the electronic structure is best described as a high-spin Ni(II) center antiferromagnetically coupled with a ligand-based radical. This complex reacts with water at room temperature to generate the dimeric complex [Tol,PhDHPy]Ni(μ-OH)Ni[Tol,PhDHPyH] which has been thoroughly characterized by SXRD, NMR, IR and deuterium-labeling experiments. Addition of simple ligands such as phosphines or pyridine displaces water and demonstrates the reversibility of water activation in this system. The water activation step has been examined by kinetic studies and DFT calculations which suggest an unusual homolytic reaction via a bimetallic mechanism. The ΔH‡, ΔS‡ and KIE (kH/kD) of the reaction are 5.5 kcal mol−1, −23.8 cal mol−1 K−1, and 2.4(1), respectively. In addition to the reversibility of water addition, this system is capable of activating water towards net O-atom transfer to substrates such as aromatic C–H bonds and phosphines. This reactivity is facilitated by the ability of the dihydrazonopyrrole ligand to accept H-atoms and illustrates the utility of metal ligand cooperation in activating O–H bonds with high bond dissociation energies.
Introduction
Water is an attractive reagent for chemical transformations due to its ubiquity, low cost, and low toxicity. Indeed, many syntheses use water as a key feedstock, reagent, or solvent. Selected examples include the water–gas shift reaction for industrial H2 production, aldehyde–water shift reactions for H2 and carboxylic acid production, hydration reactions for functionalization of unsaturated organics, and water splitting for O2 and H2 generation.1 The cleavage of the O–H bonds in water is typically a central feature of these reactions. While the transfer of protons to or from water or alcohols is a relatively facile process,2 the homolytic activation of water to generate H-atom or O-atom equivalents is substantially more challenging due to the high bond dissociation energies (BDEs) of O–H bonds as compared to organic substrates (119 kcal mol−1 for water).3 Oxidative addition is a powerful tool for net homolytic activation of chemical bonds in homogeneous systems; however, examples of O–H bond oxidative addition are uncommon due largely to the unfavorable thermodynamics mentioned above.4 This challenge motivates studies aimed at the mild activation of water in a homolytic fashion.As a potential strategy to address this issue, there have recently been numerous reports demonstrating that transition metal coordination can promote the homolysis of N–H or O–H bonds.5 For example, the BDE of water can be lowered by >50 kcal mol−1 when ligated to centers such as Ti(III) or [Ga2Mg2O5˙]+.6 We have been interested in integrating both redox non-innocence and pendant proton relays into ligand scaffolds.7 This approach should be kinetically advantageous for the homolytic activation of E–H bonds as the H-atom abstracting agent is chelated to the transition metal. Storage of an H-atom on the ligand periphery also provides an attractive strategy for the mild and reversible homolytic activation of water by substituting a comparatively weak M–H bond with a ligand–H bond that may be closer in strength to an O–H bond. There have been several well-defined examples where redox-activity and ligand-based protonation sites have been combined in a single scaffold,8 but this strategy has been under-utilized for the activation of O–H bonds.
We recently reported that complexes of Ni bound by the dihydrazonopyrrole Tol,PhDHPyH3 (1, Tol,PhDHPyH3 = 2,5-bis((2-phenylhydrazono)(p-tolyl)methyl)-pyrrole) can reversibly store H-atom equivalents at a pendant ligand site.7a We rationalized that this system would be attractive for the activation of E–H bonds, and those in water in particular. While some Ni complexes have been shown to activate water,9 the ability of the dihydrazonopyrrole ligand to accept H-atom equivalents should enable a low-energy and hence reversible homolytic process. Herein we report the synthesis and characterization of a T-shaped Ni(II) complex and its reversible activation of water. Kinetic, structural, and computational analysis supports a bimolecular activation mechanism with movement of both protons and electrons via net H-atom transfer. The resulting activated complex shows unusual reactivity with O-atom acceptors including C–H bonds and phosphines.
Results and discussion
Synthesis of T-shaped nickel complexes
The synthesis of [Tol,PhDHPyH]Ni(Py) (2-Py; Py = pyridine, Scheme 1) was carried out in an analogous manner to our previously reported procedure for preparing [Tol,PhDHPyH]Ni(PMe3) (2-PMe3) by sequential deprotonation and metalation of the pre-ligand 1.7a The formation of 2-Py was confirmed by 1H NMR spectroscopy which shows two sets of resonances assigned to the pyrrole backbone and two sets of resonances assigned to the p-tolyl functional group arising from asymmetric ligand binding (Fig. S1†). In addition, a resonance located at 5.17 ppm is assigned to the NH functional group. This NH functionality is also confirmed by a peak at 3343 cm−1 in the infrared (IR) spectrum of 2-Py (Fig. S19†). The dihydrazonopyrrole ligand in 2-Py coordinates to the Ni center with one pyrrole nitrogen, one α hydrazone nitrogen, and one β hydrazone nitrogen, resulting in one five- and one six-membered chelate ring as determined by single crystal X-ray diffraction (SXRD, Fig. 1).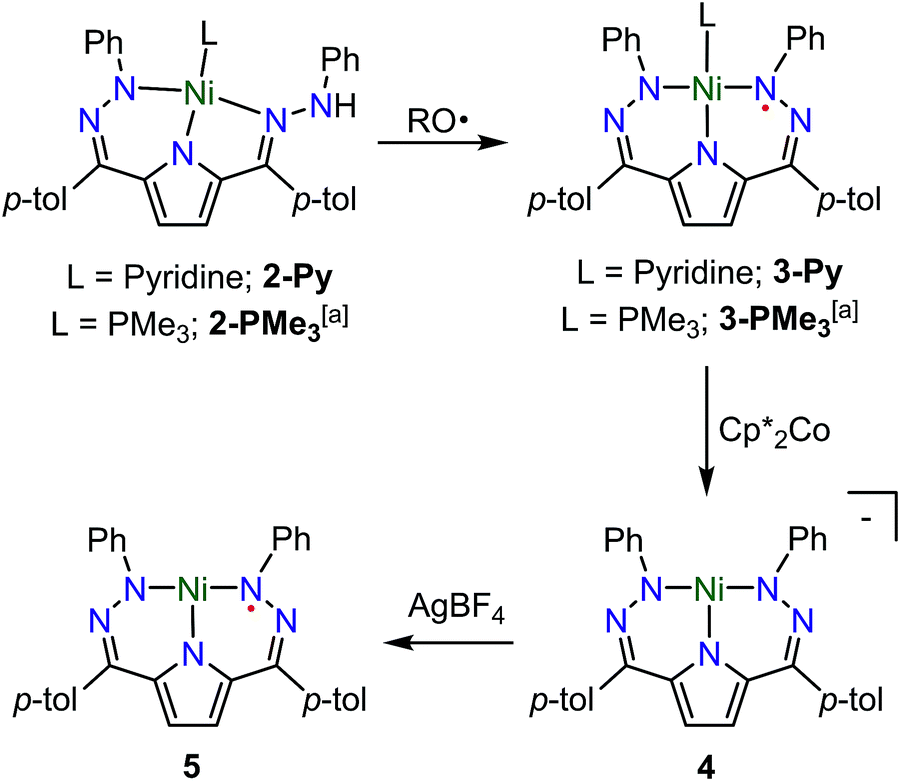 | ||
