
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemplating metastable Pd2 carboxylate aggregates†
Chen-Hao
Wang
 a,
Wen-Yang
Gao
a,
Qing
Ma
b and
David C.
Powers
*a
a,
Wen-Yang
Gao
a,
Qing
Ma
b and
David C.
Powers
*a
aTexas A&M University, Department of Chemistry, College Station, TX 77843, USA. E-mail: david.powers@chem.tamu.edu
bDND-CAT, Northwestern Synchrotron Research Centre at the Advanced Photon Source, Argonne, IL 60439, USA
First published on 30th November 2018
Abstract
Evaluation of the potential for metal–metal (M–M) cooperation to enable catalysis requires access to specific polynuclear aggregates that display appropriate geometry and size. In many cases, exerting synthetic control over the aggregation of simple metal salts is a challenge. For example, Pd(II) acetate self assembles as a trimer (i.e. Pd3(OAc)6) both in the solid state and in solution and does not feature close Pd–Pd interactions. Related carboxylate-supported Pd2 aggregates (i.e. Pd2(OAc)4), which would feature close Pd–Pd interactions, are thermodynamically metastable in solution phase and thus largely unavailable. Here we demonstrate ion metathesis within pre-formed metal–organic frameworks (MOFs) to prepare metastable Pd2 tetracarboxylates sites. The newly synthesized materials are characterized by elemental analysis, PXRD, SCXRD, EXAFS, XANES, and gas adsorption analysis. In addition, the critical role of network solvation on the kinetics of ion metathesis was revealed by coupled TGA-MS and ICP-MS experiments. The demonstration of templated ion metathesis to generate specific metastable coordination sites that are inaccessible in solution phase chemistry represents a new opportunity to interrogate the chemistry of specific polynuclear metal aggregates.
Introduction
Inspired by the facility of small molecular activation at multinuclear enzyme active sites1 and the enhanced reactivity of high nuclearity defect sites in heterogeneous catalysts,2 substantial effort has been directed towards developing multinuclear synthetic catalysts.3 Leveraging the potential opportunities for chemical catalysis that arise from cooperative redox chemistry between transition metal ions (i.e. M–M cooperation) requires synthetic access to polynuclear aggregates that feature specific nuclearity and geometry that engender efficient metal–metal (M–M) cooperation.4 Extant polynuclear catalysts are accessed by either (1) design and synthesis of multidentate ligands that enforce specific aggregation size and geometry,5 or (2) by self-assembly of transition metal ions with simple bridging ligands. These strategies are inherently limiting with respect to the structures that can be evaluated as potential catalysts because (1) the ligands required to stabilize targeted complexes may not be compatible with catalytic applications,6 and (2) the self-assembled structures may not give rise to the geometry required for efficient M–M interaction.In the context of developing new oxidation catalysts, we have become interested in controlling the aggregation state of transition metal ions in oxidatively resistant ligand scaffolds, such as all-oxygen fields (i.e. metal carboxylates). Examination of the self-assembled aggregates of metal carboxylates highlights both the diversity of aggregation sizes and geometries available to simple combinations of metals and ligands, and the challenge inherent in relying on self-assembled aggregates in catalysis. Rh(II) acetate self-assembles as a dimer (i.e. Rh2(OAc)4) due to the presence of a Rh–Rh bond (Fig. 1a).7 The activity of Rh2(OAc)4 in carbene group transfer catalysis has been ascribed to M–M cooperation mediated through the M–M bond.8 Isostructural complexes of Group 10 metals are unavailable: Ni(OAc)2 is a solvated monomer under many conditions,9 Pd(OAc)2 is an acetate-bridged timer in both solution- and solid-state (i.e. Pd3(OAc)6),10,11 and Pt(OAc)2 is tetrameric (i.e. Pt4(OAc)8).10a,12–14 The self-assembled, thermodynamically preferred aggregates of these salts do not provide platforms that engender cooperative M–M interactions (Fig. 1b).
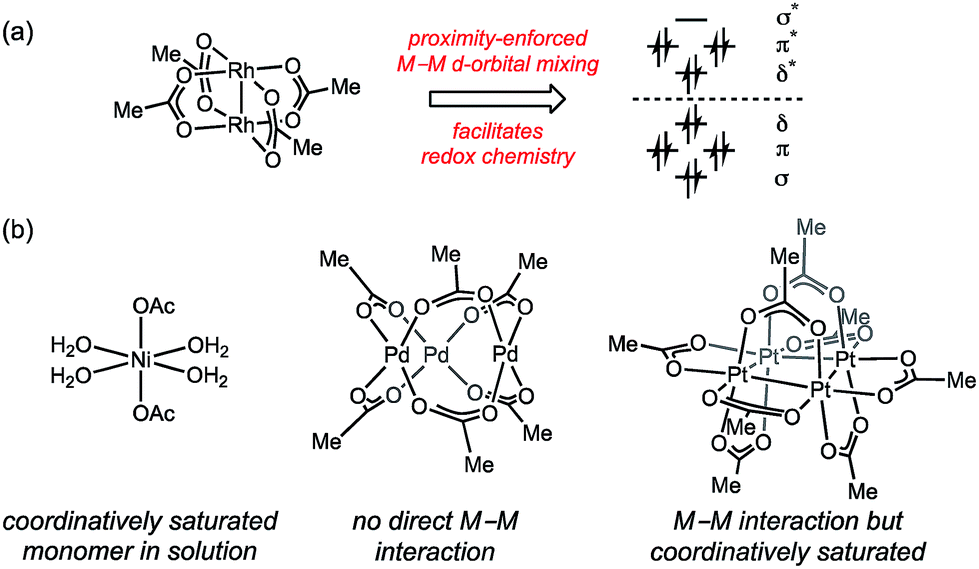 | ||
| Fig. 1 (a) Rh2(OAc)4 features a Rh–Rh single bond, which both enforces formation of a binuclear complex and mediates the redox cooperation between the metal centers responsible for M–M cooperation. (b) Group 10 metal acetates display a range of aggregation sizes and geometries, none of which supports both substrate binding and cooperative redox chemistry. | ||
Pd2 intermediates supported by oxygen-rich ligand environments have been proposed in electrochemical methane oxidation,15 directed C–H oxidation chemistry,16 and are of interest as models for PdO combustion catalysts.17Fig. 2 illustrates the small family of acetate-rich Pd2 aggregates that have been described. Stahl18 and Sanford19 have independently reported that pyridyl donors can support acetate-bridged complexes (1–3). Acetate-bridged cyclometallated complexes (i.e.4) have been observed during, and isolated from, directed C–H oxidation reactions.16 Pd2(OAc)2(acac)2 (5) has been obtained by treatment of Pd3(OAc)5(NO2) with acetylacetone (acac = acetylacetonate).20 Related acetate-bridged Pd2 species supported by γ-Keggin silicodecatungstate ligands have been characterized.21 In the presence of exogenous acetate ion—conditions relevant to the Wacker-type conversion of ethylene to vinyl acetate—concentration-dependent Pd aggregation has been observed. At intermediate acetate concentration, the dimer [Pd2(OAc)62−] (6) has been characterized by UV-vis and Raman spectroscopies; at high acetate concentrations, [Pd(OAc)42−] speciation has been suggested.22 While Pd2(OAc)4 structures that are isostructural to Rh2(OAc)4 have been the subject of numerous theoretical investigations,19,23 this aggregation mode has not been observed or characterized experimentally.
