
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective recognition of choline phosphate by tripodal hexa-urea receptors with dual binding sites: crystal and solution evidence†
Wei
Zuo‡
,
Chuandong
Jia‡
,
Huizheng
Zhang
,
Yanxia
Zhao
,
Xiao-Juan
Yang
 * and
Biao
Wu
*
* and
Biao
Wu
*
Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an 710069, China. E-mail: yangxiaojuan@nwu.edu.cn; wubiao@nwu.edu.cn
First published on 27th December 2018
Abstract
Two tripodal hexa-urea receptors functionalized with aromatic terminal groups are capable of binding choline phosphate (CP). Crystal structures of the host–guest complexes reveal that the zwitterion CP is efficiently encapsulated in the tripodal hosts in a dual-site binding mode. The phosphate tail of CP is coordinated by the urea groups and the quaternary ammonium head is bound in a ‘composite aromatic box’ through cation–π and hydrogen-bonding interactions. Such a partial aromatic binding environment for the Me3N–+ cation mimics that of most enzymes catalyzing the conversion of quaternary ammonium substrates. Moreover, NMR, ESI-MS, and fluorescence studies demonstrate the selective binding and sensing of CP over other competing species such as ADP, ATP, choline and derivatives.
Introduction
Inorganic and organic phosphate species play pivotal roles in biological systems.1 Natural phosphocholine (PC) derivatives (phospholipids) are a major component of cell membranes. The simple choline phosphate, Me3NCH2CH2OPO3− (CP), which contains a trimethylammonium head and a divalent phosphate tail (monoester), is an important phosphorylated compound in many biological processes. It is generated during the conversion of ATP to ADP (ATP + choline → ADP + CP) by choline kinase (ChoK), with the transfer of a phosphate group from ATP to choline, and serves as an intermediate for the biosynthesis of phosphatidylcholine (PC). The overexpression of an isoform of ChoK has been associated with human cancers, resulting in an increase of the CP levels required for the growth of cancer cells.2,3 Hence, in order to get deep insight into these bio-processes, it is essential to study the complexation and sensing of CP by synthetic receptors.Due to the zwitterionic and strongly solvating nature of choline phosphate, receptors for this guest and related derivatives need to be deliberately designed, e.g. by installing synergetic dual binding sites, to mimic biological systems. In general, the quaternary ammonium head is bound through cation–π interactions by a variety of macrocyclic arenes, such as calix[6]arene,4,5 cavitand,6 and hemicryptophane moieties,7 while the phosphate tail is bound by either hydrogen bonding or metal coordination.4–7 For example, de Mendoza et al.4a designed a receptor for dioctanoyl-L-α-phosphatidylcholine (DOPC), which contains a guanidinium moiety for H-bonding with the phosphate monoanion and a calix[6]arene for the Me3N–+ head, by mimicking the antibody–antigen complex McPC603-PC.8 On the other hand, phospholipase C, a zinc-containing metallo-enzyme that binds PC via simultaneous zinc-phosphate coordination and cation–π interactions,9 also promoted the design of hosts for selective binding of DOPC.6a,7 Nevertheless, such hosts often suffer from difficulties such as complicated synthetic routes, low yield, low selectivity (ion–ion interactions or metal-coordination), and lack of crystal structures for direct evidence of the host–guest interactions. Moreover, contrary to the commonly focused PC (di-ester) derivatives, studies targeted on CP are scarce. To our knowledge, only two examples of artificial receptors for CP have been reported.7,10 Thus, exploration of new strategies for selective binding and sensing of CP is still highly desirable.
