DOI:
10.1039/C8SC04169E
(Edge Article)
Chem. Sci., 2019,
10, 1831-1835
Specific recognition of proteins and peptides via controllable oriented surface imprinting of boronate affinity-anchored epitopes†
Received
19th September 2018
, Accepted 30th November 2018
First published on 3rd December 2018
Abstract
Molecularly imprinted polymers (MIPs) are chemically synthesized materials mimicking the recognition of antibodies towards antigens. Epitope imprinting has been an effective strategy, making imprinting of proteins flexible to a great extent. However, so far there is apparently a lack of facile and versatile epitope imprinting approaches. Herein, we present a new method called controllable oriented surface imprinting of boronate affinity-anchored epitopes. In this method, a C-terminus nonapeptide epitope was glycated and anchored as a template onto a boronic acid-functionalized substrate, followed by controllable oriented surface imprinting via the polycondensation of multiple silylating reagents containing functionalities capable of interacting with the epitope. The developed imprinting approach allowed for precise control of the thickness of the imprinting layer through adjusting the imprinting time, generating excellent binding properties. This method was verified to be versatile and efficient. Thus, it could greatly facilitate the preparation of MIPs for specific recognition of proteins and peptides.
Introduction
Due to the presence of imprinted cavities complementary to the shape and functionalities of template molecules, molecularly imprinted polymers (MIPs)1–7 can provide affinity and specificity toward the template molecules and thereby have found promising applications in many areas such as sensing,8,9 separation10,11 and catalysis.12,13 As compared with antibodies and enzymes, MIPs are easier to prepare, more cost-efficient and more stable. So far, a large number of imprinting approaches have been reported. However, the imprinting of biomacromolecules, especially proteins, still remains challenging. Under harsh polymerization conditions for imprinting, a severe conformational change of proteins often occurs. In addition, the large size of proteins makes them difficult to be removed from highly cross-linked polymer networks. Epitope imprinting for peptide recognition was pioneered by Rachkov and Minoura.14,15 In epitope imprinting, short characteristic peptides, rather than entire proteins, are used as templates. A milestone study in epitope imprinting from Shea and coworkers16 extended this strategy to protein recognition, in which C-terminal nonapeptides were selected as epitopes. Since then, epitope imprinting has been widely adopted to overcome the above issues in protein imprinting.17–22 However, conventional epitope imprinting still suffers from some drawbacks. In some methods, prior to the imprinting the epitopes are immobilized on substrates via a functionality on the terminal amino acids such as –SH17 and –NH2.6,18 However, the co-existence of the same functionality on other amino acids may lead to an unfavourable configuration of the epitope on the substrate. Also, there is a lack of generally applicable substrates for facile and efficient epitope immobilization. In some approaches, epitopes are not immobilized19–22 and the imprinting direction is random, which results in a low imprinting efficiency since a large portion of the template is buried in the polymer. More importantly, many methods are only applicable to limited cases, and tedious conditional selection is usually required for imprinting different epitopes.
Boronic acids, which can reversibly bind with cis-diols,23,24 are important functional monomers for the imprinting of cis-diol-containing compounds such as saccharides and glycoproteins.25–29 Recently, we developed a versatile, facile and efficient approach called boronate affinity controllable oriented surface imprinting30–33 for the imprinting of glycoproteins, glycans and monosaccharides. The prepared MIPs exhibit high specificity, strong affinity and excellent imprinting efficiency. When applying to different targets, the change in the imprinting procedure is only to adjust the imprinting time according to the molecular size of the template. Clearly, an epitope imprinting approach that features the above merits, including versatility, facility, efficiency and excellent binding properties, will be of significant importance.
