
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe inverted ketene synthon: a double umpolung approach to enantioselective β2,3-amino amide synthesis†
Mahesh
Vishe
 and
Jeffrey N.
Johnston
*
and
Jeffrey N.
Johnston
*
Department of Chemistry, Vanderbilt Institute of Chemical Biology Vanderbilt University, Nashville, Tennessee 37235, USA. E-mail: jeffrey.n.johnston@vanderbilt.edu
First published on 12th November 2018
Abstract
A stereocontrolled synthesis of β2,3-amino amides is reported. Innovation is encapsulated by the first use of nitroalkenes to achieve double umpolung in enantioselective β-amino amide synthesis. Step economy is also fulfilled by the use of Umpolung Amide Synthesis (UmAS) in the second step, delivering the amide product without intermediacy of a carboxylic acid or activated derivative. Molybdenum oxide-mediated hydride reduction provides the anti-β2,3-amino amide with high selectivity.
Introduction
The β-amino amide motif is featured in a wide range of natural products and marketed drugs. β-Peptides with repeating β-amino amide residues can exhibit helix and sheet secondary structures with a predictable range of stability.1,2 Structurally, these homologues of α-amino amides provide an additional point of side chain attachment and both α/(C2) and β/(C3) positions have been evaluated for their ability to affect secondary structure in β-peptides.1,3 In order to increase proteolytic stability, α-amino amide leads in therapeutic development are strategically modified to their β-congener.4 β-Amino amides are further classified by substitution: β3 (e.g. 3-alkyl: sitagliptin5), β2,3 (2-oxy, 3-alkyl: bestatin,6 amastatin). Approaches to β-amino amides often focus on β-amino acid preparation and subsequent conversion to an amide,7 including Arndt–Eistert homologation from the α-amino acid chiral pool,8 enantioselective Mannich additions (A, Scheme 1),9 and the enantioselective conjugate addition of carbon nucleophiles to vinylogous carbamates10 or unsaturated amides.11 Functionalization reactions include reduction of vinylogous carbamates12,13 and allylic amines,14 reduction of imines,15 and stereospecific aziridine ring-opening.16 Less common are methods that reverse the polarity (umpolung)17,18 of the C–C bond formed, but use the typical polarity of amide formation (B, Scheme 1).19,20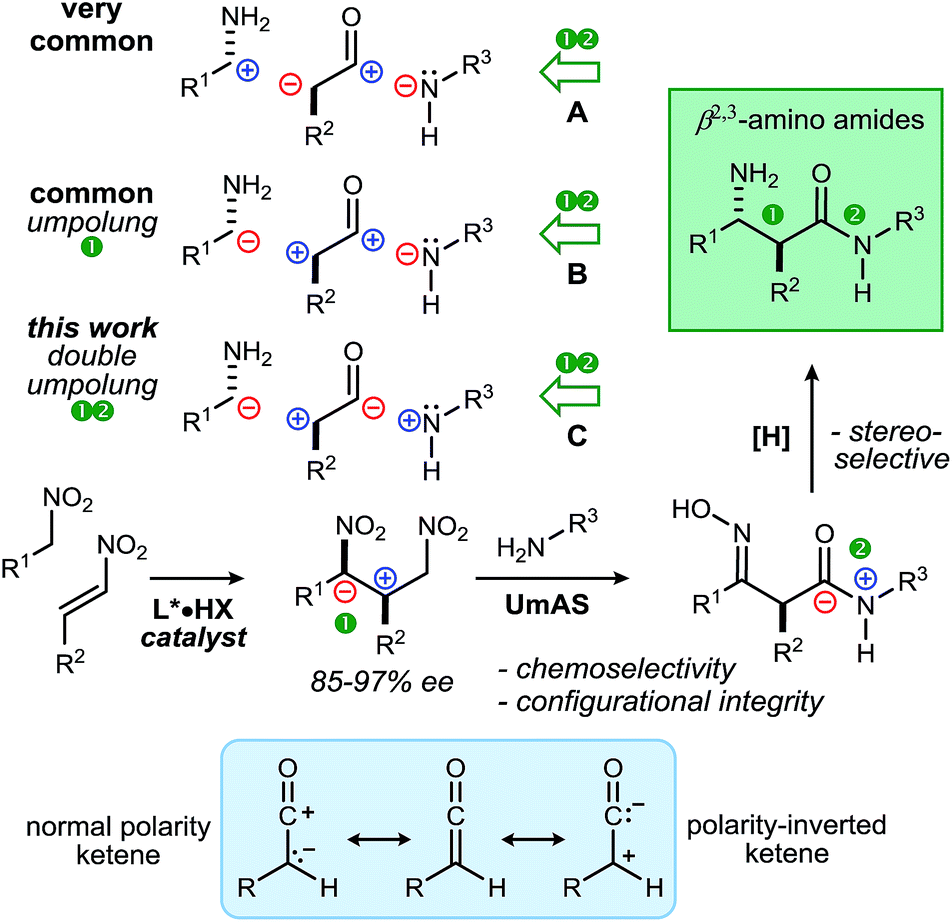 | ||
| Scheme 1 β2,3-Amino amides: retrosynthesis through carbon–carbon bond-forming approaches, mapped by polarity, highlighting an unusual ‘double umpolung’ and its realization through enantioselective catalysis and Umpolung Amide Synthesis (UmAS) (this work). | ||
Despite this attention paid to β-amino amide synthesis and its importance,21 no approaches currently reverse both polarities of the bonds formed to the central acyl fragment (C, Scheme 1), which is an ‘inverted ketene’. We hypothesized that Umpolung Amide Synthesis (UmAS) could enable a new stereocontrolled synthesis of β2,3-amino amides when preceded by nitroalkane addition to a β-substituted nitroalkene (Scheme 1). Owing to the polarity with which the carbon–carbon bond is formed, the strategy constitutes a double umpolung: both C–C and C–N bonds are formed umpolung relative to the most common approaches to construction of the acyl component.22 Importantly, this umpoled C–N bond formation could allow enantioenriched β-amino amides with acidic α-chiral carbons to be prepared, endowing polarity-inverted ketene equivalence to nitroalkenes.23
Several reports of nitroalkane additions to nitroalkenes have appeared since Du's 2006 Zn(II)/Ti(IV) catalyzed additions of nitroethane,24 including organocatalyzed25 and metal-mediated transformations.23,26,27 The reaction's development has been driven mostly by interest in simple 1,3-diamines,28 and focused applications are all but absent.23,29 We reasoned that the use of β-functionalized terminal nitroalkanes in UmAS would not only extend their impact by connecting directly with amides, but also provide a rare double umpolung22 at the strategy-level, culminating in a concise and modular enantioselective β-amino amide synthesis.