Anion coordination chemistry is an important branch in supramolecular chemistry.11 We have developed a series of oligo-urea ligands for anion coordination.12 The tripodal hexa-urea L1 (Scheme 1)13 and related ligands14 display efficient coordination with the divalent sulfate ion. Recently, the triple anion helicate formed from a bis–bis(urea) ligand with a methylene-diphenylene spacer was found to exhibit an ‘aromatic box’ that can selectively bind choline and its derivatives.15 Therefore, we reasoned that the hexa-urea moiety with aromatic terminal groups may provide the necessary dual binding sites for the anionic choline phosphate. Compared to the charge neutral PC (phosphatidylcholine), the CP carries an overall negative charge −1 with a dianionic mono-ester ROPO32− tail, which may require a higher binding affinity from the receptor than the monoanionic di-ester [(RO)2PO2]− (in PC). Nevertheless, this divalent character of ROPO32− is similar to that of SO42− and should match the hexa-urea binding cleft. Meanwhile, the aromatic systems can bind the trimethylammonium head [Me3N–+] by cation–π interactions.
 | ||
| Scheme 1 Schematic representation of (a) L1 and L2 and (b) different guests including CP, Ch, BP, BTA, DBP, taurine, betaine, ADP and ATP. | ||
To this end, we modified ligand L1 and synthesized L2 by incorporating naphthyl groups into the tripodal scaffold to enhance the binding with the Me3N–+ head and also to enable fluorescence sensing of the binding event. Encouragingly, both L1 and L2 are capable of recognizing CP, wherein the phosphate tail is encapsulated by the hexa-urea cavity and the trimethylammonium moiety is bound by a ‘composite aromatic box’.16 Herein we report the binding and fluorescent sensing of CP by ligands L1 and L2, including crystal structures of the host–guest complexes and solution studies on the selectivity toward CP over other analogues.
Results and discussion
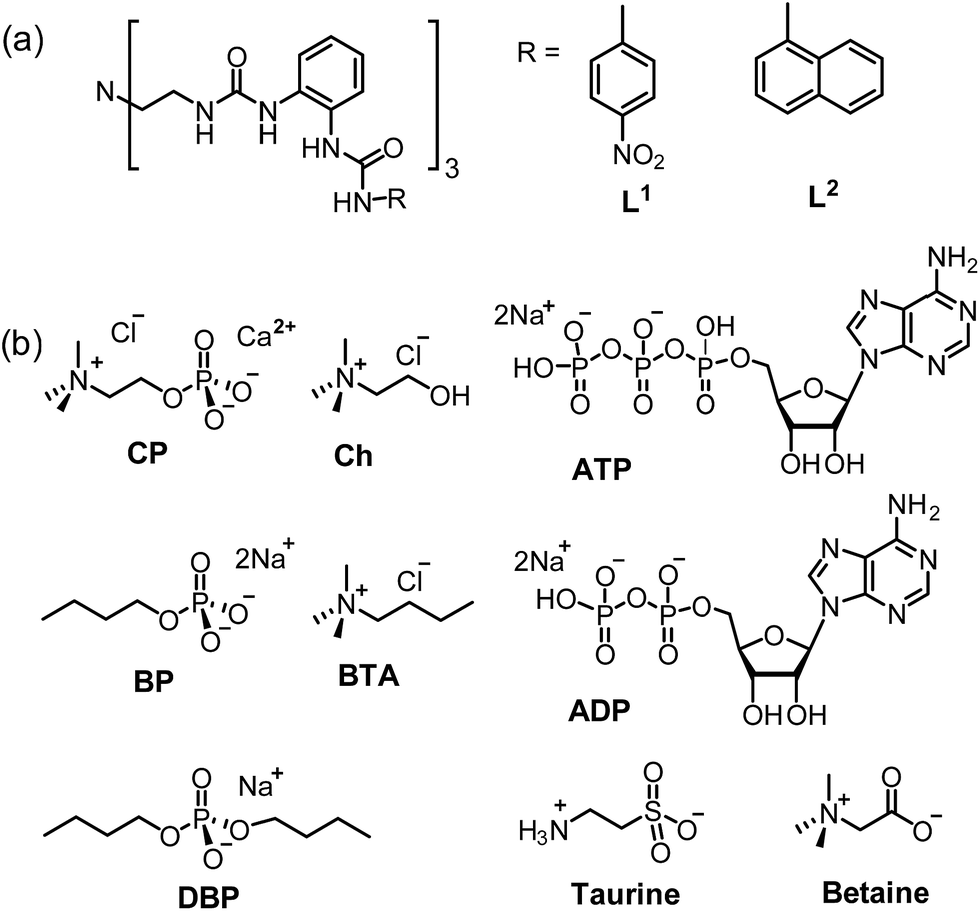