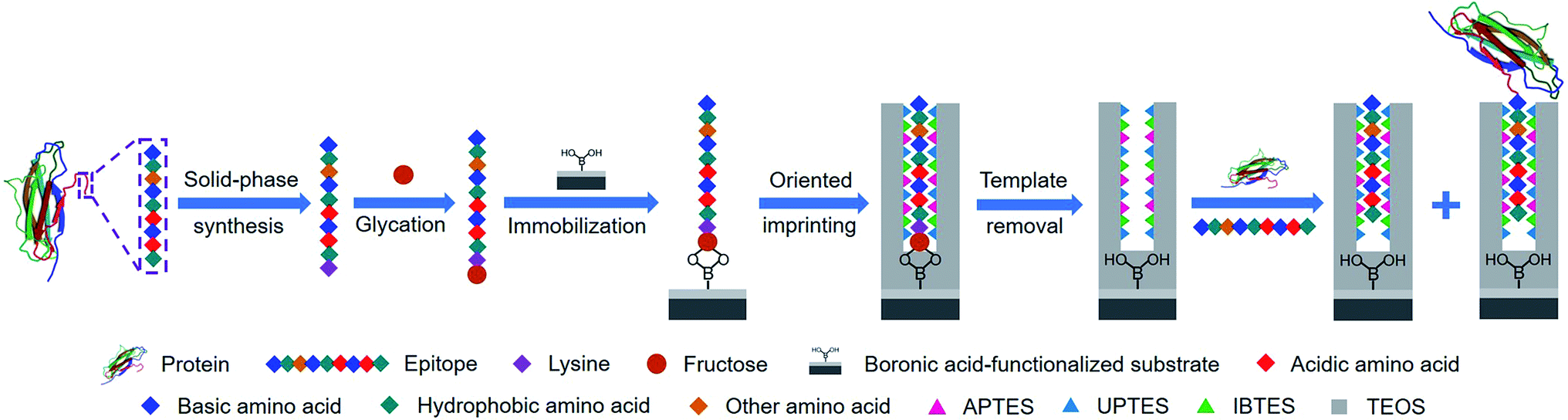
Herein, we present a new approach, called controllable oriented surface imprinting of boronate affinity-anchored epitopes, for the imprinting of proteins. This approach relies on the combination of glycation of epitopes, immobilization of glycated epitopes on boronic acid-functionalized substrates, and controllable oriented surface imprinting. The procedure of the imprinting is shown in Fig. 1. Based on the amino acid sequence of a specific protein, a C-terminus nonapeptide is selected as the epitope. Its C-terminus is attached with an additional lysine, followed by glycation with fructose. The glycated epitope is then immobilized onto a boronic acid-functionalized substrate by virtue of boronate affinity. After that, a silica coating is formed on the substrate surface to cover the template to an appropriate thickness, via the polycondensation of multiple silylating reagents containing functionalities capable of interacting with the template, including aminopropyltriethoxysilane (APTES), 3-ureidopropyl-triethoxysilane (UPTES), isobutyltriethoxysilane (IBTES) and tetraethyl orthosilicate (TEOS). Finally, the glycated epitope is removed by rinsing with an acidic acetonitrile solution to disrupt non-covalent and boronate affinity interactions, leaving imprinted cavities on the substrate surface. The obtained MIPs can rebind the intact target proteins and the epitope peptides. The highly attractive features of this approach, including versatility, facility, efficiency and excellent binding properties, were experimentally confirmed. Thus, it can be a promising method for the preparation of protein-specific MIPs for important applications.
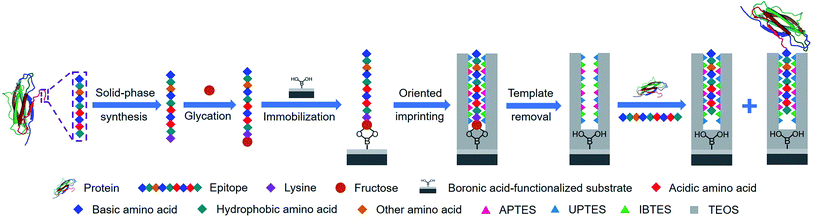 |
| | Fig. 1 Schematic of the principle and procedure of controllable oriented surface imprinting of boronate affinity-anchored epitopes. | |
Results and discussion
Selection and glycation of the epitope
β2-Microglobulin (B2M) and myoglobin (Mb) were used as the target proteins while boronic acid-functionalized magnetic nanoparticles (MNPs) were employed as the substrate in this work. B2M, which consists of 99 amino acids, is one of the major histocompatibility complex class I molecules. The abnormal level of B2M can indicate different diseases such as multiple myeloma.34 Mb, containing 153 amino acids, is a potential marker for heart attack in patients with chest pain.35 From protein structure databases such as UniProt, C-terminal nonapeptides for human B2M and Mb were found to be KIVKWDRDM and NYKELGFQG, respectively. The general procedure for the preparation of glycated epitope-imprinted MNPs is shown in Fig. S1.