| Scheme 1 Synthesis of complexes 2–5. RO˙ = 2,4,6-tri-tert-butylphenoxy radical. a Previous work.7a | ||
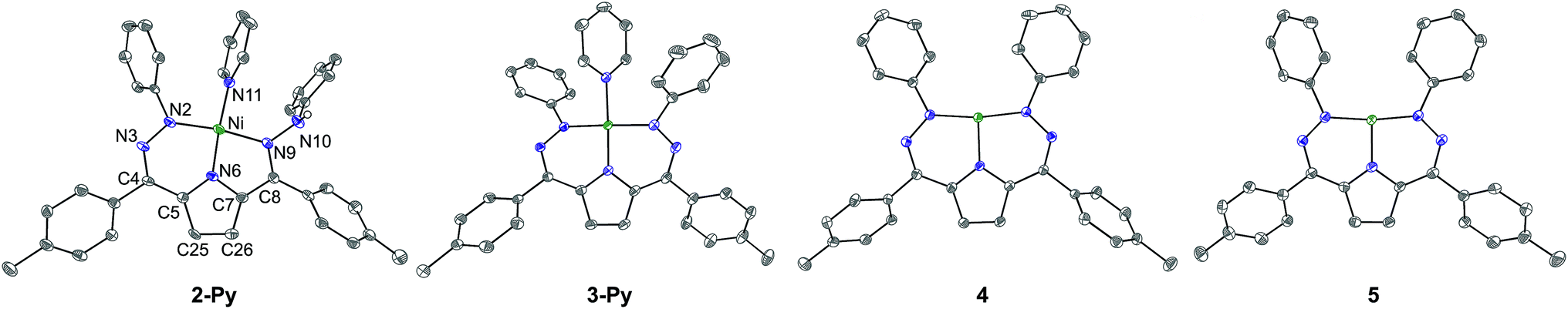 | ||
| Fig. 1 Crystal structures of 2-Py, 3-Py, 4, and 5. Ellipsoids are shown at 50% and hydrogen atoms, solvent molecules, and counterions have been omitted for clarity (with the exception of the N–H in 2-Py which was located in the difference map). C atoms shown in gray, N in blue, and Ni in green. The labeling scheme for all complexes is as shown for 2-Py. | ||
Similarly to the transformation from 2-PMe3 to [Tol,PhDHPy˙]Ni(PMe3) (3-PMe3), [Tol,PhDHPy˙]Ni(Py) (3-Py) can be prepared by abstracting a H-atom from 2-Py with 2,4,6-tri-tert-butylphenoxy radical with an isolated yield of 85% (Scheme 1, Fig. S3†). Electron paramagnetic resonance (EPR) spectroscopy confirms that 3-Py possesses an S = 1/2 ground state with a signal at g = 2.01 (Fig. S28 and 29†), which is again similar to 3-PMe3 and supports the assignment of ligand-based radical character in 3-Py. The formation of 3-Py has also been confirmed by SXRD (Fig. 1, Table 1). The metrical parameters of both 2-Py and 3-Py are comparable to the previously reported phosphine ligated congeners and furthermore support the ability of this system to store H-atom equivalents via a combination of pendant protonation sites and redox activity.
| 2-Py | 3-Py | 4 | 5 | |
|---|---|---|---|---|
| a Only one of the two independent molecules are listed. b Ni1–N9. c N2–Ni1–N9. | ||||
| Ni–N2 | 1.868(2) | 1.889(1) | 1.858(5) | 1.844(2) |
| Ni–N6 | 1.807(2) | 1.836(1) | 1.869(5) | 1.877(3) |
| Ni–N10 | 1.924(2)b | 1.883(2) | 1.858(4) | 1.848(3) |
| Ni–N11 | 1.902(2) | 1.921(1) | — | — |
| N2–N3 | 1.356(3) | 1.343(1) | 1.370(6) | 1.324(4) |
| N3–C4 | 1.315(4) | 1.322(2) | 1.314(7) | 1.337(3) |
| C25–C26 | 1.376(5) | 1.369(2) | 1.395(7) | 1.353(5) |
| C8–N9 | 1.334(4) | 1.322(2) | 1.317(7) | 1.342(5) |
| N9–N10 | 1.412(3) | 1.342(2) | 1.366(7) | 1.323(3) |
| N2–Ni1–N10 | 171.1(1)c | 177.18(6) | 170.5(2) | 171.8(1) |
| N2–Ni1–N6 | 89.5(1) | 89.31(6) | 94.7(2) | 94.0(1) |
| N10–Ni1–N6 | 82.0(1) | 88.97(6) | 94.8(2) | 94.1(1) |
We also probed whether the pyridine ligated system would support other ligand based redox events. When 3-Py was reduced by Cp*2Co (Cp* = pentamethylcyclopentadienyl; Scheme 1), the color of the reaction mixture immediately changed from deep green to deep red, which is similar to the color of [3-PMe3][Cp*2Co] potentially suggesting the formation of [3-Py][Cp*2Co].7a When we attempted to purify this putative complex by recrystallization, however, an unusual T-shaped complex [[Tol,PhDHPy]Ni][Cp*2Co] was isolated as the product (4, Scheme 1, Fig. 1, Table 1). Concurrently, we also noted the formation of 4 as an additional product formed from crystallizations of [3-PMe3][Cp*2Co]. The T-shaped geometry in 4 is unexpected as structurally rigid supporting ligands with bulky substituents are typically required to prevent the formation of Y-shaped or four-coordinate metal centers. Most reported T-shaped Ni complexes have Ni(I) centers instead of Ni(II),9b,10 however a Ni(I) coupled to a radical ligand in 4 is unlikely. Changes in the ligand bond lengths, magnetic moment, and density functional theory (DFT) calculations are all consistent with ligand based reduction resulting in a high-spin Ni(II) center coordinated by a trianionic dihydrazonopyrrole ligand (Fig. S34 and S43†). The high-spin nature of 4 is likely due to the lowering of the dx2−y2 orbital in a T-shaped geometry.
The reaction of deep green 4 with one equivalent of AgBF4 results in the formation of the deep blue complex [Tol,PhDHPy˙]Ni (5, Scheme 1). The crystal structure of 5 shows a T-shaped geometry as well (Fig. 1, Table 1) and the 1H NMR spectrum of 5 shows five paramagnetic resonances ranging from 12 to 2 ppm (Fig. S4†). Unlike 3-Py, the EPR spectrum of 5 displays a signal at g = 2.15 (Fig. S30–32†), which suggests that 5 is an S = 1/2 system but is not a simple ligand centered radical. The electronic structure of 5 was further clarified by DFT calculations, which suggest that 5 has an S = 1 Ni(II) center antiferromagnetically coupled with a ligand-based S = 1/2 radical ([Tol,PhDHPy˙]−2) (Fig. S43†) resulting in an overall S = 1/2 complex with a radical feature shifted from g = 2 in its EPR spectrum.11 Therefore complex 5 is best thought of as a ligand radical coupled to a high-spin Ni(II) center where the oxidation of 4 has occurred on the ligand.