Since ligand L1 can readily bind the sulfate ion by complementary coordination, we first examined if L1 or L2 could coordinate with the phosphate ion (PO43−), which has a similar size and shape to sulfate, and then further bind choline (Ch), which has a Me3N–+ head and a hydroxyl tail (Scheme 1). Interestingly, a ternary complex Ch2[L2·PO4·Ch] (1) was obtained by mixing the naphthyl-substituted ligand L2 with Ch3PO4 (generated in situ from choline hydroxide and H3PO4). X-ray diffraction analysis of complex 1 (Fig. 1) reveals a [PO4·L2]3− moiety that is similar to the previous sulfate complex of L1.13 The PO43− ion is encapsulated in the tripodal cavity by all six urea groups through twelve hydrogen bonds (black dashed lines, N⋯O distances range from 2.708 to 3.038 Å, av. 2.813 Å; N–H⋯O angles from 142° to 173°, av. 156°). Further to this anion capsule, a choline cation ([Me3NCH2CH2OH]+) is readily attached at the open end of the tripodal ligand. The hydroxyl tail of Ch points inwards and associates to one oxygen atom of PO43−via a strong hydrogen bond (green dashed line, O⋯O distance: 2.707 Å, O–H⋯O angle: 163°). This is similar to the binding of α-methylcholine by a phosphate-coordination triple helicate cage reported recently by us.15b The other end of choline, i.e. the trimethylammonium head, is bound by cation–π interactions with one naphthyl group (purple dashed line, N⋯centroid distance: 4.673 Å). The other two choline ions, which serve as counter cations, are located nearby by hydrogen bonding and electrostatic interactions (Fig. S3, ESI†).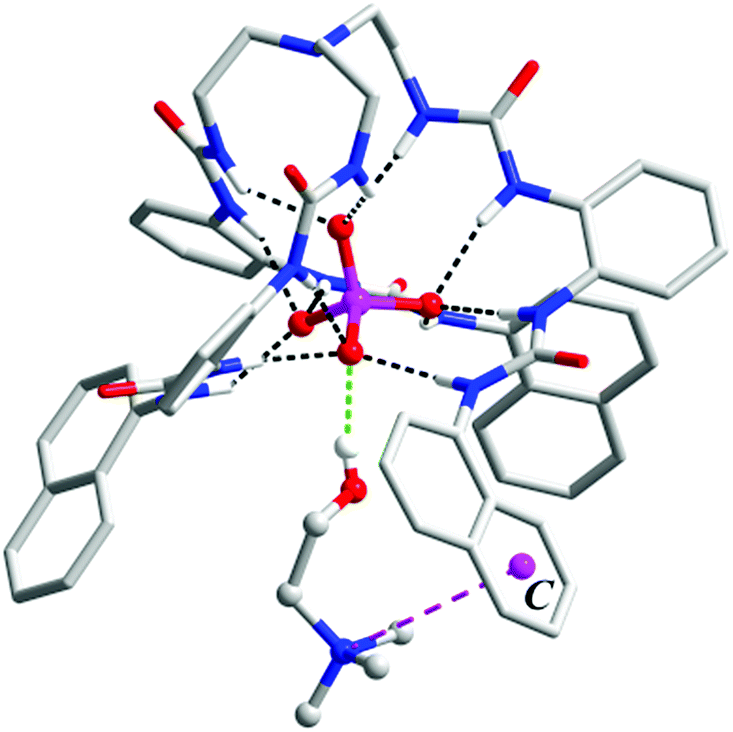 | ||
| Fig. 1 Crystal structure of the ternary complex [L2·PO4·Ch]2− (1) showing the binding of the PO43− anion and Ch+ cation, respectively. Non-acidic hydrogen atoms, solvents, and counter cations are omitted for clarity. The centroid of one aryl ring of a terminal naphthyl unit is shown as C. | ||
The hydrogen-bonded phosphate⋯choline combination in complex 1 implies possible binding of the host toward choline phosphate (CP), wherein the phosphate group is linked to choline via a direct O–P bond. To test this, ligands L1 and L2 were treated with the monoanionic choline phosphate [Me3NCH2CH2OPO3]− (CP, as TBA+ or TMA+ salt generated in situ from calcium phosphorylcholine chloride [Cl·Me3NCH2CH2OPO3·Ca] and TBAOH or TMAOH, respectively). Fortunately, in both cases single crystals of the inclusion complex, with the composition (TBA)[L1⊃CP] (complex 2) and (TMA)[L2⊃CP] (complex 3), were obtained by diffusion of diethyl ether to an MeCN solution.