†A lysine is introduced at the C-end, because its amino group can be linked with a monosaccharide through the reductive amination. Fructose was selected as the sugar moiety and 2,4-difluoro-3-formyl-phenylboronic acid (DFFPBA) was selected as the ligand, because of their strong affinity.36 The synthesis of the peptide sequences and the glycation were achieved through solid-phase synthesis. In the solid-phase synthesis of the sequence of nonapeptide plus terminal lysine, functionalities at the side chains except for the terminal lysine were all protected. Thus, the sugar moiety was only linked to the amino group of the terminal lysine. The structures of the glycated B2M epitope and glycated Mb epitope are shown in Fig. S2.†
Optimization of monomer composition and imprinting time
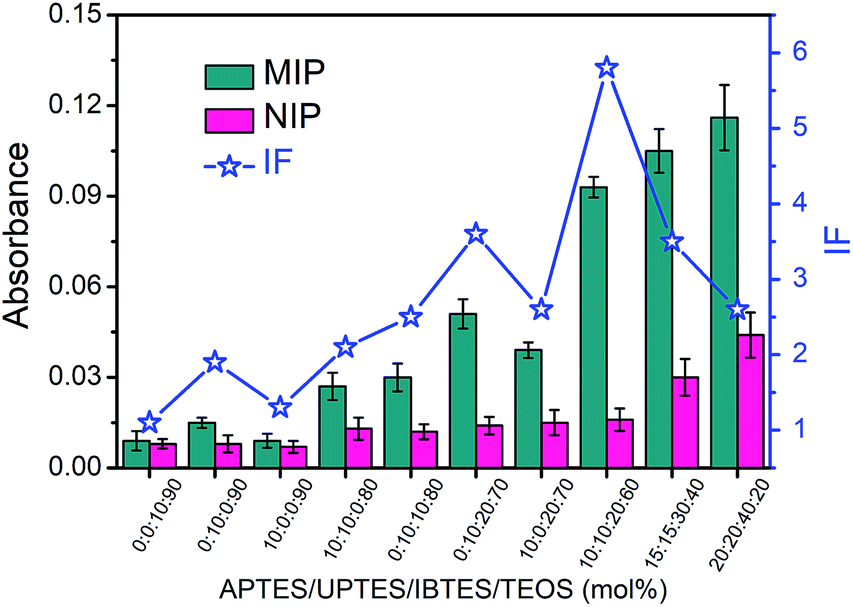
The boronate affinity of Fe3O4@SiO2@DFFPBA MNPs was verified, as shown in Fig. S3 and S4.† Unlike the boronate affinity controllable oriented surface imprinting,30–33 the boronic acid moiety introduced herein functioned only as an anchor for the facile immobilization and removal of the template, rather than participating in the rebinding with the epitopes. Because of the absence of boronate affinity in the rebinding, the use of multiple silylating reagents containing functionalities capable of interacting with the epitopes was essential to gain high rebinding affinity. The combination of the silylating reagents was selected according to the common features of amino acids of peptide epitopes. As shown in Fig. S5†, APTES, which contains an amino group, can interact with class I amino acids via electrostatic attraction as well as class IV amino acids via hydrogen bonding; UPTES, which contains a carbamido group, can interact with class II amino acids and class IV amino acids mainly via hydrogen bonding; IBTES, which contains a hydrophobic chain, can interact with class III amino acids via hydrophobic interaction. Although TEOS contains no functional groups, it can function as a crosslinker to form a hydrophilic silica skeleton, which can not only crosslink other monomers but also help to reduce non-specific adsorption. The ratio of the monomers and the imprinting time was optimized in terms of the imprinting factor (IF), which was calculated according to the ratio of the amount of epitope captured by glycated epitope-imprinted MNPs over that by non-imprinted MNPs (NIPs). As shown in Table S1† and Fig. 2, the composition of monomers played a key role. When an unsuitable composition (such as 1–5) was used, the overall IF value was rather poor, whatever the imprinting time was. In contrast, at an appropriate composition (such as 6, 8 and 9), the overall IF value was high and the imprinting time became critical. Imprinting with composition 8 (the ratio of APTES/UPTES/IBTES/TEOS was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:20:60) for 60 min produced the highest IF value (5.8).
:10:20:60) for 60 min produced the highest IF value (5.8).