In both 4 and 5, the N–Ni–N angles are ∼170°, 95° and 95° indicating nearly perfect T-shaped structures for these complexes (Table 1). The observed geometry of 4 and 5 is likely due to the sterics imposed by the six-membered chelate rings in the planar conjugated Tol,PhDHPy ligand but may also arise from more subtle electronic effects. Our previous studies on 3-PMe3 and the CV of 5 (Fig. S35†) suggest that complex 5 may be oxidized again to generate a cationic complex of the form [Tol,PhDHPy]Ni+. However, attempts to chemically oxidize 5 to generate a putative T-shaped cationic complex have thus far proven unsuccessful and typically result in the formation of anion or solvent bound products. Despite this, complexes 4 and 5 represent rare examples of T-shaped complexes with unusual electronic structures and enabled us to undertake further studies on substrate activation in this system.
Water activation
The open coordination site at the Ni center of 4 and 5 provides a unique opportunity to study the reactivity of this system towards small molecule activation. The ionic character and packing of 4 leads to low solubility in common organic solvents. We therefore have focused our initial efforts on the more soluble complex 5. We have observed that complex 5 reacts cleanly with excess water at room temperature resulting in a new purple hydroxide-bridged dimeric complex [Tol,PhDHPy]Ni(μ-OH)Ni[Tol,PhDHPyH] (6; Scheme 2 top) within 30 minutes. The dimeric structure of 6 in solid and solution states was confirmed by SXRD and NMR spectroscopy, respectively.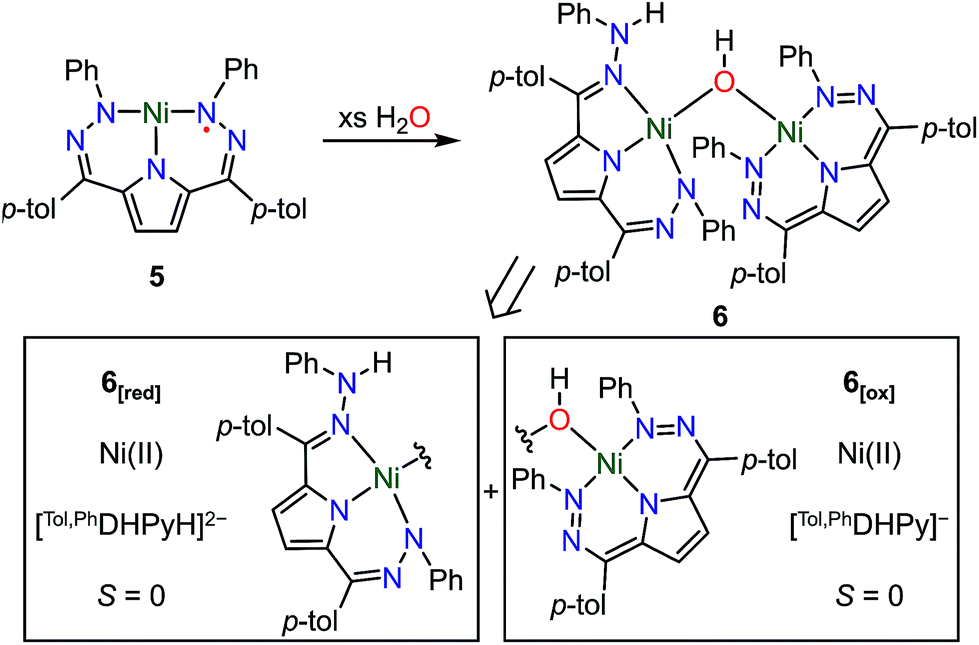 | ||
| Scheme 2 Top: Synthesis of [Tol,PhDHPy]Ni(μ-OH)Ni[Tol,PhDHPyH] (6) and Bottom: Constituent oxidized (6[ox]) and reduced (6[red]) halves of the dimer. | ||
The crystal structure of 6 (Fig. 2, Table 2) shows a dimeric structure with two Ni centers bridged by a hydroxide anion and differing coordination modes for each Tol,PhDHPy ligand. The Ni–O bond lengths are 1.923(4) and 1.891(3) Å and the Ni–O–Ni bond angle is 135.7(2)°. For the purposes of clarity, we will discuss this dimer in terms of its two halves (Scheme 2). One of the Tol,PhDHPy ligands binds in an asymmetric manner. We denote this half of the molecule as 6[red] and assign it as a [Tol,PhDHPyH]2− ligand bound to a Ni(II) center. The other half of the molecule, denoted 6[ox], features the ligand bound in a symmetric manner and we assign this half as a [Tol,PhDHPy]− ligand bound to Ni(II). The 6[ox] fragment uses two β hydrazone nitrogens to coordinate the Ni center resulting in two six-membered chelate rings. In the case of the 6[red] fragment, the dihydrazonopyrrole ligand binds through one α hydrazone nitrogen and one β hydrazone nitrogen. These formal oxidation states imply that the net reaction to form 6 involves transfer of one H-atom to a Tol,PhDHPy ligand to generate 6[red]. The balance of this reaction requires that the resulting formal hydroxyl radical binds to Ni and is reduced by the other Tol,PhDHPy radical to generate the 6[ox] fragment and subsequently dimeric 6. It is unlikely that the reaction proceeds with this exact mechanism (see below), but this formal accounting of atoms and electrons illustrates the net homolytic cleavage of water that occurs upon formation of 6.