Results and discussion
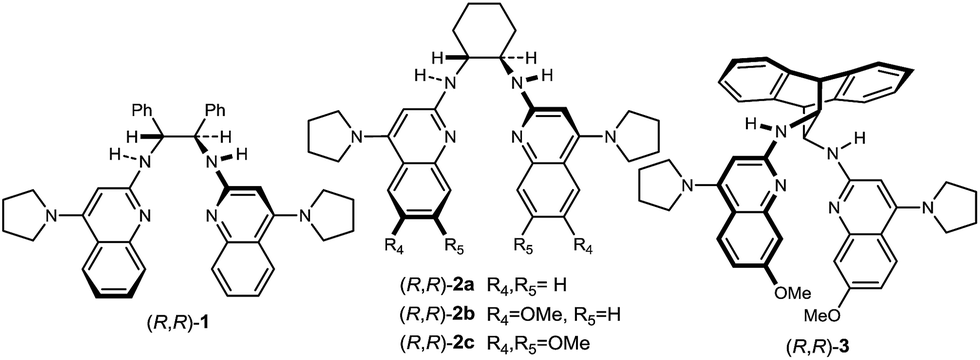
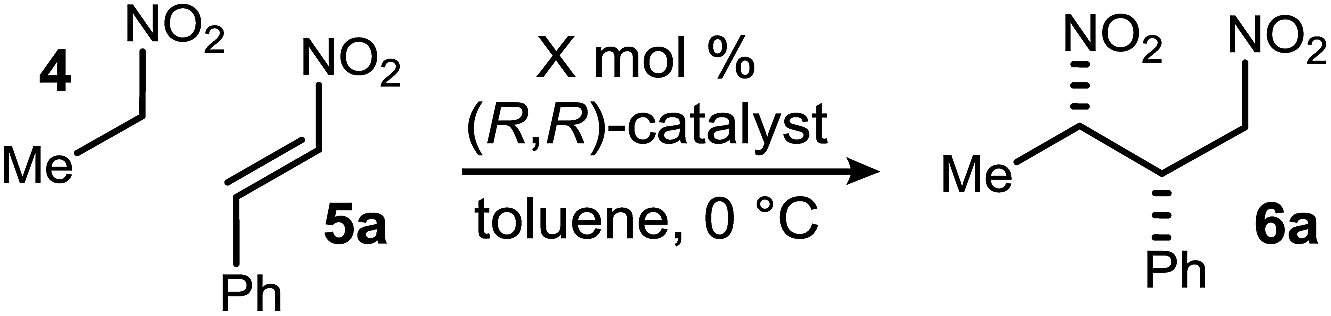
Reports of enantioselective nitroalkane–nitroalkene conjugate additions use catalysts derived from the chiral pool, or diamine backbones that similarly limit utility, and they have noted pronounced concentration effects on reactivity and selectivity.25b Moreover, some require the use of neat conditions that restrict the use of some solid nitroalkanes such as derivatives of phenyl nitromethane (i.e. p-Cl, p-NO2).25a This state of the art was anticipated to limit the impact of the approach outlined in Scheme 1. Bis(AMidine) [BAM] catalysis might remedy these shortcomings, but is unproven with electrophiles other than azomethines30 and nucleophilic alkenes.31,32 Initial investigations probed the suitability of electron-deficient alkene 5a using the leading BAM ligands. Nitroethane (4) and nitrostyrene (5a) were combined with StilbPBAM (1) (10 mol%), and the desired addition product was obtained in low dr and low ee (Table 1, entry 1). Its triflimidic acid salt (1·HNTf2) salt positively impacted the outcome, resulting in higher dr and ee (Table 1, entry 2). This correlation between protonation state, reactivity, and selectivity normally indicates that bifunctional catalysis is operative.33 PBAM (2a)34 and its salt (Table 1, entries 3–4) behaved similarly. Further attempts to manipulate the chiral ligand through substituents at the periphery of the presumed amidine binding pocket did not improve reactivity and selectivity (Table 1, entries 5–6). Lowering the temperature to −20 °C improved dr and ee to near-useful levels (Table 1, entry 7), but still impractical for broad utility. We reasoned that the substrate binding pocket may not adequately accommodate the nitroalkene electrophile. Derivatives of the conformationally rigid diamine derived from anthracene were therefore sought (i.e.3),35 as this diamine is reported to have an N–C–C–N dihedral angle of 114–117°, compared to trans-stilbene and trans-cyclohexane diamines (52° and 69°, respectively).36 The selectivity of the free base (3) trended with the analogous free base–salt pairs (Table 1, entry 8) when considering ee, but not dr (Table 1, entry 9). However, its improved reactivity allowed for lower temperature and time to completion while enhancing dr (Table 1, entry 10). The catalyst loading could be further lowered to 5 mol% provided that nitroethane is used at higher equivalents, delivering the addition product in 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 94% ee in 85% yield (Table 1, entry 11). Given the improved, but still similar selectivity of 3 relative to 1 and 2 might suggest that the latter diamines use their conformational mobility to expand the binding pocket while 3 is locked into the more productive and selective diamine conformation.