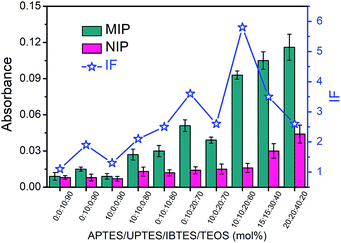 |
| | Fig. 2 The amount of the B2M epitope captured by glycated B2M epitope-imprinted MNPs and non-imprinted MNPs prepared using different proportions of monomers in an optimal imprinting time and their IF values. The error bars represent the standard deviation of three parallel experiments. | |
Characterization and performance of glycated epitope-imprinted MNPs
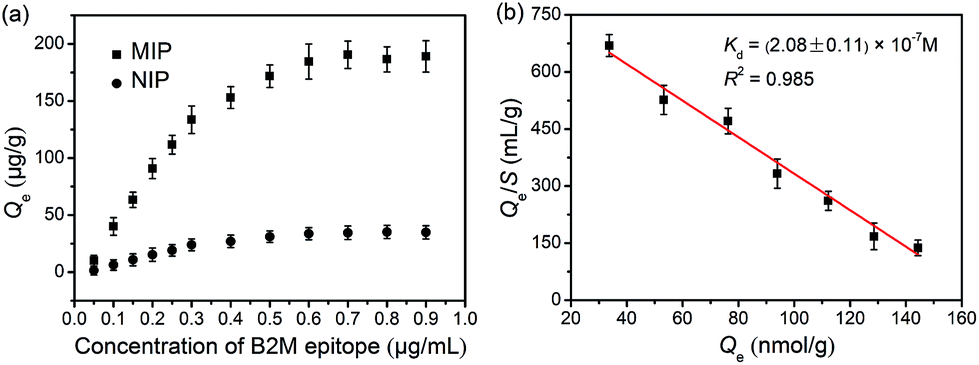
As indicated by the transmission electron microscopic (TEM) images shown in Fig. S6,† the glycated epitope-imprinted MNPs exhibited a diameter of ca. 150 nm, and a thin silica layer could be seen on the surface of the MNPs. Fourier transform infrared (FT-IR) characterization confirmed that the monomers were successfully modified on the MNPs (Fig. S7†). The binding isotherm of the glycated B2M epitope-imprinted MNPs toward the B2M epitope was evaluated. The imprinted MNPs exhibited much stronger affinity toward the B2M epitope as compared with the non-imprinted MNPs (Fig. 3a). Scatchard plot analysis (Fig. 3b) gave a dissociation constant (Kd) of (2.08 ± 0.11) × 10−7 M. The imprinting efficiency was measured to be 54.2% (see the ESI†), which was excellent for the imprinting of proteins. The extraction of the B2M epitope and the B2M intact protein by the glycated B2M epitope-imprinted MNPs was found to reach equilibrium within 20 min (Fig. S8†). Such fast equilibrium kinetics benefited from the accessible binding sites on the surface of the imprinted MNPs. Moreover, the glycated B2M epitope-imprinted MNPs could be stably stored for at least three months in a dry and sealed tube at room temperature. Even after storage for three months, the amount of B2M captured by the glycated B2M epitope-imprinted MNPs decreased only by 13.4% (Fig. S9†).
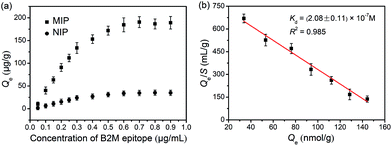 |
| | Fig. 3 (a) Binding isotherms for the binding of glycated B2M epitope-imprinted MNPs and non-imprinted MNPs to the B2M epitope. (b) Scatchard plot for the binding between glycated B2M epitope-imprinted MNPs and the B2M epitope. | |
Specificity test
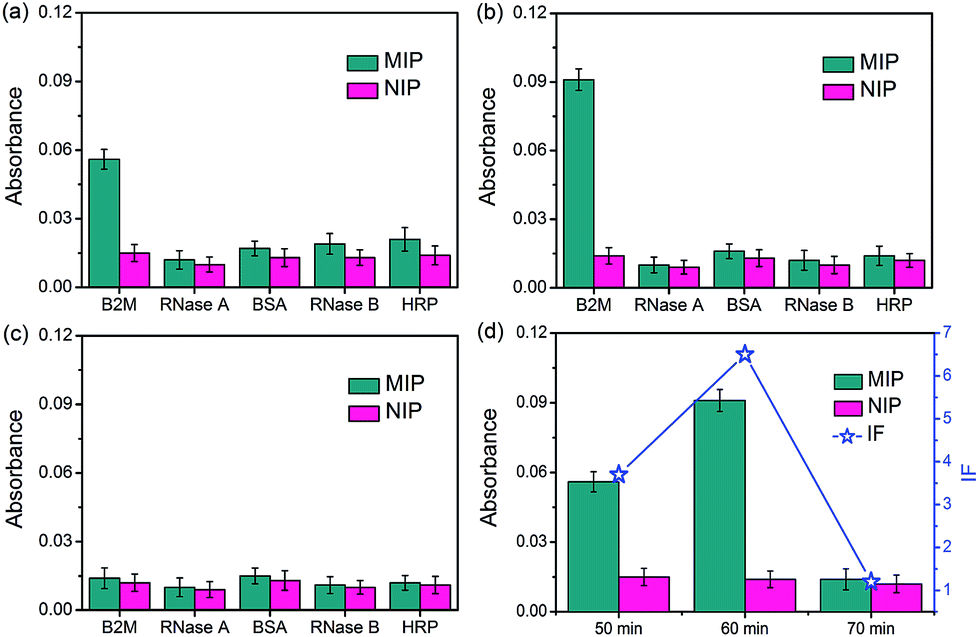
The specificity of the glycated epitope-imprinted MNPs toward intact proteins was examined by using B2M as the target protein, with ribonuclease A (RNase A), ribonuclease B (RNase B), horseradish peroxidase (HRP), and bovine serum albumin (BSA) as the competing proteins. As shown in Fig. 4, when the imprinting time was 60 min, the glycated B2M epitope-imprinted MNPs exhibited the highest IF value (6.5) and the best specificity, which were consistent with the results using the B2M epitope as the target analyte. Furthermore, the glycated B2M epitope-imprinted MNPs could endure 6 consecutive uses, with cross-reactivity less than 30% (Fig. S10†). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometric (MALDI-TOF MS) analyses also confirmed the specificity of the glycated epitope-imprinted MNPs (Fig. S11–S13†).