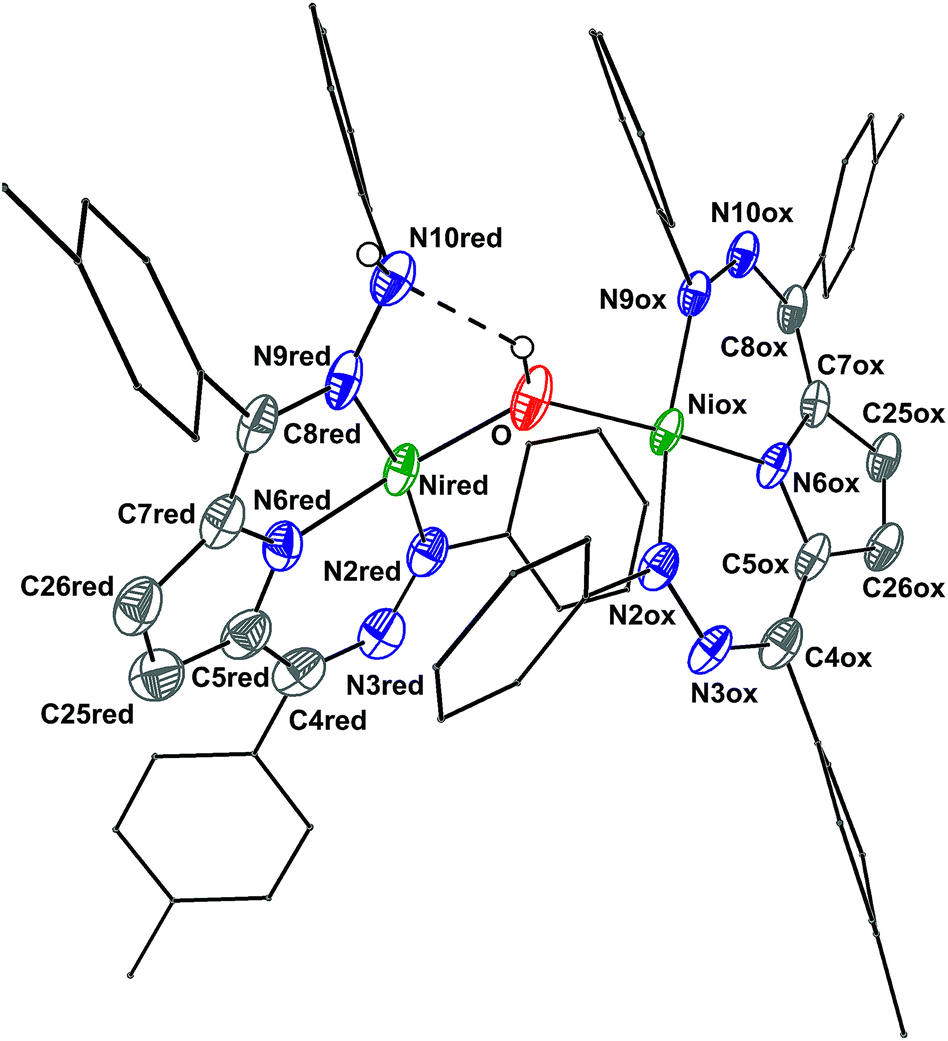 | ||
| Fig. 2 Crystal structure of 6. Ellipsoids are shown at 50% except for aryl rings which are shown in wireframe. Hydrogen atoms bound to C have been omitted for clarity, and only one of the two molecules in the asymmetric unit is shown. The dashed line indicates a hydrogen bonding interaction. C atoms shown in gray, N in blue, and Ni in green. See Table 2 for bond distances and angles. | ||
| 6[red] | 6[ox] | [Tol,PhDHPy]Ni(PMe3)+ | |
|---|---|---|---|
| Ni–O | 1.925(3) | 1.895(3) | — |
| Ni–N2 | 1.897(4) | 1.888(4) | 1.864(2) |
| Ni–N6 | 1.835(4) | 1.864(3) | 1.860(2) |
| Ni–N10 | 1.908(4) (N9) | 1.880(4) | 1.869(2) |
| N2–N3 | 1.369(6) | 1.301(5) | 1.302(2) |
| N3–C4 | 1.332(6) | 1.367(5) | 1.348(2) |
| C25–C26 | 1.385(6) | 1.342(7) | 1.346(3) |
| C8–N9 | 1.362(6) | 1.373(6) | 1.342(3) |
| N9–N10 | 1.422(5) | 1.311(5) | 1.314(2) |
| N2–Ni–N10 | 168.2(2) (N9) | 163.4(2) | 165.85(7) |
| N2–Ni–N6 | 89.7(2) | 91.5(2) | 92.65(7) |
| N10–Ni–N6 | 81.8(2) (N9) | 90.5(2) | 89.24(7) |
| Ni–O–Ni | 135.8(2) | 135.8(2) | — |
The metrical parameters in 6 lend support to the assigned oxidation state of the two halves. In 6[ox], the average lengths of the N–N bonds and the C25–C26 bond are 1.306 and 1.342 Å, respectively (Table 2). These bond lengths are similar to the previously reported cationic complex [Tol,PhDHPy]Ni(PMe3)+ (Table 2) suggesting that this half of the complex is best described as a monoanionic ligand ([Tol,PhDHPy]−). In the case of 6[red], the N2–N3 and N9–N10 bond lengths are similar with 2-Py and 2-PMe3 suggesting that this unit is a protonated trianionic ligand ([Tol,PhDHPyH]2−). These bond lengths are different from those in 5 ([Tol,PhDHPy˙]2−) demonstrating different formal ligand oxidation states and supporting that a redox process has occurred in the conversion from 5 to 6.
The O–H and N–H protons in 6 were located in the difference map. Notably, the O–H proton is directed towards the protonated N on the [Tol,PhDHPyH]2− ligand. The O–H⋯N distance is ∼2.2 Å and the O–N distance is ∼2.83 Å. These distances support the presence of a hydrogen bond in 6.12 Both the O–H and the N–H protons can also be observed spectroscopically. The 1H NMR spectrum of 6 (Fig. S6†) shows two sharp singlet resonances at 5.07 and −4.36 ppm. The former signal is close to the NH resonance of 2-Py (5.17 ppm, Fig. S1†) and we have assigned it as the NH group on the 6[red] portion of the dimer. The resonance at −4.36 ppm is similar to other reported Ni–OH species9a,13 and more downfield than reported Ni–H species;14,15 we therefore assign it to the bridging hydroxide functional group in 6. When 6 is generated with D2O both of these signals disappear suggesting that the NH and OH functional groups in 6 are transferred from water molecules (Fig. S5†). In addition, the presence of the NH and OH functional group were further confirmed by IR spectroscopy (NH/D: 3321/2460; OH/D: 3594/2651 cm−1, Fig. S23†). Finally, the NOESY spectrum of 6 shows a cross peak correlation between the NH and OH signals suggesting that the dimeric structure of 6 is maintained in solution (Fig. S7†). The methyl proton resonances of the p-tolyl groups in the aliphatic region show an integral ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:3 which also supports this assignment.
:6:3 which also supports this assignment.