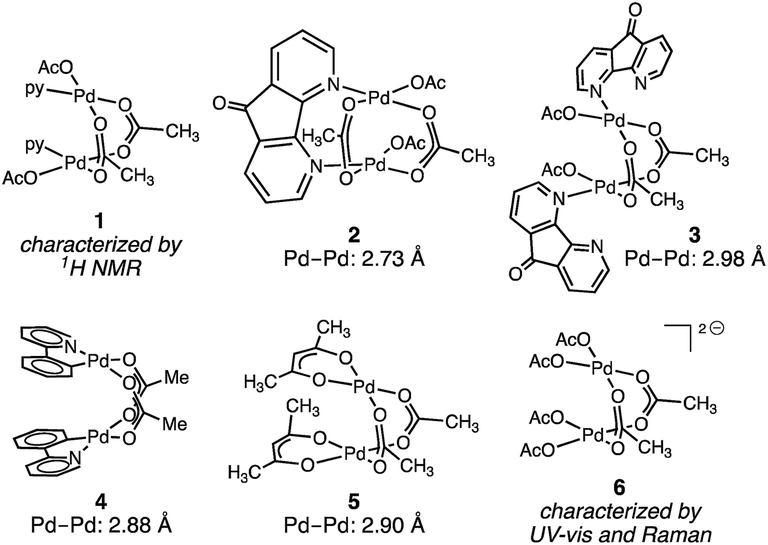 | ||
| Fig. 2 Available oxygen-rich Pd2 complexes. The structure of complex 1 was assigned by 1H NMR spectroscopy. Complexes 2–5 have been characterized crystallographically and the structure of [Pd2(OAc)62−] (6) has been proposed based on UV-vis and Raman spectroscopies. | ||
We are interested in Pd2 tetracarboxylate sites as potential redox catalysts based on the dual hypotheses that d-orbital mixing will give rise to cooperative redox chemistry in these structures4 and that the unavailability of Pd2(OAc)4 aggregates has prevented evaluation of the chemistry of this structural motif. Post-synthetic ion metathesis in metal–organic frameworks (MOFs)—a process in which metal sites in a template porous lattice are exchanged with exogenous ions—has been demonstrated to be a method that can enable access to novel coordination geometries.24,25 Ion metathesis chemistry has not been demonstrated for the introduction of 2nd- or 3rd-row transition metals.26 Here, we demonstrate that ion metathesis of binuclear transition metal sites in pre-formed porous crystalline materials provides a targeted strategy to rationally access Pd2 sites that are metastable in solution-phase chemistry. The new Pd2-based lattice is characterized by a combination of powder- (PXRD) and single-crystal X-ray diffraction (SCXRD), X-ray absorption spectroscopy (X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS)), and gas adsorption analysis. We demonstrate that proximity-enforced Pd–Pd interaction gives rise to cooperative binding of substrates, such as CS2, which is not observed for Pd3(OAc)6.
Results
Computational evaluation of Pd carboxylate aggregation
We have evaluated the isodesmic aggregation equilibria illustrated in Fig. 3. Geometry optimizations were carried out with B3LYP functional27 with LANL2TZ + f (1.472) basis set and corresponding ECP for Pd28 and 6-31G(d) basis set for other atoms (coordinates of optimized structures are tabulated in Tables S1–S4†).29 Frequency calculations at this level of theory confirmed that optimized geometries represent ground state structures. Single-point energy calculations were carried out using the M06 functional30 with sdd basis set for Pd31 and the 6-311++G(d,p) basis set for light atoms.32 Single-point solvation energies in acetonitrile were calculated using the SMD solvation model.33 These methods reproduce available structural data for Pd3(OAc)6 and have been demonstrated to provide reliable energies for related Pd carboxylate systems.23b Initial geometries for Pd2 and Pd4 aggregates were supplied as Pd substitution of known Rh2(OAc)4 and Pt4(OAc)8 structures, respectively.7,12 The geometry of Pd3(OAc)6 was based on its reported X-ray structure.10b Consistent with solid-state and solution-phase measurements, these calculations indicate that trimeric Pd3(OAc)6 (Pd–Pd: 3.23 Å) is the thermodynamically preferred aggregate. Pd2(OAc)4 (Pd–Pd: 2.62 Å), the next-most stable aggregate, is +5.0 kcal mol−1 relative to Pd3(OAc)6. The highest occupied molecular orbitals (HOMOs) of Pd2(OAc)4 and Pd3(OAc)6 are dominated by 4dz2 contributions from each of the Pd centers (Fig. 3b and c). The σ* interaction highlighted in the HOMO of Pd2(OAc)4 aggregates are responsible for the relative instability of Pd2(O2CR)4 sites and alleviation of these interactions drives the observed trimeric structure of Pd carboxylates.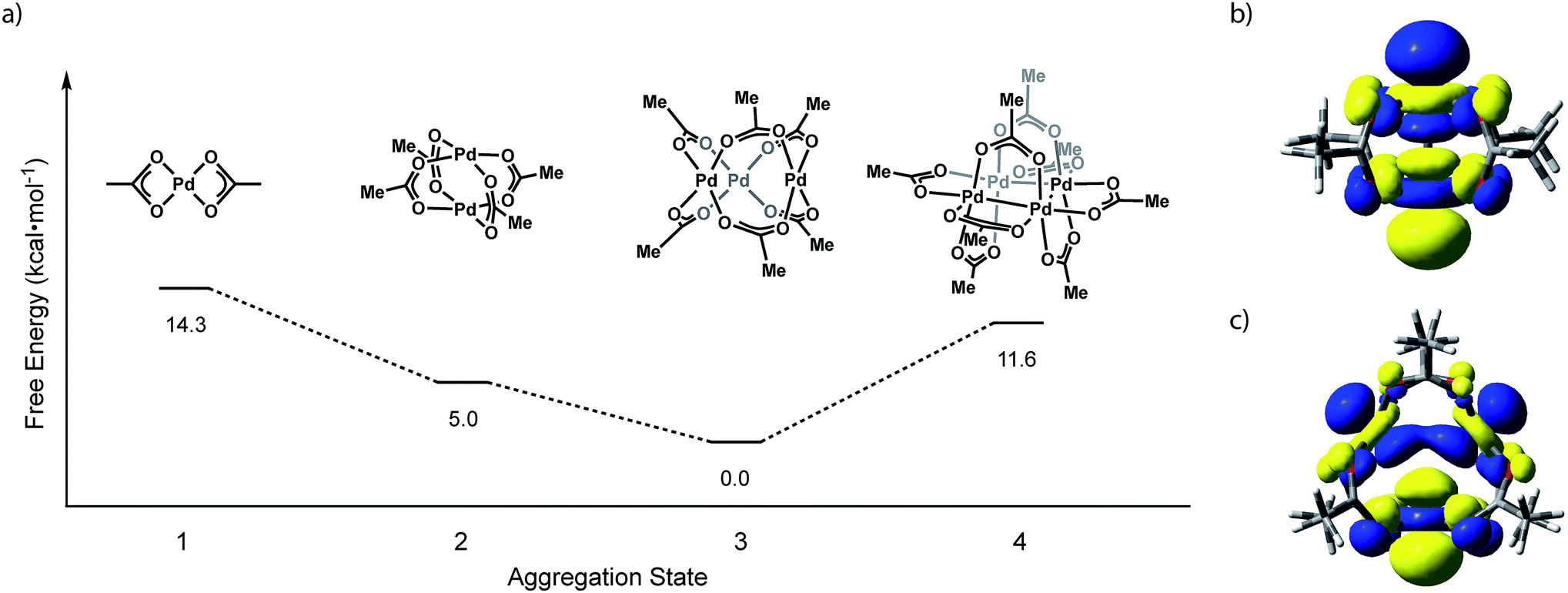 | ||
| Fig. 3 (a) Plot of free energy versus aggregate size for the aggregation equilibria available to Pd(OAc)2. Illustrations of the HOMO of (b) Pd2(OAc)4 and (c) Pd3(OAc)6. | ||
Synthesis of Pd2-based materials by ion exchange