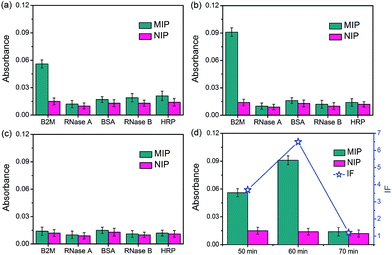 |
| | Fig. 4 The amount of different proteins captured by glycated B2M epitope-imprinted MNPs and non-imprinted MNPs prepared in the monomer ratio of APTES/UPTES/IBTES/TEOS = 10:10:20:60 with different imprinting times (a–c) and the dependence of IF on imprinting time (d). Imprinting time: (a) 50 min; (b) 60 min; (c) 70 min. | |
The specificity of the glycated epitope-imprinted MNPs at the peptide level was investigated. Tryptic digests of HRP and BSA were employed as the competing species. A mixture of the B2M epitope and the tryptic digests of HRP and BSA (molar ratio, 1:1:1) was extracted with the glycated B2M epitope-imprinted and non-imprinted MNPs. Without extraction, the B2M epitope, 10 peptides from BSA and 2 peptides and 8 glycopeptides from HRP were detected. However, with extraction by the glycated B2M epitope-imprinted MNPs, only the B2M epitope was detected. In contrast, nearly nothing was extracted by the non-imprinted MNPs (Fig. S14 and Table S2†). Clearly, the glycated epitope-imprinted MNPs exhibited high specificity at the peptide level.
Thickness controllability characterization of the imprinting layer
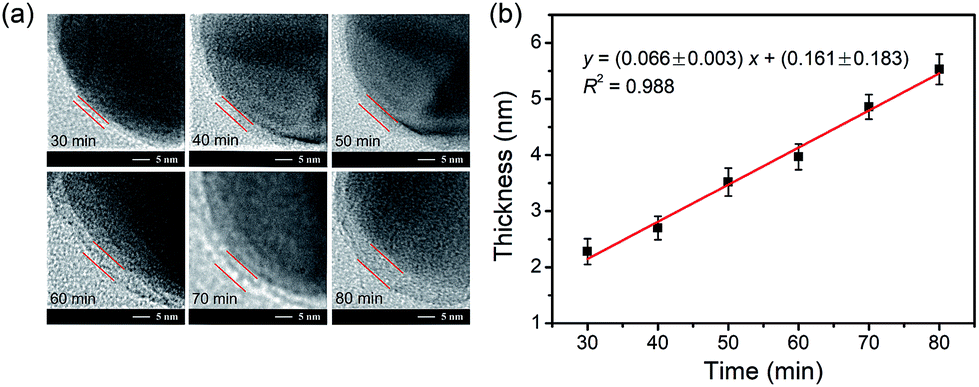
The dependence of the thickness of the imprinting layer on the imprinting time was examined. As shown in Fig. 5, the thickness of the imprinting layer linearly increased with increase in the polymerization time (y = (0.066± 0.003)x + (0.161 ± 0.183), R2 = 0.988). This indicates good controllability of the polymerization system used. The performance of the glycated epitope-imprinted MNPs prepared at different imprinting times can be rationalized in terms of the template coverage, which is the ratio of the thickness of the imprinting layer over the total length of the template and the ligand. With the software ChemBio 3D, the length of the glycated B2M epitope plus the boronic acid ligand was calculated to be ca. 4.44 nm. Using the above linear equation, the template coverage was calculated to be 78, 93, and 108% for the imprinting times of 50, 60 and 70 min, respectively. At relatively low coverage (such as 78%), the thickness of the imprinting layer was insufficient, resulting in relatively poor affinity and specificity. When the coverage was over 100%, the template molecules were all buried within the imprinting layer, making the MIP function like a NIP and thereby showing no affinity and specificity. Theoretically, the best imprinting time should be the value at which the thickness of the imprinting layer is close to but less than 100%. Clearly, the coverage at the imprinting time of 60 min was close to the theoretical optimum. The above rationalization suggests that the imprinting system used herein allowed for precision control of the imprinting process and thereby provided the best binding performance.