Mechanism of water activation
The unusual activation of water to form 6 prompted us to examine this reaction in greater detail. Complexes 2-Py, 3-Py, 5, and 6 all have different colors and UV-vis spectra (see Fig. S24–S27†). The distinct difference in absorbance features between 5 and 6 provides a useful tool to examine the activation of H2O. We initially note that purified complex 6 is not stable in THF solution; the purple color of 6 turns to a dark blue reminiscent of solutions of 5 within 30 minutes at room temperature. We propose that this color change is due to competitive THF binding and reversal of the water activation process.When water is added to a solution of 5 a color change is observed with new absorption bands located at 594 and 644 nm. Both of these features are very similar to the absorption spectra of 3-Py (592 and 650 nm, Fig. S25†) and previously reported 3-PMe3 (585 and 645 nm).7a We hypothesized that a water molecule may occupy the open coordination site in 5 resulting in a water adduct (3-H2O, Scheme 3). The tentative assignment of 3-H2O as an intermediate species is supported by EPR spectroscopy where the addition of water to 5 results in the loss of the signal at g = 2.15 and the appearance of a new signal at g = 2.01 (Fig. S33†). This shift is consistent with the shift observed upon binding of pyridine to form 3-Py and supports a similar water bound adduct. The EPR features of 3-H2O suggest that it has a low-spin Ni(II) center coordinated by a radical ligand analogous to 3-Py. 1H NMR spectroscopy further supports this tentative assignment of 3-H2O as a new paramagnetic feature similar to that observed for 3-Py is observed when 5 is mixed with excess water (Fig. S5†).
 | ||
| Scheme 3 Proposed mechanism of water activation by 5 and calculated reaction free energies (kcal mol−1) of different activation pathways. Note that Ar is the same for all complexes as shown for 3-H2O. | ||
To avoid complications from equilibria or competitive ligand binding we performed kinetic studies by examining the initial rates of formation of 6 (Fig. 3, Table S4†). We undertook these studies by reacting 5 with excess water in THF and then monitoring the reaction for 15–20 minutes by UV-vis spectroscopy (Fig. 3). By conducting kinetic studies at varied concentration and temperature, we have determined that the formation of 6 is second-order in [Ni] with an observed rate constant (kobs) of 3.4(6) × 103 M−2 s−1. The ΔH‡ and ΔS‡ for this reaction are 5.5 kcal mol−1 and −23.8 cal mol−1 K−1, respectively. Using D2O as the substrate reveals a kinetic isotopic effect (KIE) of kH/kD = 2.4(1). The second-order kinetics and negative value of the entropy of activation demonstrate that the water activation step is a bimolecular process.
 | ||
| Fig. 3 UV-vis kinetic trace of the reaction of 5 with excess H2O at room temperature. Each line represents a one-minute increment. The inset shows a plot of the dependence of rate on temperature from which the activation parameters have been obtained. | ||
In addition to the experimental approaches mentioned above, DFT calculations were used to further understand the details of the water activation promoted by 5 to generate 6 (Scheme 3). The DFT calculations reveal that generation of the water adduct 3-H2O from 5 and water is energetically downhill (−1.2 kcal mol−1) suggesting water coordination is the first step of the reaction. This step is also consistent with our observations by UV-vis, EPR, and 1H NMR spectroscopy. The calculated spin density of 3-H2O further supports our assignment that 3-H2O has a low-spin Ni(II) center coordinated by a radical ligand also as suggested by EPR spectroscopy. Subsequent to water coordination, the net formation of 6 from two molecules of 3-H2O is also energetically favorable (−0.8 kcal mol−1). The small energy difference between 3-H2O and 6 suggests that the water activation might be reversible which is also consistent with our experimental observations.
Concerted formation of 6 from two equivalents of 3-H2O seems unlikely so we also have used DFT calculations to examine the feasibility of other reaction intermediates (Scheme 3). The observed 2nd order dependence on [Ni] led us to investigate three bimolecular reactions: heterolytic proton transfer, bimolecular oxidative addition, and homolytic H-atom transfer. Bridging aquo complexes are an additional possibility, but we have been unable to locate any minima in the optimizations of these types of species. In addition to these bimolecular pathways we have also considered an intermediate wherein intramolecular H-transfer from water to a pendant N has occurred as well as the transition state for this process. This intermediate is 4.2 kcal mol−1 uphill in energy and the transition state is 17.7 kcal mol−1 higher in energy than 3-H2O (Table S6†). These values are substantially higher than those observed experimentally leading us to consider this pathway as unlikely.
The net products from heterolytic proton transfer between two equivalents of 3-H2O, [[Tol,PhDHPyH˙]Ni(H2O)]+ and [[Tol,PhDHPy˙]Ni(OH)]−, are calculated to be 73.7 kcal mol−1 uphill in energy from 3-H2O. Similarly, the expected products from bimolecular oxidative addition, [Tol,PhDHPy]Ni(OH) and [Tol,PhDHPy]Ni(H), are also high in energy at 24.5 kcal mol−1. The high calculated reaction energies of these two putative pathways are inconsistent with the fast rate of reaction we observe at room temperature or the experimentally measured activation parameters. Our previous studies suggest that complexes of the form [[Tol,PhDHPyH˙]Ni(L)]+ are not stable and will decompose into [[Tol,PhDHPy]Ni(L)]+ by formal loss of an H-atom which also argues against a proton transfer pathway.7a
Unlike the two pathways mentioned above, intermediates along a homolytic pathway are calculated to be energetically accessible. The reaction of a molecule of 3-H2O to formally abstract a H-atom from another molecule of 3-H2O to generate 2-H2O and [Tol,PhDHPy]Ni(OH) is only 1.2 kcal mol−1 uphill. Subsequent dimerization with loss of water to form 6 is then only 2.0 kcal mol−1 downhill which suggests that fragmentation of 6 in the presence of excess water or another ancillary ligand is energetically reasonable. The low energies of these proposed intermediates are consistent with the reversibility of this process in the presence of additional ligands or coordinating solvents.
The exact transition state for the formation of 6 is currently unknown. We have attempted to locate transition state structures computationally but have thus far been unsuccessful (Fig. S44†). Experimentally, the reaction is 2nd order in [Ni] and displays a negative entropy of activation. Both of these facts strongly support a bimolecular transition state. The KIE value of 2.4(1) also suggests that the transfer of hydrogen is involved in the transition state. The magnitude of the KIE is consistent with a PCET process, however, it is difficult to definitively interpret the KIE as tunneling character and proton transfer distance can dramatically impact the magnitude of this value.16 DFT calculations also support a bimolecular homolytic process as more classic proton transfer or oxidative addition intermediates are calculated to be high in energy. The net products of homolytic activation are very low in energy (1.2 kcal mol−1) and are likely in equilibrium with 6. Regardless, all of the experimental and computational data supports a bimolecular transition state involving hydrogen transfer in the rate determining step. What these combined studies suggest is that the activation of water by 5 may best be described as a homolytic process wherein both proton and electron transfer is required for facile reactivity.