In the crystal structure of complex 2 (Fig. 2), the phosphate group of CP is coordinated by all six urea units through 12 hydrogen bonds (black dashed lines, N⋯O distances range from 2.748 to 3.053 Å, av. 2.894 Å; N–H⋯O angles from 145° to 166°, av. 153°). However, in comparison to complex 1, only the three ‘free’ oxygen atoms of ROPO32− are bound, each through four (instead of three) H-bonds from urea NH donors. The fourth O atom, which is esterified by the choline arm, is not involved in H-bonding. Meanwhile, the orientation of the tetrahedral phosphate is reversed, with one vertex (attached by the choline arm) pointing to the outer substituents so that the trimethylammonium fragment can contact with the aryl rings.
 | ||
| Fig. 2 (a) Crystal structure of complex [L1⊃CP]− (2) (non-acidic H atoms, solvents, and the counter cation are omitted for clarity); (b) interactions of the zwitterion CP with the receptor: hydrogen bonds around the phosphate tail (black dashed lines) and the ‘composite aromatic box’ (green dashed circle) for the Me3N–+ head. The centroid of one nitrophenyl group is shown as C. | ||
Within the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complex, the Me3N–+ head forms an ‘intramolecular’ cation–π interaction with one nitrophenyl group (purple dashed line, N⋯centroid distance: 5.454 Å; Fig. 2b). However, a closer inspection revealed that the quaternary ammonium head is also bound by ‘intermolecular’ C–H⋯O hydrogen bonds from a urea carbonyl and a nitro group of other surrounding [L1⊃CP] units (blue dashed lines, C⋯O distances range from 3.168 to 3.434 Å, av. 3.264 Å, Fig. 2b). Overall, the CP guest is accommodated in a ‘composite aromatic box’ (illustrated as a green dashed circle in Fig. 2b).16 It is known that quaternary ammonium is a very important motif in biomolecules (such as choline and its derivatives), and the cation–π interactions are recognized to be extensively present for the binding of this cationic group. However, it was found that, although synthetic receptors frequently (78%) employ a highly hydrophobic, complete aromatic box containing three or four (or more) aromatic rings for binding, in related enzymes the majority (93%) of the Me3N–+ binding site adopts a combination of both aromatic (one or two aromatic residues) and charged or polar interactions. This latter feature is termed a ‘composite aromatic box’ by Nagy and Vértessy and it is proposed that such a general architecture may be advantageous for assisting multiple steps of enzyme action.16