We initiated our investigations of ion metathesis to generate metastable Pd2 aggregates using M3btc2 (btc = 1,3,5-benzenetricarboxylate) networks because (1) these networks feature carboxylate-bridged binuclear sites, (2) a variety of metal ions have been demonstrated to give rise to isostructural networks,34 and (3) we have an ongoing interest in the group-transfer chemistry of M2 sites housed in this class of materials.35 We specifically identified Zn3- and Cu3btc2 networks based on the similarity of the ionic radii of Zn, Cu, and Pd within carboxylate ligand frameworks.36 Molecular heterobimetallic complexes PdZn(OAc)4·H2O and PdCu(OAc)4·H2O are known,37 and the Pd–O distances in these structures are well matched: Pd–O for PdZn(OAc)4·H2O = 2.007 Å, Pd–O for PdCu(OAc)4·H2O = 2.000 Å, Cu–O = 1.997 Å, and Zn–O = 2.057 Å.Ion metathesis experiments were carried out by soaking pre-formed template lattices in solutions containing Pd(II) sources. The soaking solution was refreshed weekly. The extent of ion exchange was evaluated by inductively coupled plasma-mass spectrometry (ICP-MS) analysis of solutions prepared by exhaustively washing exchanged materials with both the solvent used in the soaking experiment and MeCN followed by digestion of the solid material with aqueous HNO3.38 We initiated our experiments by examining potential ion exchange chemistry into Cu3btc2. We examined a variety of Pd sources (Pd(OAc)2, Pd(O2CCF3)2, PdCl2, and [Pd(CH3CN)4][BF4]2) in a series of reaction solvents (CHCl3, CH3CN, MeOH, EtOH, and DMF) between 23–80 °C. Under no condition were we able to detect substantial Pd(II) incorporation into the Cu-based network (<1% exchange; these experiments are summarized in Table S5†).36 Based on the demonstration that Zn sites can be fluxional in the Zn4O nodes of MOF-5,39 we turned our attention to exchange experiments with Zn3btc2. Treatment of Zn3btc2 with CHCl3 solutions of Pd3(OAc)6 for 20 d results in ∼35% exchange of Zn for Pd. While further soaking with Pd2+ solutions did not result in increased Pd incorporation, the partially exchanged material was isomorphous with Zn3btc2 (for PXRD, see Fig. S1a†) which indicated the viability of Pd exchange into lattice sites of M2-based materials. Fig. S1b† compares the PXRD patterns obtained for Zn3btc and Pd-exchanged btc-based networks with the PXRD pattern that would be following network reorganization accommodate trigonal prismatic nodes (i.e. analogous to the structure of Pd3(OAc)6), and demonstrates that the extended M2-based lattice is not changed upon ion metathesis.
To access materials with greater Pd incorporation, we examined the ion metathesis chemistry of other Zn2-based porous materials (data summarized in Tables S6 and S7†). Large families of metal–organic frameworks are available in which carboxylate linkers support the targeted M2 coordination sites, and the extent of ion exchange has previously been reported to correlate with linker flexibility.25b Among the Zn-based porous frameworks that were investigated, Zn3btei (btei = 5,5′,5′′-(benzene-1,3,5-triyltris(ethyne-2,1-diyl))triisophthalate)40 was identified as participating in the most efficient ion metathesis chemistry, affording 79% Pd exchange after 112 d (Fig. 4a, black trace). The Pd-exchanged material is isomorphous to Zn3btei based on powder X-ray diffraction analysis (Fig. 4b). Soaking the exchanged materials in CHCl3 for 14 days resulted in no Pd leaching (ICP-MS analysis). Consistent with our observations with Zn3btc3 and Cu3btc2, as well as observations of others regarding the relative facility of ion metathesis of Cu and Zn sites,25b exchange into Cu3btei afforded 20% exchange after 56 d.