Reactivity of 6
We have also examined the reactivity of this unusual complex with several different reagents (Scheme 4). We observe a color change from purple to green when complex 6 is treated with pyridine. The generation of water and 3-Py as the major Ni-containing product were supported by 1H NMR (Fig. S12†). The formation of 3-Py with concomitant extrusion of water shows that the water activation reaction to form 6 is reversible. When 6 is reacted with trimethylphosphine (PMe3), a similar reaction is observed where 6 is converted into 3-PMe3. We also noted, however, the formation of 2-PMe3 and OPMe3 although the yields of these species were small and variable (Fig. S13–14†). This reaction suggests that complex 5 can formally homolytically split water into H-atom and O-atom equivalents. The H-atom equivalents are trapped by the Tol,PhDHPy ligand and the O-atom equivalents can be trapped by phosphines. We postulated that the variable amounts of H- and O-atom transfer when using PMe3 as an O-atom acceptor are due to competitive formation of 3-PMe3 arising from the strong donor properties and small size of PMe3. We therefore investigated triphenylphosphine (PPh3) as an O-atom acceptor which would be less likely to coordinate to the Ni center.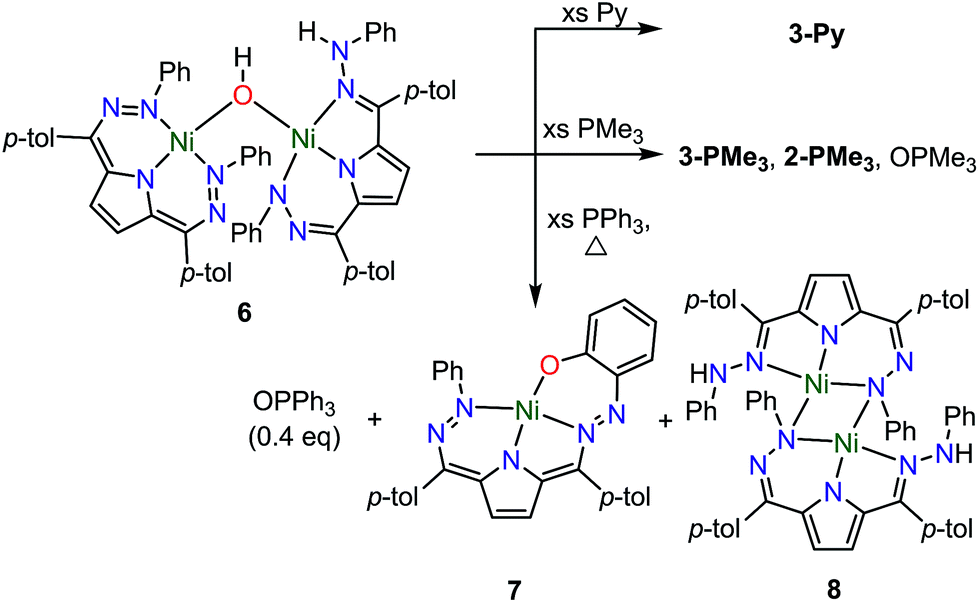 | ||
| Scheme 4 Reactivity of 6. | ||
The reaction of 6 and PPh3 was sluggish at room temperature and was therefore carried out at higher temperature. When 6 and PPh3 were heated to 85 °C in toluene overnight, ∼0.4 equivalents of PPh3 were oxidized to OPPh3 as observed by 31P NMR (Fig. S18†). The source of the O in the OPPh3 product was confirmed as water by mass spectrometry of the reaction mixture of PPh3 and 6 generated from H218O (Fig. S36†). The 1H NMR spectrum of the crude reaction mixture shows the formation of multiple species resulting from the decomposition of 6, from which the previously reported C–H activated product 7 and ([Tol,PhDHPyH]Ni)2 (8, Scheme 4) were identified as two major species (Fig. S15 and S17†).7a While we have not been able to generate 8 in bulk, the identity of this complex was confirmed by SXRD and 1H NMR analysis on a small amount of crystalline material isolated from this reaction mixture (Fig. 4, Fig. S9†). Complex 8 is also observed when 5 is reacted with H2 at 85 °C in C6D6 (Fig. S10†).
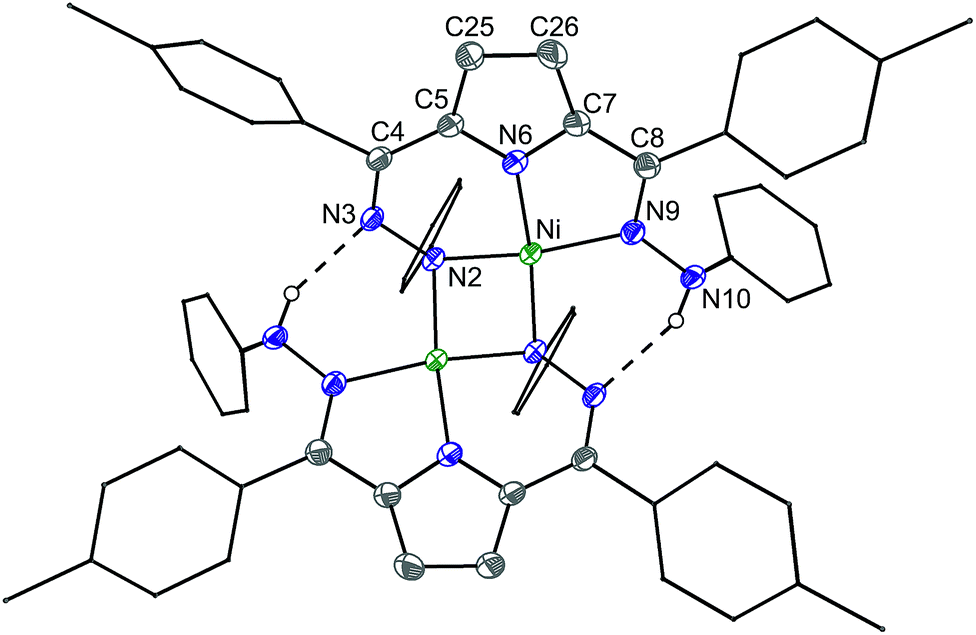 | ||
| Fig. 4 Crystal structure of 8. Note that the dimeric structure is shown although the asymmetric unit only contains one Ni center. Ellipsoids are shown at 50% except for aryl rings which are shown in wireframe. Hydrogen atoms bound to C have been omitted for clarity. Dashed lines indicate a hydrogen bonding interaction. C atoms shown in gray, N in blue, and Ni in green. Selected bond distances (Å) and angles (°) (* indicates a bond to the symmetric equivalent): Ni–N2 = 1.908(1), Ni–N6 = 1.813(1), Ni–N9 = 1.929(1), Ni–Ni* = 2.624(5), Ni–N2* = 1.929(1), N2–N3 = 1.451(1), N3–C4 = 1.302(2), C25–C26 = 1.453(2), C8–N9 = 1.317(2), N9–N10 = 1.413(1), N10–H⋯N3* = 2.02(2), N3–N10* = 2.815(2), N2–Ni–N10 = 171.45(5), N2–Ni–N6 = 91.41(5), N10–Ni–N6 = 82.13(5). | ||
A general mechanistic scheme that explains this observed reactivity is shown in Scheme 5. An equilibrium between 6 and 3-H2O/3-L is likely. Simple ligand substitution of 3-H2O with L would then result in the observed products 3-L (for L = PMe3 and Py). Alternatively, 6 may react to transfer its O-atom. This O-atom equivalent may be transferred to a suitable substrate such as a phosphine. In the absence of a substrate, or with a less electron rich substrate, C–H activation on the ligand to form 7 is competitive. We anticipate that this ligand activation process is sluggish, and only occurs under forcing conditions. Formal O-atom transfer from 6 would result in the formation of two equivalents of [Tol,PhDHPyH]Ni which could then be trapped by incoming L to form two equivalents of 2-L. In the absence of an incoming ligand, or if the incoming ligand is too large to bind effectively, the [Tol,PhDHPyH]Ni fragment may dimerize to form 8. This simple mechanistic scheme illustrates that the equilibrium of 6 sets up two competing reaction pathways, either ligand exchange or oxidation. The relative preference for each of these pathways will then depend on the properties of the added ligand, specifically its ability to bind or be oxidized. Relatively weakly reducing ligands, such as pyridine, will favor binding and displacement of water. Strongly binding and reducing ligands such as PMe3 will lead to both oxidation and water displacement to form a mixture of OPMe3, 2-PMe3, and 3-PMe3. Bulky ligands like PPh3 which are unlikely to bind due to steric constraints will only be oxidized. However, this oxidation is slow and C–H activation is competitive, resulting in the observation of OPPh3, 7, and 8. Heating in the absence of substrate can only result in C–H activation to form 7 and 8. This simple mechanistic picture is consistent with the observed product selectivity and explains the reactivity of 6.