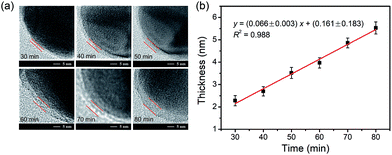 |
| | Fig. 5 (a) TEM images for the imprinting layer of AgNPs at the monomer ratio of APTES/UPTES/IBTES/TEOS = 10:10:20:60 at different polymerization times. Scale bar = 5 nm. (b) Relationship between the thickness of the imprinting layer and polymerization time. The ratio of APTES/UPTES/IBTES/TEOS was 10:10:20:60. The error bars represent the standard deviation of five parallel experiments. | |
Real sample application
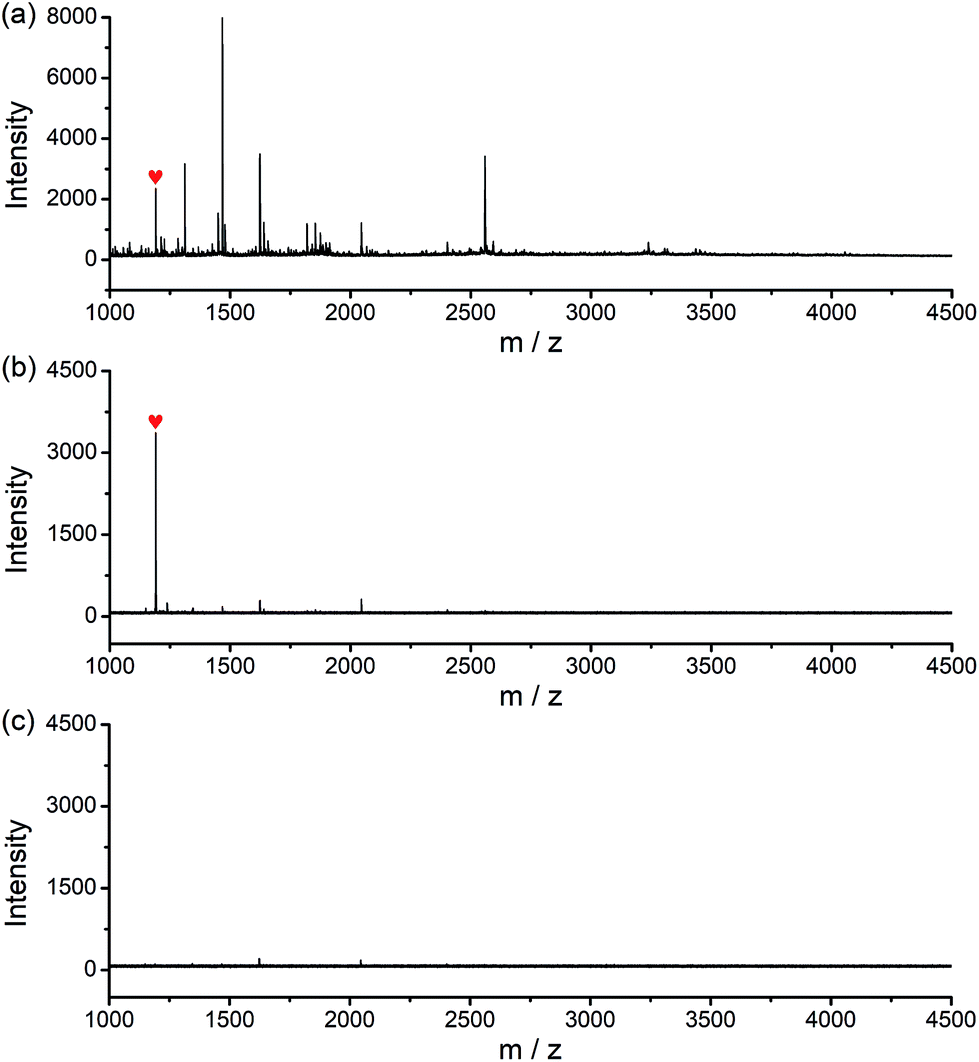
The potential of the glycated epitope-imprinted MNPs for real-world application was demonstrated by the extraction of B2M directly from human serum. MALDI-TOF MS analyses indicated that B2M existed in low abundance in human serum, accompanied by abundant interfering proteins, such as human serum albumin (HSA). As a comparison, after the serum sample was treated with the glycated B2M epitope-imprinted MNPs, only enriched B2M was detected (Fig. S15†). To further investigate the properties of the glycated epitope-imprinted MNPs, the tryptic digests of human serum samples spiked with the B2M epitope were analyzed. Even though there were plenty of interfering species, only the B2M epitope was detected by MALDI-TOF MS (Fig. 6). Apparently, the glycated epitope-imprinted MNPs exhibited excellent specificity toward both the intact target protein and its epitope even in a complex sample matrix.