:1 dr and 94% ee in 85% yield (Table 1, entry 11). Given the improved, but still similar selectivity of 3 relative to 1 and 2 might suggest that the latter diamines use their conformational mobility to expand the binding pocket while 3 is locked into the more productive and selective diamine conformation.
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Catalyst | 4 (equiv.) | X mol% | Yieldb (%) | dr | eec (%) |
| a All reactions were performed on a 0.10 mmol scale (0.2 M) using nitroethane (5 equiv.) and a standard 24 h reaction time at 0 °C. b Isolated yield. c Enantiomeric ratios were measured by HPLC using a chiral stationary phase. See ESI for details. d Reaction conditions: −20 °C for 4 d. e Reaction conditions: −20 °C for 45 h. | ||||||
| 1 | 1 | 5 | 10 | 70 | 3:1 |
30/34 |
| 2 | 1·HNTf2 | 5 | 10 | 80 | 14:1 |
80 |
| 3 | 2a | 5 | 10 | 75 | 1.6:1 |
25/25 |
| 4 | 2a·HNTf2 | 5 | 10 | 80 | 6:1 |
85/77 |
| 5 | 2b·HNTf2 | 5 | 10 | 65 | 5.5:1 |
80/73 |
| 6 | 2c·HNTf2 | 5 | 10 | 65 | 3.3:1 |
79/70 |
| 7 | 2a·HNTf2d | 5 | 10 | 60 | 7:1 |
89 |
| 8 | 3 | 5 | 10 | 65 | 2:1 |
25/30 |
| 9 | 3·HNTf2 | 5 | 10 | 73 | 3.5:1 |
92/90 |
| 10 | 3·HNTf2e | 5 | 10 | 55 | 5:1 |
95/95 |
| 11 | 3·HNTf2e | 20 | 5 | 85 | 20:1 |
94 |
|
||||||
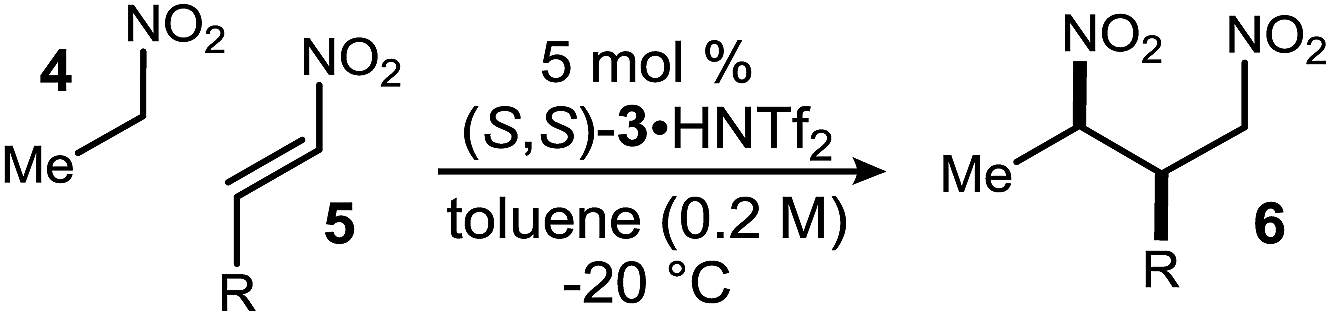
These conditions, optimized for 6a, were then applied to additional β-nitro styrenes (Table 2). β-Nitrostyrenes bearing electron-donating substituents at different positions gave products in uniformly high yield and selectivity (Table 2, entries 2–5). 4-Me-substituted nitrostyrene converted to product with high yield (88%), dr (20:1), and 95% ee (Table 2, entry 6), as did halogen substituents (Table 2, entries 7–12), trifluoromethoxy (Table 2, entry 13), and large naphthyl substituents (Table 2, entries 14–15). The 2-furanyl derivative gave lower dr (9:1) and ee (85%) (Table 2, entry 16), similar to an aliphatic substituent (Table 2, entries 17–18).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | R | 6 | drb | Yieldc (%) | eed (%) |
| a All reactions were performed on a 0.30 mmol scale (0.2 M) using nitroethane (20 equiv.) and a standard 45 h reaction time at −20 °C. b Measured by 1H NMR. c Isolated yield. d Enantiomeric ratios were measured by HPLC using a chiral stationary phase. See ESI for details. | |||||
| 1 | Ph | a | 20:1 |
85 | 94 |
| 2 | 2-MeO-C6H4 | b | 20:1 |
94 | 92 |
| 3 | 3-MeO-C6H4 | c | 20:1 |
85 | 93 |
| 4 | 4-MeO-C6H4 | d | 20:1 |
81 | 95 |
| 5 | 3,4-(MeO)2-C6H3 | e | 20:1 |
83 | 93 |
| 6 | 4-Me-C6H4 | f | 20:1 |
88 | 95 |
| 7 | 2-F-C6H4 | g | 20:1 |
85 | 94 |
| 8 | 4-F-C6H4 | h | 20:1 |
88 | 95 |
| 9 | 2-Cl-C6H4 | i | 20:1 |
88 | 95 |
| 10 | 3-Cl-C6H4 | j | 18:1 |
80 | 97 |
| 11 | 4-Cl-C6H4 | k | 20:1 |
80 | 96 |
| 12 | 3-Br-C6H4 | l | 15:1 |
78 | 97 |
| 13 | 4-CF3O-C6H4 | m | 20:1 |
81 | 94 |
| 14 | 1-Naphthyl | n | 20:1 |
93 | 95 |
| 15 | 2-Naphthyl | o | 20:1 |
88 | 95 |
| 16 | 2-Furanyl | p | 9:1 |
82 | 85 |
| 17 | PhCH2CH2 | q | 10:1 |
80 | 90 |
| 18 | i Pr | r | 11:1 |
25 | 91 |