:1 host–guest complex, the Me3N–+ head forms an ‘intramolecular’ cation–π interaction with one nitrophenyl group (purple dashed line, N⋯centroid distance: 5.454 Å; Fig. 2b). However, a closer inspection revealed that the quaternary ammonium head is also bound by ‘intermolecular’ C–H⋯O hydrogen bonds from a urea carbonyl and a nitro group of other surrounding [L1⊃CP] units (blue dashed lines, C⋯O distances range from 3.168 to 3.434 Å, av. 3.264 Å, Fig. 2b). Overall, the CP guest is accommodated in a ‘composite aromatic box’ (illustrated as a green dashed circle in Fig. 2b).16 It is known that quaternary ammonium is a very important motif in biomolecules (such as choline and its derivatives), and the cation–π interactions are recognized to be extensively present for the binding of this cationic group. However, it was found that, although synthetic receptors frequently (78%) employ a highly hydrophobic, complete aromatic box containing three or four (or more) aromatic rings for binding, in related enzymes the majority (93%) of the Me3N–+ binding site adopts a combination of both aromatic (one or two aromatic residues) and charged or polar interactions. This latter feature is termed a ‘composite aromatic box’ by Nagy and Vértessy and it is proposed that such a general architecture may be advantageous for assisting multiple steps of enzyme action.16
Thus, complex 2 (and the analogue 3, vide infra) represents a nice artificial receptor showing not only a complementary binding cavity for the phosphate mono-ester but also a ‘composite’ (or ‘partial’) aromatic environment for Me3N–+ that mimics the proteins in the solid state (Fig. S5†). Moreover, it is worth noting that the current complexes are rare examples of X-ray crystal evidence of a host–guest complex with CP. The only other known example is the complex of a pyrogallol[4]arene receptor, in which the cationic Me3N–+ head of CP forms cation–π interactions while the phosphate is involved in hydrogen bonding with the hydroxyl groups of the receptor and lattice water molecules.10
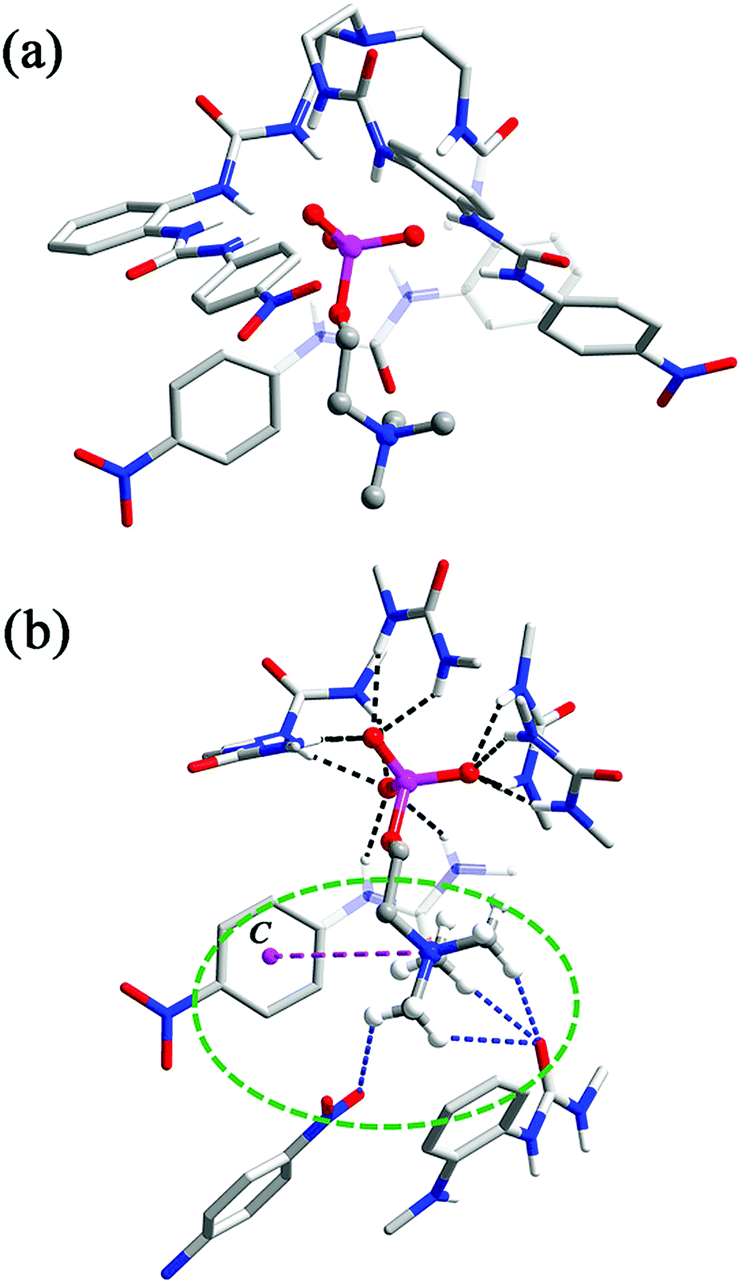