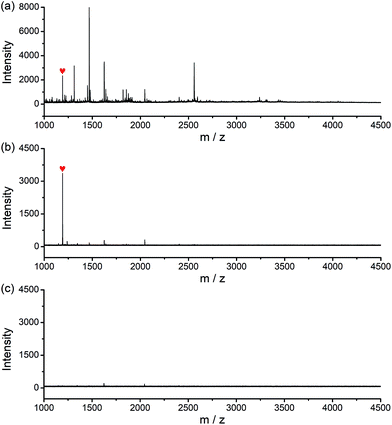 |
| | Fig. 6 MALDI-TOF MS spectra for the analysis of the B2M epitope spiked tryptic digest of human serum. (a) Without, (b) with extraction by glycated B2M epitope-imprinted MNPs and (c) with extraction by the corresponding non-imprinted MNPs ( : B2M epitope). : B2M epitope). | |
Method versatility
The versatility of this approach was demonstrated by applying the developed method to the imprinting of Mb. Although the ratio of the silylating monomers optimized for the B2M epitope might not be the best for the imprinting of the Mb epitope, the combination should be applicable to the imprinting of the Mb epitope and other epitopes, since the combination of the silylating monomers was selected according to the common features of amino acids. On the other hand, for nonapeptide epitopes glycated with the same monosaccharide through the linkage of the same additional amino acid, the molecular lengths are the same. Therefore, if the same composition of the polymerization system is used, the imprinting time optimized for one epitope can be applied to another. Thus, composition 8 (APTES/UPTES/IBTES/TEOS = 10:10:20:60) was used and the imprinting time was set at 60 min. The prepared glycated Mb epitope-imprinted MNPs exhibited excellent specificity at both the intact protein level and the peptide level (Fig. S16–S18, Table S3†). The IF value was 5.6, and the imprinting efficiency was 52.6% (see the ESI†).
Conclusions
In summary, we developed a versatile, facile and efficient approach for the imprinting of proteins. Through the introduction of a sugar moiety into the epitope, the immobilization and release of the templates became straightforward through manipulating the molecular interactions involved. With the immobilized templates and the silane-based imprinting system, the imprinting process was performed in a manner of precisely controllable oriented surface imprinting, providing strong affinity, high specificity and excellent efficiency. Thus, the proposed imprinting approach provided new access to the facile preparation of MIPs that allow for the specific recognition of proteins and peptides.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support from the National Science Fund for Distinguished Young Scholars from the National Natural Science Foundation of China (21425520 and 21834003), the “333” Talents Project from the Jiangsu Provincial Government, China (BRA2016351), the Open Grant from the State Key Laboratory of Analytical Chemistry for Life Science (5431ZZXM1706), and the program A for Outstanding PhD candidate of Nanjing University (201801A006).
Notes and references
- G. Wulff and A. Sarhan, Angew. Chem., Int. Ed. Engl., 1972, 11, 341–344 CAS.
- G. Vlatakis, L. I. Andersson, R. Müller and K. Mosbach, Nature, 1993, 361, 645–647 CrossRef CAS PubMed.
- L. Ye and K. Mosbach, J. Am. Chem. Soc., 2001, 123, 2901–2902 CrossRef CAS PubMed.
- J. Svenson, N. Zheng and I. A. Nicholls, J. Am. Chem. Soc., 2004, 126, 8554–8560 CrossRef CAS PubMed.
- A. Cutivet, C. Schembri, J. Kovensky and K. Haupt, J. Am. Chem. Soc., 2009, 131, 14699–14702 CrossRef CAS PubMed.
- Y. Hoshino, H. Koide, T. Urakami, H. Kanazawa, T. Kodama, N. Oku and K. J. Shea, J. Am. Chem. Soc., 2010, 132, 6644–6645 CrossRef CAS PubMed.
- J. K. Awino and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 12552–12555 CrossRef CAS PubMed.
- A. Kugimiya and T. Takeuchi, Biosens. Bioelectron., 2001, 16, 1059–1062 CrossRef CAS PubMed.
- Y. Fuchs, O. Soppera, A. G. Mayes and K. Haupt, Adv. Mater., 2013, 25, 566–570 CrossRef CAS PubMed.
- B. R. Hart, D. J. Rush and K. J. Shea, J. Am. Chem. Soc., 2000, 122, 460–465 CrossRef CAS.