As noted above, a limited number of nitroalkanes have been evaluated in additions of this type. Only fluorinated phenyl nitromethane derivatives are precedented in enantioselective conjugate additions to nitroalkenes, perhaps due to the acidity of the phenyl nitromethane adducts.37 After optimization of the conditions (see ESI† for details), a single equivalent of the nucleophile was sufficient for the reaction at 24 h at −20 °C. Addition of phenyl nitromethane gave 15:1 dr and 92% ee of 8a in high yield (Table 3, entry 1). An assortment of substituted aryl nitromethanes displayed unremarkable behavior (Table 3, entries 2–4). It was noted, however, that the dr observed in the crude reaction mixture (10:1) degraded to 8:1 after column chromatography for 8b. The effect of silica gel exposure was similar in the case of 8c (dr = 12:1 in crude reaction mixture), which changed to 7:1 after chromatography (96% yield, 91% ee for 8c, Table 3, entry 3). Only in the case of the strongest electron withdrawing group was selectivity modest (Table 3, entry 5).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | R | 8 | drb | Yieldc (%) | eed (%) |
| a All reactions were performed on a 0.10 mmol scale (0.05 M) using the aryl nitromethane (1 equiv.) and a standard 24 h reaction time. b dr after purification on silica gel column chromatography. c Isolated yield. d Enantiomeric ratios were measured by HPLC using a chiral stationary phase. See ESI for details. | |||||
| 1 | C6H5 | a | 15:1 |
95 | 92 |
| 2 | 4-F-C6H4 | b | 8:1 |
98 | 92 |
| 3 | 4-Cl-C6H4 | c | 7:1 |
96 | 91 |
| 4 | 4-Me-C6H4 | d | 16:1 |
98 | 93 |
| 5 | 4-NO2-C6H4 | e | 3:1 |
97 | 75/70 |
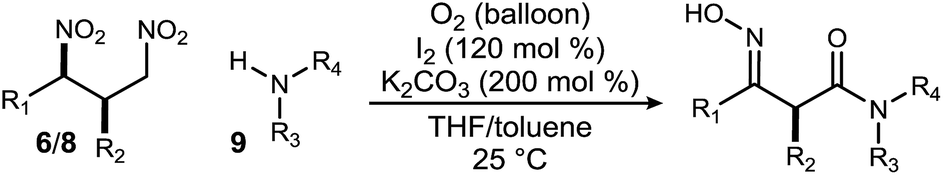
In order to complete the vision for a β2,3-amino amide synthesis, and explore the use of β-functionalized donors in UmAS,38 the one-pot strategy for conversion of primary nitroalkanes to amide was applied.39 Use of I2 as the halogen source was necessary to achieve the desired amide formation, presumably to generate the α-iodo-nitroalkane intermediate necessary for UmAS. Despite the formally oxidative conditions, the β-nitro group was concomitantly reduced to an oxime. Dinitroalkane 6a converted to 10a in 70% yield without change in ee (Table 4, entry 1), as expected due to the reversed polarity characteristic of C–N bond formation in UmAS. Nitroalkane 6a was assayed using a range of primary mono- (Table 4, entries 2–4) and disubstituted amines (Table 4, entries 5–7), each recording unchanged ee. (S)-α-Methyl benzyl amine delivered amide 10h in 67% yield without detectable epimerization (Table 4, entry 8). In these studies, α-aryl amides that might be particularly prone to epimerization under the basic conditions were examined (Table 4, entries 9–14). At room temperature degradation of ee was observed through post-reaction amide epimerization. Lowering the reaction temperature to 0 °C required a longer reaction time (2.5 d) but gave 14 in 90% ee (Table 4, entry 12). Similar was the case with substrates 8c and 8d whose products 15 and 16 were obtained in 78% and 76% yield with 89 and 92% ee respectively (Table 4, entries 13–14).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | R1/R2 | 6/8 | ee (6/8)b | ee (amide)b | Amine | Amide | Yieldc (%) |
| a All reactions were performed on a 0.1 mmol scale (0.1 M) using amine (2 equiv.) and standard 24 h reaction time at 25 °C. b Enantiomeric ratios were measured by HPLC using a chiral stationary phase. See ESI for details. c Isolated yield. d Diastereomeric excess, determined by 1H NMR. e Reaction conditions: 0 °C for 2.5 d. | |||||||
| 1 | Me/Ph | a | 94 | 94 |
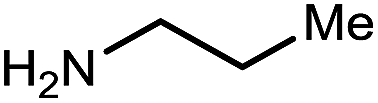
|
10a | 70 |
| 2 | Me/Ph | a | 94 | 94 |