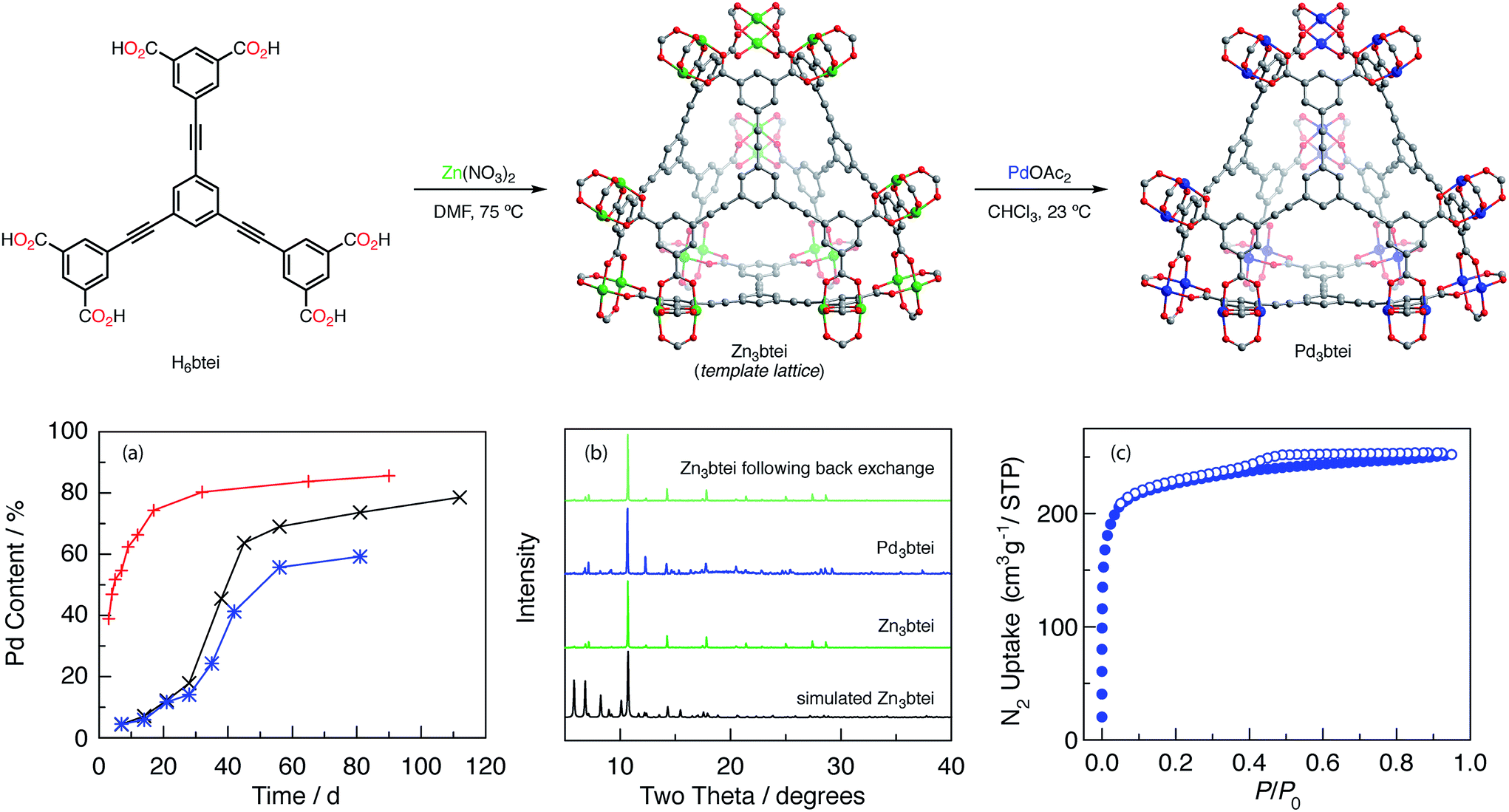 | ||
Fig. 4 Synthesis of Zn3btei and ion exchange with Pd(II) to generate Pd3btei. (a) Plot of Pd incorporation into Zn3btei as a function of time. Data plotted for soaking into a microcrystalline powder of Zn3btei ( ), crystalline Zn3btei ( ), crystalline Zn3btei ( ), and Zn3btei that was solvent-activated with CHCl3 ( ), and Zn3btei that was solvent-activated with CHCl3 ( ). (b) From bottom to top: simulated PXRD of Zn3btei, experimental PXRD of as-synthesized Zn3btei, experimental PXRD of Pd3btei, and PXRD of Zn3btei obtained by back exchange of Pd3btei with Zn(II). (c) N2 adsorption isotherm of Pd3btei (adsorption ( ). (b) From bottom to top: simulated PXRD of Zn3btei, experimental PXRD of as-synthesized Zn3btei, experimental PXRD of Pd3btei, and PXRD of Zn3btei obtained by back exchange of Pd3btei with Zn(II). (c) N2 adsorption isotherm of Pd3btei (adsorption ( ), desorption ( ), desorption ( )) obtained at 77 K. )) obtained at 77 K. | ||
Ion metathesis of Pd2+ into Zn3btei is reversible. Exposure of the Pd-exchanged material to CH3CN solutions of Zn(NO3)2 led to the back exchange to regenerate Zn3btei (Fig. 4b).
Impact of solvation on ion metathesis
Ion metathesis reactions are often stymied by slow exchange kinetics.24e We were intrigued by the observed (and reproducible) sigmoidal exchange profile pictured in Fig. 4a. Fig. 4a illustrates that exchange is slow for the first ∼30 days, after which point substantially more rapid Pd incorporation is observed. The sigmoidal kinetics behavior is not related to sample morphology: two preparations, one using Zn(NO3)2 and the other based on ZnBr2, have been reported and afford Zn3btei as either a microcrystalline powder or as single crystals.40 Examination of these two samples resulted in similar exchange vs. time plots (Fig. 4a, black vs. blue traces).The IR spectrum of as-synthesized Zn3btei displays a stretching frequency at 1650 cm−1, which has been assigned as Zn-bound DMF ligands (Fig. S2†).36,41 As exchange chemistry is carried out in CHCl3, the intensity of this feature diminishes over the course of ∼30 days. Thermogravimetric analysis-mass spectrometry (TGA-MS) analysis of samples obtained following exposure to CHCl3 for varying lengths of time confirms that concurrent with the disappearance of the carbonyl stretching mode from the IR spectrum, DMF solvent is removed from the Zn material; after 27 days of soaking, DMF is no longer observed (Fig. S3†). Similar activation of metal sites by chlorinated solvents has been observed by treating DMF-solvated Cu3btc2 with CH2Cl2.42
We hypothesized that the DMF ligated Zn sites that are present in as-synthesized Zn3btei participate in exchange more slowly than sites with weak donors, such as CHCl3, that are generated by solvent activation of Zn3btei. To interrogate this hypothesis, we soaked a sample of Zn3btei in CHCl3 for 30 days, after which time we exposed the solvent-activated sample to Pd(II) sources (Fig. 4a, red trace). In this exchange reaction, no induction period for Zn-to-Pd exchange is observed and 86% Pd incorporation is achieved after 90 days (Fig. S4a†). Using solvent-exchanged materials, the rate of Pd incorporation is dependent on the Pd salt used for exchange. For example, ion exchange with Pd(NO3)2 is much faster than with Pd(OAc)2 (88% incorporation is observed after 10 days, Fig. S4b†). The faster uptake is associated with a loss of material crystallinity (Fig. S4c†) so in subsequent experiments we have utilized Pd3btei prepared from exchange with Pd(OAc)2.