The analogous complex 3 also features 1:1 complexation of CP by ligand L2, but its structure is somewhat different from that of 2. In this case, five of the urea groups are convergent and participate in the binding to the ROPO32− tail of CP, through a total of ten N–H⋯O hydrogen bonds to the three non-esterified O atoms (one of them receives only two H-bonds because a urea group is twisted away from the binding pocket (N8, N9, Fig. 3a)). The N⋯O distances range from 2.736 to 3.151 Å (av. 2.940 Å) and N–H⋯O angles are from 134° to 172° (av. 151°, black dashed lines, Fig. 3b). The ‘missing’ urea group, instead, forms two intermolecular hydrogen bonds with a urea carbonyl of another inclusion complex. A pair of symmetry-related urea⋯urea interactions dimerizes two [L2⊃CP]− units in close proximity (Fig. S4†). This twisting of the urea arm may be caused by both of the steric and electronic reasons: the larger naphthyl groups may force one arm away and the lack of electron-withdrawing groups may weaken the hydrogen bonding ability of the urea NH donors. Nonetheless, this deformation facilitates a further cation–π interaction between the phenylene linker (centroid C1 in Fig. 3b) of this bis-urea arm and the trimethylammonium fragment of CP (purple dashed line, N⋯C1 distance: 4.524 Å) in addition to that provided by a terminal naphthyl ring (centroid C2, N⋯C2: 4.717 Å, Fig. 3b). As in complex 2, there are also interactions between the Me3N–+ cation and nearby carbonyl functionalities (blue dashed lines, C⋯O distances range from 3.191 to 3.450 Å, av. 3.306 Å; Fig. 3b).
 | ||
| Fig. 3 (a) Crystal structure of complex [L2⊃CP]− (3) (non-acidic H atoms, solvents, and the counter cation are omitted for clarity); (b) N–H⋯O hydrogen bonds (black dashed lines), cation–π (purple dashed lines), and C–H⋯O (blue dashed lines) interactions between L2 and CP. C1 and C2 represent the centroids of aryl groups. | ||
The interactions between L1/L2 and CP in solution were studied by 1H NMR experiments in DMSO-d6/2% H2O. With the addition of CP, all of the NH signals of L1 and L2 experienced considerable downfield shifts (Fig. S9 and S10†) due to H-bonding of phosphate by the NH groups. The addition of 2.0 equiv. of CP resulted in saturated binding with no further changes when more guests were added. The change of the NH chemical shifts of L2 (Δδav = 1.33 ppm) is slightly larger than that of L1 (Δδav = 1.27 ppm). Meanwhile, the signal of methyl groups of encaged CP in L2 experienced larger (Δδ = −0.9 ppm) upfield shifts than that of L1 with CP (Δδ = −0.5 ppm, Fig. 4 and Table S5†), implying a stronger shielding effect on CP from the aryl groups of L2 than L1.