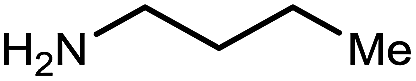
|
10b | 71 |
| 3 | Me/Ph | a | 94 | 94 |
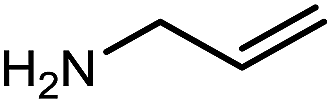
|
10c | 75 |
| 4 | Me/Ph | a | 94 | 94 |
|
10d | 74 |
| 5 | Me/Ph | a | 94 | 94 |
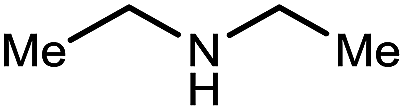
|
10e | 70 |
| 6 | Me/Ph | a | 94 | 94 |
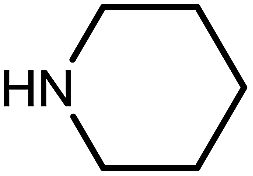
|
10f | 83 |
| 7 | Me/Ph | a | 94 | 94 |
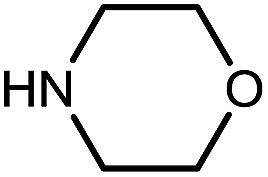
|
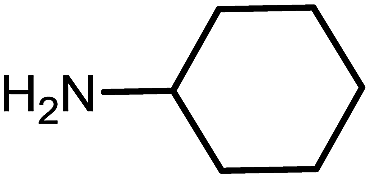
10g | 81 |
| 8 | Me/Ph | a | 94 | 94d |
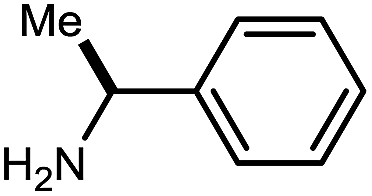
|
10h | 67 |
| 9 | Me/BnCH2 | q | 90 | 90 |
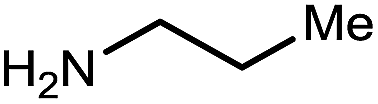
|
11 | 82 |
| 10 | Me/4-MeOC6H4 | d | 95 | 93 |
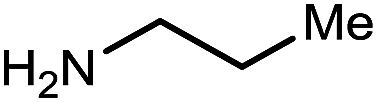
|
12 | 73 |
| 11 | Me/3-Cl-C6H4 | j | 96 | 95 |
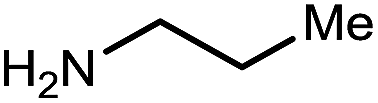
|
13 | 75 |
| 12 | Ph/Ph | a | 92 | 90 |
|
14 | 75 |
| 13 | 4-Cl-C6H4/Ph | c | 91 | 89 |
|
15 | 78 |
| 14 | 4-Me-C6H4/Ph | d | 93 | 92 |
|
16 | 76 |
These observations suggest all but complete conservation of configuration during the UmAS step. Consistent with prior mechanistic studies, we hypothesize that the secondary nitro effects redox amidation by oxygen atom transfer to an α-aminomethyl radical (or cation) intermediate.40,41 This mechanism would also suggest post-reaction epimerization. Both events are consistent with the outcomes detailed in Table 4, and the need to lower the reaction temperature for 8a–8d to slow base-mediated racemization. The overall strategy for enantioselective β-amino amide synthesis was realized by diastereoselective reduction of the β-oximino-amide using molybdenum-modified borohydride as described Scheme 2.42 These were later functionalized to their corresponding acyl, Cbz, and Fmoc derivatives (Scheme 2). Compound 10a was reduced to β2,3-amino amide 17 which was then converted to a dihydropyrimidinone,43 which confirmed the relative stereochemical assignment (NOE).44
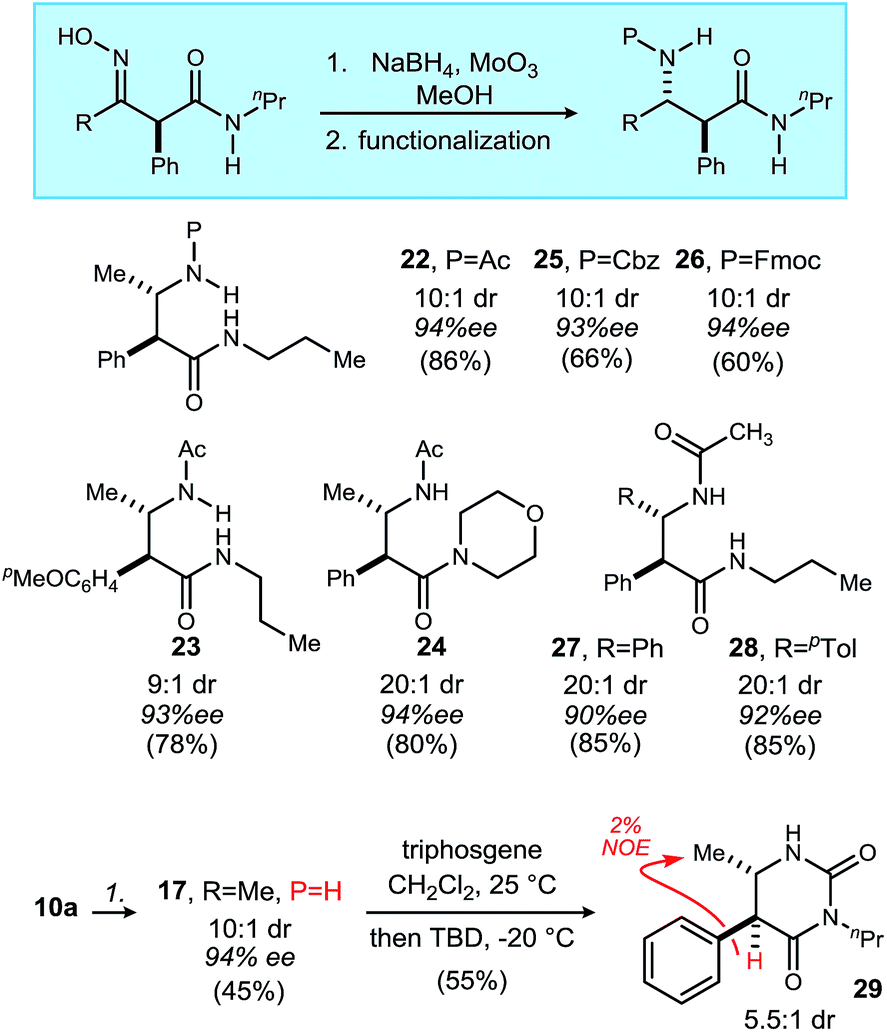 | ||
| Scheme 2 Diastereoselective reduction of oximes to anti-β2,3-amino amides, and subsequent derivatizations. | ||
Conclusions
In conclusion, the sequencing of nitroalkane–nitroalkene conjugate addition/UmAS/reduction provides an enantioselective synthesis of anti-β2,3-amino amides. Use of a new conformationally restricted BAM Brønsted acid-base catalyst delivered key intermediates with high enantiomeric excess. An unprecedented redox-UmAS delivered secondary and tertiary amides that can be reduced to their corresponding anti-β-amino amides. Retrosynthetically, this approach and protocol provides a solution to the unusual double umpolung involving an acyl retron – an inverted ketene – wherein both C–C and C–N bonds are formed with unusual polarities. These findings suggest that other β-chiral amides might be prepared by deploying the double umpolung strategy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Institute of General Medical Sciences of the National Institutes of Health (GM 063557 & GM 084333) for financial support.Notes and references
- Reviews: (a) D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015–2022 RSC; (b) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed; (c) L. Kiss, M. Cherepanova and F. Fülöp, Tetrahedron, 2015, 71, 2049–2069 CrossRef CAS; (d) M. Ashfaq, R. Tabassum, M. M. Ahmad, N. A. Hassan, H. Oku and G. Rivera, Med. Chem., 2015, 5, 295–309 Search PubMed.