Gas adsorption
Zn3btei has been reported to be a non-porous material, which has been attributed to network collapse upon desolvation;40 see Fig. S5† for gas adsorption analysis of Zn3btei. The N2-adsorption isotherm obtained for a sample of Pd3btei activated under the same conditions used for Zn3btei (evacuation at 23 °C, see Fig. S5† for other activation protocols) provided a BET surface area of 857 m2 g−1 (a Langmuir surface area of 991 m2 g−1; Fig. 4c). While substantially higher than the surface area measured for Zn3btei, the BET surface area of Pd3btei is less than that of the isostructural Cu3btei material (reported BET surface area of 3730 m2 g−1;40a theoretical surface area is 3677 m2 g−1, see ESI† for the details of calculation).Single-crystal X-ray diffraction
Zn3btei (rht-topology) crystallizes in the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and the asymmetric unit contains two Zn ions.40 We have examined a single crystal that was isolated following partial Pd exchange (the Pd content of the sample from which the crystal was isolated was 49% (ICP-MS); crystallography details are compiled in Table S8†). Refinement of metal site occupancy in this partially exchanged crystal indicated exchange of one of the symmetry inequivalent Zn sites: Zn(1) has not undergone any exchange while Zn(2) has been ∼40% exchanged. Concurrent with exchange, the occupancy of the apical ligand associated with the site undergoing exchange decreases. This observation is consistent with five-coordinate Zn(II) ions being replaced by four-coordinate, square planar Pd(II) ions. While PXRD measurements of fully exchange Pd3btei indicated that bulk crystallinity is maintained during exchange, single crystallinity was not maintained during complete exchange, which prevented acquisition of SCXRD data for fully exchanged btei networks.
m space group and the asymmetric unit contains two Zn ions.40 We have examined a single crystal that was isolated following partial Pd exchange (the Pd content of the sample from which the crystal was isolated was 49% (ICP-MS); crystallography details are compiled in Table S8†). Refinement of metal site occupancy in this partially exchanged crystal indicated exchange of one of the symmetry inequivalent Zn sites: Zn(1) has not undergone any exchange while Zn(2) has been ∼40% exchanged. Concurrent with exchange, the occupancy of the apical ligand associated with the site undergoing exchange decreases. This observation is consistent with five-coordinate Zn(II) ions being replaced by four-coordinate, square planar Pd(II) ions. While PXRD measurements of fully exchange Pd3btei indicated that bulk crystallinity is maintained during exchange, single crystallinity was not maintained during complete exchange, which prevented acquisition of SCXRD data for fully exchanged btei networks.
X-ray absorption spectroscopy
Extended X-ray absorption fine structure (EXAFS) analysis was pursued to interrogate the coordination environment of the metal sites during and after ion exchange. We have analyzed Zn3btei, Pd3btei, as well as partially exchanged sample (∼50% exchange). In addition, we have examined PdZn(OAc)4·H2O as a molecular model of partially exchange heterobimetallic Pd–Zn sites. Data and fitting parameters are collected in Fig. S6–S10 and Tables S9–S13† and M–M distances obtained from these data sets are tabulated in Table 1.The Zn K-edge data obtained for Zn3btei indicate that the coordination environment about Zn is comprised of four Zn–O distances at 2.00 Å, one Zn–O distance at 2.17 Å, and a Zn–Zn distance of 2.86 Å, which is consistent with the known pentacoordinate Zn environment in solvated Zn3btei.
EXAFS data collected following partial Pd exchange were fit as a mixture of Zn2, PdZn, and Pd2 sites (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio). The Zn K-edge data indicated the Zn coordination environment in partially exchanged samples displays four Zn–O distances at 2.01 Å and one Zn–O distance at 2.18 Å. The Zn2 sites display a Zn–Zn distance of 2.90 Å and the PdZn sites display a Pd–Zn separation of 2.60 Å. The Pd K-edge data collected for the same sample were fit with four Pd–O distances of 2.01 Å. The PdZn sites display a Pd–Zn separation of 2.65 Å and the Pd2 sites show a Pd–Pd distance of 2.70 Å. The Pd–Zn distance determined from this sample is consistent with the Pd–Zn separation in the molecular site mimic PdZn(OAc)4·H2O (2.576 Å (SCXRD), 2.61 Å (EXAFS)).37
:2:1 ratio). The Zn K-edge data indicated the Zn coordination environment in partially exchanged samples displays four Zn–O distances at 2.01 Å and one Zn–O distance at 2.18 Å. The Zn2 sites display a Zn–Zn distance of 2.90 Å and the PdZn sites display a Pd–Zn separation of 2.60 Å. The Pd K-edge data collected for the same sample were fit with four Pd–O distances of 2.01 Å. The PdZn sites display a Pd–Zn separation of 2.65 Å and the Pd2 sites show a Pd–Pd distance of 2.70 Å. The Pd–Zn distance determined from this sample is consistent with the Pd–Zn separation in the molecular site mimic PdZn(OAc)4·H2O (2.576 Å (SCXRD), 2.61 Å (EXAFS)).37
The Pd K-edge data obtained for Pd3btei indicated the coordination environment of Pd is comprised of four Pd–O distances at 1.98 Å and one Pd–Pd distance at 2.72 Å. The obtained EXAFS data set is dominated by Pd–Pd scattering and the best fit does not incorporate a Pd–Zn interaction.
Analysis of the X-ray absorption near edge structure (XANES) data for both partially and completely exchanged samples indicate that Pd is incorporated into the M3btei network in the +II oxidation state (Fig. S11†).43 Based on comparison of the edge energy for Pd3(OAc)6, Pd3btei, PdZn(OAc)4, and Pd metal, the edge energy is inconsistent with the presence of Pd(0) in the exchanged samples.
Reactivity of lattice-confined Pd2 sites
We examined the hypothesis that proximity-enforced Pd–Pd interaction would raise the HOMO and result in a more nucleophilic and more easily oxidized Pd site in comparison to Pd carboxylates in which such Pd–Pd proximity is not enforced (i.e. the trinuclear structure of Pd3(OAc)6). To evaluate this hypothesis we examined the interaction of Pd3btei with CS2. Electron donation into CS2 is expected to manifest as a weakening of the C–S π-bonds and a low-energy shift in the associated IR stretching frequency.44 The IR spectrum of CS2-impregnated Pd3btei displays an 11 cm−1 red shift in the IR stretching frequency (Fig. S12†). No similar changes were observed upon CS2 update in Zn3btei or upon treatment of Pd3(OAc)6 with CS2.We have evaluated the reactivity of Pd3btei towards a variety of reaction conditions. The Pd–O linkages in Pd3btei are susceptible to protonation: treatment of Pd3btei with weak acids such as AcOH leads to the digestion of the extended lattice and the evolution of free H6btei and soluble Pd salts. The protolytic instability of the Pd–O bonds in Pd3btei can give rise to reaction chemistry with protic substrates: treatment of Pd3btei with benzyl alcohol resulted in the evolution of benzaldehyde with the concurrent formation of Pd(0). This observation is consistent with protonation of the Pd–O bond followed by β-hydride elimination of the incipient Pd(II) alkoxide. We have also carried out a series of experiments in which Pd3btei was treated with oxidants, such as peracetic acid and 2-tert-butylsulfonyl iodosylbenzene. Despite attempts to introduce exogenous substrates that might participate in C–H functionalization of olefin oxidation, in all conditions examined, the exclusive oxidation product obtained is 1,3,5-benzene tricarboxylic acid, which is the product of oxidative cleavage of the alkynyl groups in btei ligand. Similar treatment of H6btei and Zn3btei also generates 1,3,5-benzene tricarboxylic acid.