 | ||
| Scheme 5 Proposed mechanisms for the reactivity of 6. L = phosphines or pyridine as shown in Scheme 4. | ||
These reactivity studies establish three important points. Firstly, the activation of water by 5 is reversible with the addition of exogenous ligands. Secondly, complex 5 is capable of net H-atom abstraction from water. Finally, the O-atom equivalent generated from this net H-atom transfer is competent to oxidize substrates such as phosphines and C–H bonds.
Conclusions
Dihydrazonopyrrole complexes of Ni can support reversible ligand-based storage of H-atoms. The reactivity of this system, however, was previously limited by coordinative saturation at the metal center. In this report, we have shown that we can isolate unusual T-shaped dihydrazonopyrrole Ni complexes. The open coordination site of these species enables a rare reversible activation of water via metal ligand cooperation. All of the evidence supports that this activation process is homolytic with the net reaction being a H-atom transfer from water to the dihydrazonopyrrole ligand. The resulting hydroxyl radical equivalent is formally reduced to hydroxide by the second Ni complex. This activation is notable due to the high BDE of the O–H bonds in water and illustrates the utility of metal ligand cooperativity in mediating thermodynamically challenging bond activations. Furthermore, the reactivity of the activated complex indicates that a classically benign reagent, water, can be activated to mediate phosphine or C–H oxidations. The reactivity reported here motivates studies aimed at related, potentially catalytic, transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Chicago for funding. We also thank the Research Computing Center at the University of Chicago for providing computing resources. ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation (NSF), under Grant No. NSF/CHE1346572. Use of the PILATUS3 X CdTe 1M detector is supported by the National Science Foundation under the grant number NSF/DMR-1531283. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DEAC02-06CH11357.Notes and references
- (a) G. G. Stanley, D. A. Aubry, N. Bridges, B. Barker and B. Courtney, Prepr. Pap.–Am. Chem. Soc., Div. Fuel Chem., 2004, 49, 712–713 CAS; (b) R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS PubMed; (c) L. Hintermann and A. Labonne, Synthesis, 2007, 2007, 1121–1150 CrossRef; (d) C. F. Nising and S. Bräse, Chem. Soc. Rev., 2008, 37, 1218 RSC; (e) M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed; (f) T. P. Brewster, W. C. Ou, J. C. Tran, K. I. Goldberg, S. K. Hanson, T. R. Cundari and D. M. Heinekey, ACS Catal., 2014, 4, 3034–3038 CrossRef CAS; (g) T. P. Brewster, J. M. Goldberg, J. C. Tran, D. M. Heinekey and K. I. Goldberg, ACS Catal., 2016, 6, 6302–6305 CrossRef CAS.
- For some examples featuring ligand protonation see: (a) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (b) C. C. Comanescu and V. M. Iluc, Organometallics, 2014, 33, 6059–6064 CrossRef CAS; (c) K.-N. T. Tseng, J. W. Kampf and N. K. Szymczak, ACS Catal., 2015, 5, 5468–5485 CrossRef CAS; (d) A. M. Tondreau, R. Michalczyk and J. M. Boncella, Organometallics, 2017, 36, 4179–4183 CrossRef CAS; (e) C. C. Comanescu and V. M. Iluc, Polyhedron, 2018, 143, 176–183 CrossRef CAS; (f) A. J. Kosanovich, W.-C. Shih and O. V. Ozerov, Inorg. Chem., 2018, 57, 545–547 CrossRef CAS PubMed.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, 2002 Search PubMed.
- (a) G. L. Hillhouse and J. E. Bercaw, J. Am. Chem. Soc., 1984, 106, 5472–5478 CrossRef CAS; (b) M. J. Burn, M. G. Fickes, J. F. Hartwig, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 5875–5876 CrossRef CAS; (c) O. Blum and D. Milstein, J. Am. Chem. Soc., 2002, 124, 11456–11467 CrossRef CAS PubMed; (d) C. M. Fafard, D. Adhikari, B. M. Foxman, D. J. Mindiola and O. V. Ozerov, J. Am. Chem. Soc., 2007, 129, 10318–10319 CrossRef CAS PubMed; (e) O. V. Ozerov, Chem. Soc. Rev., 2009, 38, 83–88 RSC; (f) W. E. Piers, Organometallics, 2011, 30, 13–16 CrossRef CAS; (g) S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640–4650 CrossRef CAS PubMed; (h) T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef CAS PubMed.
- (a) R. T. Jonas and T. D. P. Stack, J. Am. Chem. Soc., 2002, 124, 83–96 CrossRef; (b) J. P. Roth and J. M. Mayer, Inorg. Chem., 1999, 38, 2760–2761 CrossRef CAS PubMed; (c) J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357–2367 CrossRef CAS PubMed; (d) D. P. Estes, D. C. Grills and J. R. Norton, J. Am. Chem. Soc., 2014, 136, 17362–17365 CrossRef CAS PubMed; (e) K. T. Tarantino, D. C. Miller, T. A. Callon and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 6440–6443 CrossRef CAS PubMed; (f) M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730–733 CrossRef CAS PubMed; (g) P. Bhattacharya, Z. M. Heiden, E. S. Wiedner, S. Raugei, N. A. Piro, W. S. Kassel, R. M. Bullock and M. T. Mock, J. Am. Chem. Soc., 2017, 139, 2916–2919 CrossRef CAS PubMed; (h) M. J. Bezdek and P. J. Chirik, Angew. Chem., Int. Ed., 2018, 130, 2246–2250 CrossRef.
- (a) M. Paradas, A. G. Campaña, T. Jiménez, R. Robles, J. E. Oltra, E. Buñuel, J. Justicia, D. J. Cárdenas and J. M. Cuerva, J. Am. Chem. Soc., 2010, 132, 12748–12756 CrossRef CAS PubMed; (b) J. Li, S. Zhou, X.-N. Wu, S. Tang, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2015, 54, 11861–11864 CrossRef CAS PubMed.