- (a) M.-I. Aguilar, A. W. Purcell, R. Devi, R. Lew, J. Rossjohn, A. I. Smith and P. Perlmutter, Org. Biomol. Chem., 2007, 5, 2884–2890 RSC; (b) D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015–2022 RSC.
- R. Fernando, C. Francisco, A. Alberto, B. Jesus Hector, P. Jesus Manuel and Z. Maria del Mar, Curr. Top. Med. Chem., 2014, 14, 1225–1234 CrossRef.
- D. F. Hook, P. Bindschädler, Y. R. Mahajan, R. Šebesta, P. Kast and D. Seebach, Chem. Biodiversity, 2005, 2, 591–632 CrossRef CAS PubMed.
- D. Kim, L. Wang, M. Beconi, G. J. Eiermann, M. H. Fisher, H. He, G. J. Hickey, J. E. Kowalchick, B. Leiting, K. Lyons, F. Marsilio, M. E. McCann, R. A. Patel, A. Petrov, G. Scapin, S. B. Patel, R. S. Roy, J. K. Wu, M. J. Wyvratt, B. B. Zhang, L. Zhu, N. A. Thornberry and A. E. Weber, J. Med. Chem., 2005, 48, 141–151 CrossRef CAS PubMed.
- C. C. Stamper, D. L. Bienvenue, B. Bennett, D. Ringe, G. A. Petsko and R. C. Holz, Biochemistry, 2004, 43, 9620–9628 CrossRef CAS PubMed.
- Leading reference: B. Weiner, W. Szymanski, D. B. Janssen, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 1656–1691 RSC.
- (a) G. Guichard, S. Abele and D. Seebach, Helv. Chim. Acta, 1998, 81, 187–206 CrossRef CAS; (b) D. Seebach, M. Overhand, F. N. M. Kühnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913–941 CrossRef CAS.
- Reviews: (a) A. Michael, W. Bernhard and R. Nikolaus, Angew. Chem., Int. Ed., 1998, 37, 1044–1070 CrossRef; (b) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069–1094 CrossRef CAS PubMed; (c) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626–2704 CrossRef CAS PubMed; (d) A. Ting and S. E. Schaus, Eur. J. Org. Chem., 2007, 2007, 5797–5815 CrossRef; (e) N. Z. Burns and E. N. Jacobsen, Sci. Synth., Stereosel. Synth., 2011, 2, 785–834 CAS; (f) S. Salim, H. N. Ann, K. K. Keerthi and A. Gopinathan, Asian J. Org. Chem., 2018, 7, 613–633 CrossRef.
- (a) M. P. Sibi and J. Chen, Org. Lett., 2002, 4, 2933–2936 CrossRef CAS PubMed; (b) M. P. Sibi and Y. Asano, J. Am. Chem. Soc., 2001, 123, 9708–9709 CrossRef CAS PubMed.
- Enantioselective amine additions to unsaturated amides, selected examples: (a) M. P. Sibi, J. J. Shay, M. Liu and C. P. Jasperse, J. Am. Chem. Soc., 1998, 120, 6615–6616 CrossRef CAS; (b) J. K. Myers and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 8959–8960 CrossRef CAS.
- (a) Y.-G. Zhou, W. Tang, W.-B. Wang, W. Li and X. Zhang, J. Am. Chem. Soc., 2002, 124, 4952–4953 CrossRef CAS PubMed; (b) M. Yasutake, I. D. Gridnev, N. Higashi and T. Imamoto, Org. Lett., 2001, 3, 1701–1704 CrossRef CAS PubMed; (c) D. Heller, J. Holz, H.-J. Drexler, J. Lang, K. Drauz, H.-P. Krimmer and A. Börner, J. Org. Chem., 2001, 66, 6816–6817 CrossRef CAS PubMed; (d) G. Zhu, Z. Chen and X. Zhang, J. Org. Chem., 1999, 64, 6907–6910 CrossRef CAS PubMed; (e) D. Saylik, E. M. Campi, A. C. Donohue, W. R. Jackson and A. J. Robinson, Tetrahedron: Asymmetry, 2001, 12, 657–667 CrossRef CAS.
- M. P. Rainka, Y. Aye and S. L. Buchwald, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5821–5823 CrossRef CAS PubMed.
- J. Deng, X.-P. Hu, J.-D. Huang, S.-B. Yu, D.-Y. Wang, Z.-C. Duan and Z. Zheng, J. Org. Chem., 2008, 73, 2015–2017 CrossRef CAS PubMed.
- (a) D. Steinhuebel, Y. Sun, K. Matsumura, N. Sayo and T. Saito, J. Am. Chem. Soc., 2009, 131, 11316–11317 CrossRef CAS PubMed; (b) C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed; (c) D. Mtat, R. Touati and B. Ben Hassine, Tetrahedron Lett., 2014, 55, 6354–6358 CrossRef CAS.