Discussion
Pd(OAc)2 is a ubiquitous catalyst and precatalyst in organic synthesis, and as such the structure of this important molecule has been the focus of sustained interest. Single-crystal X-ray diffraction, EXAFS, and solid-state NMR experiments have confirmed that Pd(OAc)2 exhibits a trimeric structure (i.e. Pd3(OAc)6) in the solid state (Fig. 1).10b,c A combination of NMR, Raman, and IR spectroscopies as well as EXAFS data and ebulliometry measurements indicate that the trimeric structure is maintained upon dissolution in many common solvents.10a,c–e Trinuclear aggregation simultaneously satisfies the proclivity for square planar coordination about Pd(II) ions and minimizes overlap of filled 4dz2 orbitals (Fig. 3).The close Pd–Pd interactions in acetate-bridged binuclear Pd complexes, such as complex 4 (Fig. 2), are credited for Pd–Pd cooperation and facile redox events in some Pd-catalyzed C–H oxidation chemistry.16A priori, one might expect that attractive d8⋯d8 interactions, which arise from symmetry allowed mixing of the 4d orbitals with the 5s and 5p orbitals, could stabilize Pd2 aggregate.45 However, in comparison to Rh, Ir, and Pt, for which attractive d8⋯d8 interactions have been documented, Pd exhibits an anomalously large energy gap between the 4d and 5s and 5p orbitals, which attenuates the impact of symmetry-allowed orbital mixing.43 The potential stabilization of mixing of the 4d with 5p and 5s orbitals is more than compensated for by the enforced filled–filled 4dz2 interactions in Pd2 aggregates. The kinetic instability of Pd2 aggregation can be overcome with strong donor ligands such as formamidinate and guanidinate ligands,46 but the use of such basic ligands can introduce potential ligand-based redox chemistry.6 With interest in oxidation catalysis, we have been attracted to identifying strategies to stabilize Pd2 aggregates in the absence of strongly basic nitrogen donors and have focused on the potential to isolate these structures in oxidation-resistant oxygen ligand fields.
Here, we demonstrate that ion metathesis of Pd(II) into pre-formed Zn(II)-based porous materials enables the synthesis and stabilization of thermodynamically metastable Pd2 tetracarboxylate sites confined with a porous lattice (Fig. 4). Extended exposure of Zn2-based materials to Pd(II)-containing solutions results in the replacement of lattice Zn(II) with Pd(II). Based on a combination of SCXRD and EXAFS analysis, the templated Pd2 sites are installed by sequential replacement of the Zn ions in the template lattice. Pd exchange efficiency depends on both the templating metal ion—Pd exchange is more efficient in Zn2-based materials than those comprised of Cu2 sites—and the structure of the organic linkers—Pd exchange into Zn3btc2 proceeds only to ∼35% Pd incorporation while use of the larger linking group btei enabled exchange reactions to proceed to ∼80% Pd incorporation. This observation is consistent with previous reports that larger, potentially more flexible organic linkers, support greater ion metathesis chemistry, which may be due to the ability of these more flexible structures to accommodate the subtle structural differences between materials before and after ion metathesis chemistry.25b
Cation exchange in porous materials has begun to emerge as a powerful strategy to access materials that are unavailable from solvothermal synthetic methods. Based on XANES analysis, the generated Pd2-based materials are comprised of Pd(II) ions, and the isolated materials do not contain Pd(0). This observation is in contrast to previous solvothermal approaches to Pd2-containing materials, which result in formation of Pd(0) nanoparticles intercalated within the MOF structure.47 These observations highlight the importance of low-temperature cation exchange to generate Pd-based MOFs: facile reduction of Pd(II) at high temperature and in the presence of many of the coordinating solvents typically employed in solvothermal synthesis of porous materials mandates that low-temperature synthetic methods to be developed to introduce Pd(II) sites.
While cation exchange chemistry is an important approach towards designer porous materials, often, the lack of mechanistic information regarding ion metathesis chemistry prevents predictive application of cation exchange reactions to target porous structures. Analysis of the time-dependent reaction progress of Pd exchange into Zn sites has revealed the presence of a kinetic induction period for ion exchange when DMF-solvated Zn templates were employed. At early reaction times, exchange proceeds slowly. At later times, substantially faster exchange is observed (Fig. 4a). IR spectroscopy and TGA-MS experiments, carried out during the kinetic induction period indicated that during this reaction regime, Zn-bound DMF ligands were being removed. The more rapid exchange chemistry was observed once DMF ligands were completely removed. The importance of network solvation was confirmed by pre-soaking materials in CHCl3. Ion exchange on these pre-activated materials resulted in ion exchange with no induction period. We hypothesize that the observed solvation-dependent exchange chemistry reflects the increased Lewis acidity of under-ligated Zn(II) ions and the increased proclivity of these ions to bind incoming carboxylate ions delivered by Pd3(OAc)6.
The unique electronic structure of the Pd2 site that is conferred by proximity-enforced Pd–Pd interaction is evidenced by the observation of CS2 binding. The HOMO-energy-raising impact of dz2 mixing provides enhanced Lewis basicity and thus binding of Lewis acidic substrates which is not observed in either Zn3btei or Pd3(OAc)6. Oxidative lability of the alkynyl linkers in the btei ligands has thus far precluded examination of the impact of HOMO-raising on oxidation catalysis with lattice-confined substrates.
Conclusions
The scope of polynuclear complexes that can be evaluated as potential catalyst platforms would be greatly expanded if general strategies to control the size and geometry of discrete aggregates supported by simple ligands were available. Here, we have demonstrated that cation metathesis within porous MOFs can be leveraged to prepare and stabilize Pd2 tetracarboxylate sites that are metastable in solution-phase chemistry. These studies have revealed the important role that lattice solvation can exert on the course of ion metathesis chemistry. We anticipate that the development of predictive models for when cation exchange chemistry will be facile in combination with access to new templated polynuclear aggregates housed in oxidatively stable ligand environments will contribute to the development of new families of polynuclear catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Texas A&M University, the Welch Foundation (A-1907), and the U.S. Department of Energy (DE-SC0018977) for financial support. Portions of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by Northwestern University, E.I. DuPont de Nemours & Co., and The Dow Chemical Company. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank Yu-Sheng Chen for acquisition of sing-crystal X-ray diffraction. NSF's ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1346572. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.References
- (a) R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239 CrossRef CAS PubMed; (b) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS PubMed; (c) J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273 CrossRef CAS PubMed; (d) B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041 CrossRef CAS PubMed.