- A. Nematollahzadeh, W. Sun, C. S. A. Aureliano, D. Ltkemeyer, J. Stute, M. J. Abdekhodaie, A. Shojaei and B. Sellergren, Angew. Chem., Int. Ed., 2011, 50, 495–498 CrossRef CAS PubMed.
- G. Wulff, Chem. Rev., 2002, 102, 1–28 CrossRef CAS PubMed.
- Z. J. Zhang and J. W. Liu, Mater. Horiz., 2018, 5, 738–744 RSC.
- A. Rachkov and N. Minoura, J. Chromatogr. A, 2000, 889, 111–118 CrossRef CAS PubMed.
- A. Rachkov and N. Minoura, Biochim. Biophys.Acta, 2001, 1544, 255–266 CrossRef CAS.
- H. Nishino, C. S. Huang and K. J. Shea, Angew. Chem., Int. Ed., 2006, 45, 2392–2396 CrossRef CAS PubMed.
- D. Dechtrirat, K. J. Jetzschmann, W. F. M. Stöcklein, F. W. Scheller and N. Gajovic-Eichelmann, Adv. Funct. Mater., 2012, 22, 5231–5237 CrossRef CAS.
- A. N. Tang, L. P. Duan, M. J. Liu and X. C. Dong, J. Mater. Chem. B, 2016, 4, 7464–7471 RSC.
- C. H. Lu, Y. Zhang, S. F. Tang, Z. B. Fang, H. H. Yang, X. Chen and G. N. Chen, Biosens. Bioelectron., 2012, 31, 439–444 CrossRef CAS PubMed.
- A. M. Bossi, P. S. Sharma, L. Montana, G. Zoccatelli, O. Laub and R. Levi, Anal. Chem., 2012, 84, 4036–4041 CrossRef CAS PubMed.
- P. Palladino, M. Minunni and S. Scarano, Biosens. Bioelectron., 2018, 106, 93–98 CrossRef CAS PubMed.
- Y. Q. Yang, X. W. He, Y. Z. Wang, W. Y. Li and Y. K. Zhang, Biosens.Bioelectron., 2014, 54, 266–272 CrossRef CAS PubMed.
- S. D. Bull, M. G. Davidson, J. M. H. Van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y. B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Z. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef CAS PubMed.
- D. J. Li, Y. Chen and Z. Liu, Chem. Soc. Rev., 2015, 44, 8097–8123 RSC.
- L. Li, Y. Lu, Z. J. Bie, H. Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 7451–7454 CrossRef CAS PubMed.
- Y. X. Li, M. Hong, M. Miao, Q. Bin, Z. Y. Lin, Z. W. Cai and G. N. Chen, J. Mater. Chem. B, 2013, 1, 1044–1051 RSC.
- W. Zhang, W. Liu, P. Li, H. B. Xiao, H. Wang and B. Tang, Angew. Chem., Int. Ed., 2014, 53, 12489–12493 CAS.
- A. Stephenson-Brown, A. L. Acton, J. A. Preece, J. S. Fossey and P. M. Mendes, Chem. Sci., 2015, 6, 5114–5119 RSC.
- J. K. Awino, R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 9759–9762 CrossRef CAS PubMed.
- R. R. Xing, S. S. Wang, Z. J. Bie, H. He and Z. Liu, Nat. Protoc., 2017, 12, 964–987 CrossRef CAS PubMed.
- S. S. Wang, J. Ye, Z. J. Bie and Z. Liu, Chem. Sci., 2014, 5, 1135–1140 RSC.
- Z. J. Bie, Y. Chen, J. Ye, S. S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2015, 54, 10211–10215 CrossRef CAS PubMed.
- S. S. Wang, D. Y. Yin, W. J. Wang, X. J. Shen, J. J. Zhu, H. Y. Chen and Z. Liu, Sci. Rep., 2016, 6, 22757 CrossRef CAS PubMed.
- S. Gatto, G. Ball, F. Onida, H. M. Kantarjian, E. H. Estey and M. Beran, Blood, 2013, 102, 1622–1625 CrossRef PubMed.
- M. Weber, M. Rau, K. Madlener, A. Elsaesser, D. Bankovic, V. Mitrovic and C. Hamm, Clin. Biochem., 2005, 38, 1027–1030 CrossRef CAS PubMed.
- C. C. Lü, H. Y. Li, H. Y. Wang and Z. Liu, Anal. Chem., 2013, 85, 2361–2369 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04169e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Yanyan
Ma
,
Yijia
Wang
,
Yanrong
Wen
and
Zhen
Liu
,
Yanyan
Ma
,
Yijia
Wang
,
Yanrong
Wen
and
Zhen
Liu
 : B2M epitope).
: B2M epitope).