- F. A. Davis, G. V. Reddy and C.-H. Liang, Tetrahedron Lett., 1997, 38, 5139–5142 CrossRef.
- (a) D. Seebach and D. Enders, Angew. Chem., Int. Ed., 1975, 14, 15–32 CrossRef; (b) D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239–258 CrossRef.
- NHC catalysis in β-amino amide synthesis: (a) H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2007, 129, 13796–13797 CrossRef CAS PubMed; (b) Y. Kawanaka, E. M. Phillips and K. A. Scheidt, J. Am. Chem. Soc., 2009, 131, 18028–18029 CrossRef CAS PubMed.
- Review: (a) S. Norbert, Angew. Chem., Int. Ed., 2003, 42, 5794–5795 CrossRef PubMed. Leading references: (b) A. Rimkus and N. Sewald, Org. Lett., 2003, 5, 79–80 CrossRef CAS PubMed; (c) U. Eilitz, F. Leßmann, O. Seidelmann and V. Wendisch, Tetrahedron: Asymmetry, 2003, 14, 189–191 CrossRef CAS; (d) A. Duursma, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2003, 125, 3700–3701 CrossRef CAS PubMed; (e) S. Kanemasa, M. Nishiuchi, A. Kamimura and K. Hori, J. Am. Chem. Soc., 1994, 116, 2324–2339 CrossRef CAS; (f) S. Yu, H. Ishida, M. E. Juarez-Garcia and J. W. Bode, Chem. Sci., 2010, 1, 637–641 RSC.
- See also: umpolung for γ-amino acid synthesis: Y. Wu, L. Hu, Z. Li and L. Deng, Nature, 2015, 523, 445 CrossRef CAS PubMed.
- (a) S. M. Kim and J. W. Yang, Org. Biomol. Chem., 2013, 11, 4737–4749 RSC; (b) M. Liu and M. P. Sibi, Tetrahedron, 2002, 58, 7991–8035 CrossRef CAS; (c) C. Cabrele, T. A. Martinek, O. Reiser and Ł. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS PubMed; (d) L. Kiss, I. M. Mándity and F. Fülöp, Amino Acids, 2017, 49, 1441–1455 CrossRef CAS PubMed; (e) B. Huang, L. Zeng, Y. Shen and S. Cui, Angew. Chem., Int. Ed., 2017, 56, 4565–4568 CrossRef CAS PubMed; (f) J.-S. Yu, H. Noda and M. Shibasaki, Angew. Chem., Int. Ed., 2018, 57, 818–822 CrossRef CAS PubMed; (g) C. G. Goodman and J. S. Johnson, J. Am. Chem. Soc., 2015, 137, 14574–14577 CrossRef CAS PubMed.
- The term ‘double umpolung’ is used in accord with Seebach who first articulated its use (including a depiction of the inverted ketene in Scheme 1): (a) D. Seebach, R. Henning and J. Gonnermann, Chem. Ber., 1979, 112, 234–248 CrossRef CAS. However, it has also been used to describe the application of umpolung sequentially at the same site: (b) R. C. F. Jones and J. R. Nichols, Tetrahedron, 2013, 69, 4114–4119 CrossRef CAS.
- (a) S. Ranganathan, D. Ranganathan and A. K. Mehrotra, J. Am. Chem. Soc., 1974, 96, 5261–5262 CrossRef CAS; (b) Z. Peng, M. Narcis and N. Takenaka, Molecules, 2013, 18, 9982 CrossRef CAS PubMed.
- S.-F. Lu, D.-M. Du, J. Xu and S.-W. Zhang, J. Am. Chem. Soc., 2006, 128, 7418–7419 CrossRef CAS PubMed.
- (a) J. Wang, H. Li, L. Zu, W. Jiang and W. Wang, Adv. Synth. Catal., 2006, 348, 2047–2050 CrossRef CAS; (b) C. Rampalakos and W. D. Wulff, J. Am. Chem. Soc., 2008, 130, 13524–13525 CrossRef PubMed; (c) X.-Q. Dong, H.-L. Teng and C.-J. Wang, Org. Lett., 2009, 11, 1265–1268 CrossRef CAS PubMed; (d) W. Yang and D.-M. Du, Chem. Commun., 2011, 47, 12706–12708 RSC; (e) Y.-Q. Deng, Z.-W. Zhang, Y.-H. Feng, A. S. C. Chan and G. Lu, Tetrahedron: Asymmetry, 2012, 23, 1647–1652 CrossRef CAS; (f) E. Kanberoğlu and C. Tanyeli, Asian J. Org. Chem., 2016, 5, 114–119 CrossRef.
- X. Yang, X. Zhou, L. Lin, L. Chang, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2008, 47, 7079–7081 CrossRef CAS PubMed.
- Enantioselective silyl nitronate additions: O. Takashi, T. Saki, D. Kanae and M. Keiji, Angew. Chem., Int. Ed., 2006, 45, 7606–7608 CrossRef PubMed.
- X. Ji and H. Huang, Org. Biomol. Chem., 2016, 14, 10557–10566 RSC.
- R. Ballini, S. Gabrielli and A. Palmieri, Eur. J. Org. Chem., 2014, 2014, 1805–1816 CrossRef CAS.