- (a) T. Zambelli, J. Wintterlin, J. Trost and G. Ertl, Science, 1996, 273, 1688 CrossRef CAS; (b) R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465 CrossRef PubMed.
- For recent reviews, see: (a) J. P. Krogman and C. M. Thomas, Chem. Commun., 2014, 50, 5115 RSC; (b) K. P. Kornecki, J. F. Berry, D. C. Powers and T. Ritter, Prog. Inorg. Chem., 2014, 58, 223 Search PubMed; (c) I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936 CrossRef CAS; (d) D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705 RSC; (e) J. F. Berry and C. M. Thomas, Dalton Trans., 2017, 46, 5472 RSC; (f) N. P. Mankad, Chem. Commun., 2018, 54, 1291 RSC.
- D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840 CrossRef CAS PubMed.
- For examples, see: (a) A. Velian, S. Lin, A. J. M. Miller, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2010, 132, 6296 CrossRef CAS PubMed; (b) T. M. Powers, A. R. Fout, S.-L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 3336 CrossRef CAS PubMed; (c) P. A. Rudd, S. S. Liu, N. Planas, E. Bill, L. Gagliardi and C. C. Lu, Angew. Chem., Int. Ed., 2013, 52, 4449 CrossRef CAS PubMed; (d) S. Kuppuswamy, T. M. Powers, J. P. Krogman, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Chem. Sci., 2013, 4, 3557 RSC; (e) S. E. Flowers and B. M. Cossairt, Organometallics, 2014, 33, 4341 CrossRef CAS; (f) Y. Lee, K. J. Anderton, F. T. Sloane, D. M. Ermert, K. A. Abboud, R. Garcia-Serres and L. J. Murray, J. Am. Chem. Soc., 2015, 137, 10610 CrossRef CAS PubMed.
- For example, N–N coupling has been observed following oxidation in guanidinate-supported complexes: J. B. Diccianni, C. Hu and T. Diao, Angew. Chem., Int. Ed., 2016, 55, 7534 CrossRef CAS PubMed.
- (a) M. A. Porai-Koshits and A. S. Antschishkina, Dokl. Chem., 1962, 146, 920 Search PubMed; (b) F. A. Cotton, B. G. DeBoer, M. D. LaPrade, J. R. Pipal and D. A. Ucko, J. Am. Chem. Soc., 1970, 92, 2926 CrossRef CAS.
- E. Nakamura, N. Yoshikai and M. Yamanaka, J. Am. Chem. Soc., 2002, 124, 7181 CrossRef CAS PubMed.
- (a) J. N. van Niekirk and F. R. L. Schoening, Acta Crystallogr., 1953, 6, 609 CrossRef; (b) T. C. Downie, W. Harrison, E. S. Raper and M. A. Hepworth, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 706 CrossRef CAS.
- (a) T. A. Stephenson, S. M. Morehouse, A. R. Powell, J. P. Heffer and G. Wilkinson, J. Chem. Soc., 1965, 3632 RSC; (b) A. C. Skapski and M. L. Smart, J. Chem. Soc. D, 1970, 658 RSC; (c) V. I. Bakhmutov, J. F. Berry, F. A. Cotton, S. Ibragimov and C. A. Murillo, Dalton Trans., 2005, 1989 RSC; (d) V. M. Nosova, Y. A. Ustynyuk, L. G. Bruk, O. N. Temkin, A. V. Kisin and P. A. Storozhenko, Inorg. Chem., 2011, 50, 9300 CrossRef CAS PubMed; (e) L. A. Adrio, B. N. Nguyen, G. Guilera, A. G. Livingston and K. K. Hii, Catal. Sci. Technol., 2012, 2, 316 RSC; (f) W. A. Carole, J. Bradley, M. Sarwar and T. J. Colacot, Org. Lett., 2015, 17, 5472 CrossRef CAS PubMed; (g) R. J. Pakula, M. Srebro-Hooper, C. G. Fry, H. J. Reich, J. Autschbach and J. F. Berry, Inorg. Chem., 2018, 57, 8046 CrossRef CAS PubMed.
- A polymeric crystal structure has also been reported, but this structure has not been widely studied: S. D. Kirik, R. F. Mulagaleev and A. I. Blokhin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, M449 CrossRef PubMed.
- M. A. A. F. d. C. T. Carrondo and A. C. Skapski, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1857 CrossRef.
- In contrast to tetrameric Pt(II) acetate complexes, dimeric Pt(III) acetate complexes have been observed: T. G. Appleton, K. A. Byriel, J. M. Garrett, J. R. Hall, C. H. L. Kennard, M. T. Mathieson and R. Stranger, Inorg. Chem., 1995, 34, 5646 CrossRef CAS.
- Oxygen-rich d8–d8 Pt dimers have been investigated: T. V. Darnton, B. M. Hunter, M. G. Hill, S. Zális, A. Vlcek Jr and H. B. Gray, J. Am. Chem. Soc., 2016, 138, 5699 CrossRef CAS PubMed.
- M. E. O'Reilly, R. S. Kim, S. Oh and Y. Surendranath, ACS Cent. Sci., 2017, 3, 1174 CrossRef PubMed.
- (a) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed; (b) D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS PubMed; (c) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234 CrossRef CAS PubMed; (d) D. C. Powers, D. Y. Xiao, M. A. L. Geibel and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14530 CrossRef CAS PubMed; (e) D. C. Powers, E. Lee, A. Ariafard, M. S. Sanford, B. F. Yates, A. J. Canty and T. Ritter, J. Am. Chem. Soc., 2012, 134, 12002 CrossRef CAS PubMed; (f) A. J. Canty, A. Ariafard, M. S. Sanford and B. F. Yates, Organometallics, 2013, 32, 544 CrossRef CAS.
- (a) R. Yamauchi, A. Endou, M. Kubo and A. Miyamoto, Int. J. Quantum Chem., 1997, 61, 673 CrossRef; (b) S. M. Lang, T. Schnabel and T. M. Bernhardt, Phys. Chem. Chem. Phys., 2012, 14, 9364 RSC; (c) S. M. Lang, A. Frank and T. M. Bernhardt, J. Phys. Chem. C, 2013, 117, 9791 CrossRef CAS.
- P. B. White, J. N. Jaworski, C. G. Fry, B. S. Dolinar, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2016, 138, 4869 CrossRef CAS PubMed.