- (a) M.-C. Chang, A. J. McNeece, E. A. Hill, A. S. Filatov and J. S. Anderson, Chem.–Eur. J., 2018, 24, 8001–8008 CrossRef CAS PubMed; (b) A. J. McNeece, M.-C. Chang, A. S. Filatov and J. S. Anderson, Inorg. Chem., 2018, 57, 7044–7050 CrossRef CAS PubMed.
- (a) P. Chaudhuri, M. Hess, J. Müller, K. Hildenbrand, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 9599–9610 CrossRef CAS; (b) R. Hübner, S. Weber, S. Strobel, B. Sarkar, S. Záliš and W. Kaim, Organometallics, 2011, 30, 1414–1418 CrossRef; (c) F. Lu, R. A. Zarkesh and A. F. Heyduk, Eur. J. Inorg. Chem., 2012, 2012, 467–470 CrossRef CAS; (d) M. Bubrin, D. Schweinfurth, F. Ehret, S. Záliš, H. Kvapilová, J. Fiedler, Q. Zeng, F. Hartl and W. Kaim, Organometallics, 2014, 33, 4973–4985 CrossRef CAS; (e) S. P. Semproni, C. Milsmann and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 9211–9224 CrossRef CAS PubMed; (f) E. J. Thompson and L. A. Berben, Angew. Chem., Int. Ed., 2015, 127, 11808–11812 CrossRef; (g) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (h) K. T. Horak and T. Agapie, J. Am. Chem. Soc., 2016, 138, 3443–3452 CrossRef CAS PubMed; (i) J. T. Henthorn and T. Agapie, Inorg. Chem., 2016, 55, 5337–5342 CrossRef CAS PubMed; (j) T. J. Sherbow, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2017, 56, 8651–8660 CrossRef CAS PubMed; (k) G. W. Margulieux, M. J. Bezdek, Z. R. Turner and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 6110–6113 CrossRef CAS PubMed; (l) F. Schneck, M. Finger, M. Tromp and S. Schneider, Chem. - Eur. J., 2017, 23, 33–37 CrossRef CAS PubMed; (m) K. E. Rosenkoetter, M. K. Wojnar, B. J. Charette, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2018, 57(16), 9728–9737 CrossRef CAS PubMed.
- (a) D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed; (b) C. Yoo and Y. Lee, Angew. Chem., Int. Ed., 2017, 56, 9502–9506 CrossRef CAS PubMed.
- (a) N. A. Eckert, A. Dinescu, T. R. Cundari and P. L. Holland, Inorg. Chem., 2005, 44, 7702–7704 CrossRef CAS PubMed; (b) M. J. Ingleson, B. C. Fullmer, D. T. Buschhorn, H. Fan, M. Pink, J. C. Huffman and K. G. Caulton, Inorg. Chem., 2008, 47, 407–409 CrossRef CAS PubMed; (c) H. Fan, B. C. Fullmer, M. Pink and K. G. Caulton, Angew. Chem., Int. Ed., 2008, 120, 9252–9254 CrossRef; (d) B. C. Fullmer, H. Fan, M. Pink and K. G. Caulton, Inorg. Chim. Acta, 2011, 369, 49–54 CrossRef CAS; (e) B. C. Fullmer, H. Fan, M. Pink, J. C. Huffman, N. P. Tsvetkov and K. G. Caulton, J. Am. Chem. Soc., 2011, 133, 2571–2582 CrossRef CAS PubMed; (f) N. O. Andrella, A. J. Sicard, S. I. Gorelsky, I. Korobkov and R. T. Baker, Chem. Sci., 2015, 6, 6392–6397 RSC; (g) C. A. Rettenmeier, H. Wadepohl and L. H. Gade, Chem. Sci., 2016, 7, 3533–3542 RSC; (h) S. Pelties and R. Wolf, Organometallics, 2016, 35, 2722–2727 CrossRef CAS; (i) C. A. Rettenmeier, J. Wenz, H. Wadepohl and L. H. Gade, Inorg. Chem., 2016, 55, 8214–8224 CrossRef CAS PubMed; (j) J. Wenz, A. Kochan, H. Wadepohl and L. H. Gade, Inorg. Chem., 2017, 56, 3631–3643 CrossRef CAS PubMed.
- (a) J. Takaichi, Y. Morimoto, K. Ohkubo, C. Shimokawa, T. Hojo, S. Mori, H. Asahara, H. Sugimoto, N. Fujieda, N. Nishiwaki, S. Fukuzumi and S. Itoh, Inorg. Chem., 2014, 53, 6159–6169 CrossRef CAS PubMed; (b) A. Parrot, Y. Morimoto, S. Paria, H. Sugimoto, N. Fujieda and S. Itoh, Dalton Trans., 2017, 46, 8013–8016 RSC.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed., 1995, 34, 1555–1573 CrossRef CAS.
- (a) J. B. Gilroy, B. O. Patrick, R. McDonald and R. G. Hicks, Inorg. Chem., 2008, 47, 1287–1294 CrossRef CAS PubMed; (b) F. Ölscher, I. Göttker-Schnetmann, V. Monteil and S. Mecking, J. Am. Chem. Soc., 2015, 137, 14819–14828 CrossRef PubMed; (c) K. Lee, C. M. Donahue and S. R. Daly, Dalton Trans., 2017, 46, 9394–9406 RSC.
- For terminal Ni hydrides: (a) P. Fischer, K. Götz, A. Eichhorn and U. Radius, Organometallics, 2012, 31, 1374–1383 CrossRef CAS; (b) L. M. Martínez-Prieto, C. Melero, D. del Río, P. Palma, J. Cámpora and E. Álvarez, Organometallics, 2012, 31, 1425–1438 CrossRef; (c) S. J. Connelly Robinson, C. M. Zall, D. L. Miller, J. C. Linehan and A. M. Appel, Dalton Trans., 2016, 45, 10017–10023 RSC.
- For bridging Ni hydrides: (a) D. A. Vicic and W. D. Jones, J. Am. Chem. Soc., 1997, 119, 10855–10856 CrossRef CAS; (b) I. Bach, R. Goddard, C. Kopiske, K. Seevogel and K.-R. Pörschke, Organometallics, 1999, 18, 10–20 CrossRef CAS; (c) B. L. Tran, M. Pink and D. J. Mindiola, Organometallics, 2009, 28, 2234–2243 CrossRef CAS.
- (a) S. J. Edwards, A. V. Soudackov and S. Hammes-Schiffer, J. Phys. Chem. A, 2009, 113, 2117–2126 CrossRef CAS PubMed; (b) J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed; (c) T. F. Markle, I. J. Rhile and J. M. Mayer, J. Am. Chem. Soc., 2011, 133, 17341–17352 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and CIF files. CCDC 1569427, 1860789–1860792. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03719a |
| ‡ Current address: National Taiwan University Department of Chemistry, No. 1, Section 4, Roosevelt Rd, Da'an District, Taipei City, Taiwan 10. |
| This journal is © The Royal Society of Chemistry 2019 |