- (a) D. J. Sprague, A. Singh and J. N. Johnston, Chem. Sci., 2018, 9, 2336–2339 RSC; (b) B. A. Vara and J. N. Johnston, J. Am. Chem. Soc., 2016, 138, 13794–13797 CrossRef CAS PubMed; (c) V. T. Lim, S. V. Tsukanov, A. B. Stephens and J. N. Johnston, Org. Synth., 2016, 93, 88–99 CrossRef CAS PubMed; (d) S. V. Tsukanov, M. D. Johnson, S. A. May, M. Rosemeyer, M. A. Watkins, S. P. Kolis, M. H. Yates and J. N. Johnston, Org. Process Res. Dev., 2016, 20, 215–226 CrossRef CAS PubMed; (e) K. E. Schwieter and J. N. Johnston, ACS Catal., 2015, 5, 6559–6562 CrossRef CAS PubMed; (f) A. Singh, R. A. Yoder, B. Shen and J. N. Johnston, J. Am. Chem. Soc., 2007, 129, 3466–3467 CrossRef CAS PubMed; (g) B. A. Vara, A. Mayasundari, J. C. Tellis, M. W. Danneman, V. Arredondo, T. A. Davis, J. Min, K. Finch, R. K. Guy and J. N. Johnston, J. Org. Chem., 2014, 79, 6913–6938 CrossRef CAS PubMed; (h) D. M. Makley and J. N. Johnston, Org. Lett., 2014, 16, 3146–3149 CrossRef CAS PubMed; (i) M. C. Dobish, F. Villalta, M. R. Waterman, G. I. Lepesheva and J. N. Johnston, Org. Lett., 2012, 14, 6322–6325 CrossRef CAS PubMed; (j) T. A. Davis, M. W. Danneman and J. N. Johnston, Chem. Commun., 2012, 48, 5578–5580 RSC; (k) T. A. Davis and J. N. Johnston, Chem. Sci., 2011, 2, 1076–1079 RSC; (l) B. Shen and J. N. Johnston, Org. Lett., 2008, 10, 4397–4400 CrossRef CAS PubMed; (m) J. C. Wilt, M. Pink and J. N. Johnston, Chem. Commun., 2008, 4177–4179 RSC; (n) A. Singh and J. N. Johnston, J. Am. Chem. Soc., 2008, 130, 5866–5867 CrossRef CAS PubMed; (o) B. M. Nugent, R. A. Yoder and J. N. Johnston, J. Am. Chem. Soc., 2004, 126, 3418–3419 CrossRef CAS PubMed.
- (a) M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068–6071 CrossRef CAS PubMed; (b) Y. Toda, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2014, 136, 14734–14737 CrossRef CAS PubMed; (c) B. A. Vara, T. J. Struble, W. Wang, M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2015, 137, 7302–7305 CrossRef CAS PubMed; (d) M. T. Knowe, M. W. Danneman, S. Sun, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2018, 140, 1998–2001 CrossRef CAS PubMed.
- D. J. Sprague, B. M. Nugent, R. A. Yoder, B. A. Vara and J. N. Johnston, Org. Lett., 2015, 17, 880–883 CrossRef CAS PubMed.
- T. A. Davis, J. C. Wilt and J. N. Johnston, J. Am. Chem. Soc., 2010, 132, 2880 CrossRef CAS PubMed.
- T. A. Davis, M. C. Dobish, K. E. Schwieter, A. C. Chun and J. N. Johnston, Org. Synth., 2012, 89, 380–393 CrossRef CAS.
- (a) B. M. Trost, D. L. Van Vranken and C. Bingel, J. Am. Chem. Soc., 1992, 114, 9327–9343 CrossRef CAS; (b) R. Coffinier, M. E. Assal, P. A. Peixoto, C. Bosset, K. Miqueu, J.-M. Sotiropoulos, L. Pouységu and S. Quideau, Org. Lett., 2016, 18, 1120–1123 CrossRef CAS PubMed.
- H. Kim, C. Yen, P. Preston and J. Chin, Org. Lett., 2006, 8, 5239–5242 CrossRef CAS PubMed.
- J. Kwiatkowski and Y. Lu, Chem. Commun., 2014, 50, 9313–9316 RSC.
- (a) B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027 CrossRef CAS PubMed; (b) J. P. Shackleford, B. Shen and J. N. Johnston, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 44–46 CrossRef CAS PubMed; (c) K. E. Schwieter, B. Shen, J. P. Shackleford, M. W. Leighty and J. N. Johnston, Org. Lett., 2014, 16, 4714–4717 CrossRef CAS PubMed; (d) K. E. Schwieter and J. N. Johnston, Chem. Sci., 2015, 6, 2590–2595 RSC; (e) K. Tokumaru, K. Bera and J. N. Johnston, Synthesis, 2017, 49, 4670–4675 CrossRef CAS PubMed; (f) K. Tokumaru and J. N. Johnston, Chem. Sci., 2017, 8, 3187–3191 RSC.
- (a) K. E. Schwieter and J. N. Johnston, Chem. Commun., 2016, 52, 152–155 RSC; (b) J. Li, M. J. Lear, Y. Kawamoto, S. Umemiya, A. R. Wong, E. Kwon, I. Sato and Y. Hayashi, Angew. Chem., Int. Ed., 2015, 54, 12986–12990 CrossRef CAS PubMed.
- Preliminary mechanistic studies suggest intramolecular oxygen atom transfer from nitro to amide..
- R. Ballini and M. Petrini, Adv. Synth. Catal., 2015, 357, 2371–2402 CrossRef CAS.
- A. S. Demir, C. Tanyeli, Ö. Şeşenoǧlu, Ş. Demiç and Ö. Ö. Evin, Tetrahedron Lett., 1996, 37, 407–410 CrossRef CAS.
- J. Guillon, M. Daoust, D. Radulovic, M. Boulouard, P. Dallemagne, E. Legrand, S. Rault, M. A. Quermonne and M. Robba, Eur. J. Med. Chem., 1996, 31, 335–339 CrossRef CAS.
- This compound measured at 87% ee, starting from 94% ee of compound 10a, and appears to occur during base-mediated isocyanate formation or after..
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details and analytical data for all new compounds. See DOI: 10.1039/c8sc04330b |
| This journal is © The Royal Society of Chemistry 2019 |