- A. K. Cook and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 3109 CrossRef CAS PubMed.
- S. P. Khranenko, E. A. Shusharina and S. A. Gromilov, J. Struct. Chem., 2011, 52, 544 CrossRef CAS.
- T. Hirano, K. Uehara, K. Kamata and N. Mizuno, J. Am. Chem. Soc., 2012, 134, 6425 CrossRef CAS PubMed.
- (a) R. N. Pandey and P. M. Henry, Can. J. Chem., 1974, 52, 1241 CrossRef CAS; (b) D. D. Kragten and R. A. van Santen, Inorg. Chem., 1999, 38, 331 CrossRef CAS.
- (a) D. D. Kragten, R. A. van Santen, M. Neurock and J. J. Lerou, J. Phys. Chem. A, 1999, 103, 2756 CrossRef CAS; (b) Y. F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344 CrossRef CAS PubMed; (c) B. E. Haines, H. Xu, P. Verma, X.-C. Wang, J.-Q. Yu and D. G. Musaev, J. Am. Chem. Soc., 2015, 137, 9022 CrossRef CAS PubMed; (d) B. E. Haines, J. F. Berry, J.-Q. Yu and D. G. Musaev, ACS Catal., 2016, 6, 829 CrossRef CAS.
- For examples of post-synthetic metal-ion exchange, see: (a) M. Dincă and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172 CrossRef PubMed; (b) L. Mi, H. Hou, Z. Song, H. Han, H. Xu, Y. Fan and S.-W. Ng, Cryst. Growth Des., 2007, 7, 2553 CrossRef CAS; (c) C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886 CrossRef CAS PubMed; (d) E. D. Metzger, C. K. Brozek, R. J. Comito and M. Dincă, ACS Cent. Sci., 2016, 2, 148 CrossRef CAS PubMed; (e) L. Liu, L. Li, J. A. DeGayner, P. Winegar, Y. Fang and T. D. Harris, J. Am. Chem. Soc., 2018, 140, 11444 CrossRef CAS PubMed.
- For relevant reviews: (a) M. Lalonde, W. Bury, O. Karagiardi, Z. Brown, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2013, 1, 5453 RSC; (b) C. K. Brozek and M. Dincă, Chem. Soc. Rev., 2014, 43, 5456 RSC; (c) P. Dera, J. E. Mondloch, O. Karagiardi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896 RSC; (d) M. Bosch, S. Yuan, W. Rutledge and H.-C. Zhou, Acc. Chem. Res., 2017, 50, 857 CrossRef CAS PubMed; (e) S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855 CrossRef CAS PubMed.
- There are a limited number of examples of d0 and d10 ions in the second and third row that have been demonstrated to participate in ion metathesis chemistry. For Hf(IV): M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082 CrossRef CAS PubMed For Cd(II): (a) S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814 CrossRef CAS PubMed; (b) S. Huang, X. Li, X. Shi, H. Hou and Y. Fan, J. Mater. Chem., 2010, 20, 5695 RSC; (c) G. Mukherjee and K. Biradha, Chem. Commun., 2012, 48, 4293 RSC.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 1623 CrossRef.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS; (b) A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111 CrossRef CAS; (c) L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029 CrossRef CAS PubMed.
- (a) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS; (b) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1971, 54, 2257 Search PubMed; (c) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- (a) M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef CAS; (b) D. Andrae, U. Haußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chem. Acc., 1990, 77, 123 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- C. R. Wade and M. Dincă, Dalton Trans., 2012, 41, 7931 RSC.
- (a) A. Das, J. H. Reibenspies, Y.-S. Chen and D. C. Powers, J. Am. Chem. Soc., 2017, 139, 2912 CrossRef CAS PubMed; (b) C.-H. Wang, A. Das, W.-Y. Gao and D. C. Powers, Angew. Chem., Int. Ed., 2018, 57, 3676 CrossRef CAS PubMed.
- (a) S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS PubMed; (b) X. Song, S. Jeong, D. Kim and M. S. Lah, CrystEngComm, 2012, 14, 5753 RSC.
- N. S. Akhmadullina, N. V. Cherkashina, N. Y. Kozitsyna, I. P. Stolarov, E. V. Perova, A. E. Gekhman, S. E. Nefedov, M. N. Vargaftik and I. I. Moiseev, Inorg. Chim. Acta, 2009, 362, 1943 CrossRef CAS.
- Experiments in which substantial Pd(0) formation was observed, either visually or by the diagnostic PXRD signal at ∼40° were not included.
- C. K. Brozek, V. K. Michaelis, T.-C. Ong, L. Bellarosa, N. López, R. G. Griffin and M. Dincă, ACS Cent. Sci., 2015, 1, 252 CrossRef CAS PubMed.
- (a) S. Hong, M. Oh, M. Park, J. W. Yoon, J.-S. Chang and M. S. Lah, Chem. Commun., 2009, 5397 RSC; (b) D. Zhao, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 9186 CrossRef CAS PubMed.
- I. Boldog, K. V. Domasevitch, J. Sanchiz, P. Mayer and C. Janiak, Dalton Trans., 2014, 43, 12590 RSC.
- H. K. Kim, W. S. Yun, M.-B. Kim, J. Y. Kim, Y.-S. Bae, J. Lee and N. C. Jeong, J. Am. Chem. Soc., 2015, 137, 10009 CrossRef CAS PubMed.
- K.-I. Shimizu, Y. Kamiya, K. Osaki, H. Yoshida and A. Satsuma, Catal. Sci. Technol., 2012, 2, 767 RSC.
- A. Sahibed-Dine, A. Aboulayt, M. Bensitel, A. B. Mohammed Saad, M. Daturi and J. C. Lavally, J. Mol. Catal. A: Chem., 2000, 162, 125 CrossRef CAS.
- J. E. Bercaw, A. C. Durrell, H. B. Gray, J. C. Green, N. Hazari, J. A. Labinger and J. R. Winkler, Inorg. Chem., 2010, 49, 1801 CrossRef CAS PubMed.
- J. F. Berry, F. A. Cotton, S. A. Ibragimov, C. A. Murillo and X. Wang, Inorg. Chem., 2005, 44, 6129 CrossRef CAS PubMed.
- Pd incorporation into Cu3btc2 networks has been reported by the addition of Cu2+ sources to the solvothermal synthesis, but the resulting materials contain substantial Pd(0), in the form of entrapped nanoparticles: W. Zhang, Z. Chen, M. Al-Naji, P. Guo, S. Cwik, O. Halbherr, Y. Wang, M. Muhler, N. Wilde, R. Gläser and R. A. Fischer, Dalton Trans., 2016, 45, 14883 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04940h |
| This journal is © The Royal Society of Chemistry 2019 |