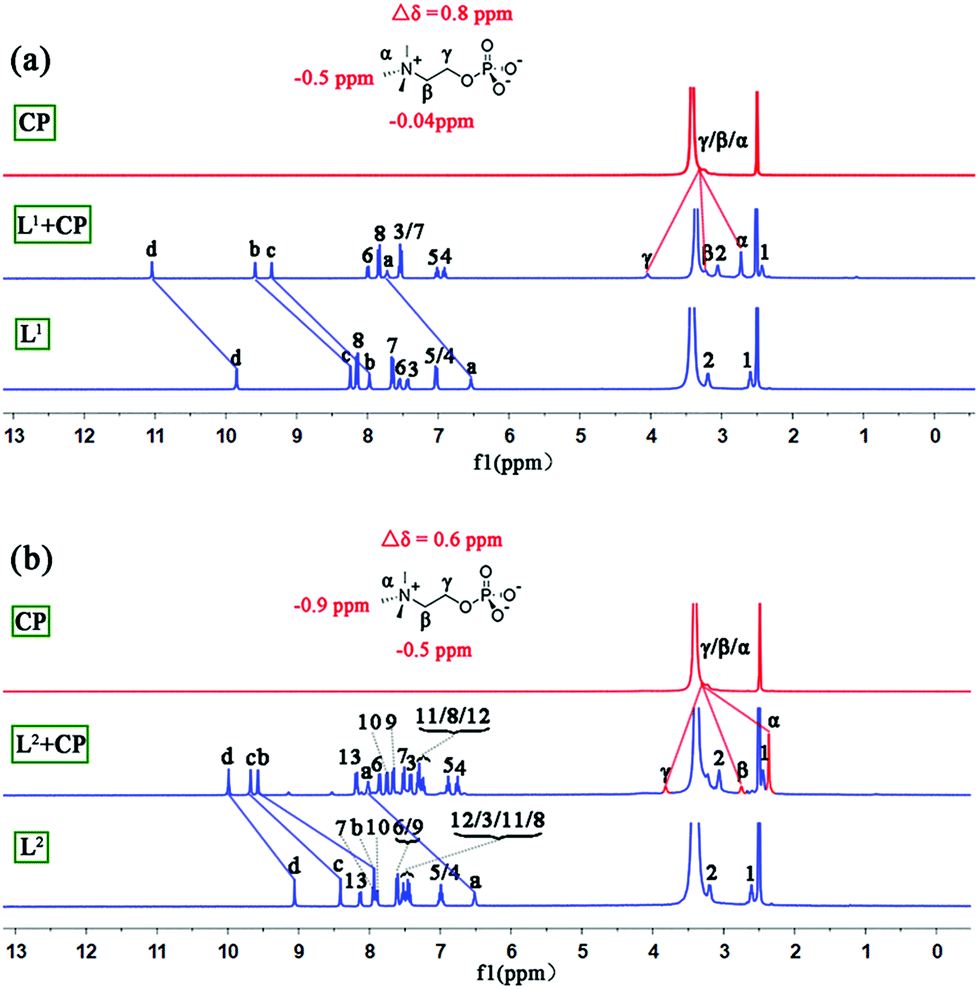 | ||
| Fig. 4 Stacking 1H NMR spectra (DMSO-d6/2% H2O, 400 MHz) of (a) CP, L1 + CP, and L1 and (b) CP, L2 + CP, and L2. | ||
Moreover, the binding ability of L2 toward CP in a more competitive solvent (DMSO-d6/D2O, 75/25, v/v; more water will cause precipitation) was investigated (Fig. S11†). Our previous work demonstrated that the tripodal cleft of L1 provides an excellent environment for the highly hydrophilic sulfate ion even in a mixed solvent containing 25% water and can reversibly extract sulfate from water to the organic phase.13 In the current work, such a water tolerance was also observed (the shielding effect on the methyl groups of CP, Δδ = −0.7 ppm, however, is slightly smaller than in the case of 2% H2O). Additionally, high-resolution mass spectrometry (HR ESI-MS) confirmed the formation of complex 1 and the 1:1 binding of L1/L2 to CP (e.g. m/z obsd 1222.4458 vs. calcd 1222.4214 for [L1 + CP]− and obsd 1237.5353 vs. calcd 1237.5131 for [L2 + CP]−; Fig. S6–S8†).
For the naphthyl-decorated L2, the binding of choline phosphate was further studied by fluorescence titrations. Upon gradual addition of CP to 10 μM solution of L2 in DMSO/2% H2O, significant fluorescence quenching of the host at 372 nm was observed, with 2.0 equiv. of CP causing the saturation of the titration (Fig. 5). A binding constant of 1.2 × 106 M−1 was obtained by fitting the titration curve with the Dynafit program17 to a 1:1 mode, which was further confirmed by Job's plot (Fig. S17†). In contrast, the binding constant (1.0 × 104 M−1) in DMSO/25% H2O was much smaller (Fig. S18†), possibly due to the decreased hydrogen bonding strength of phosphate in this aqueous solution.
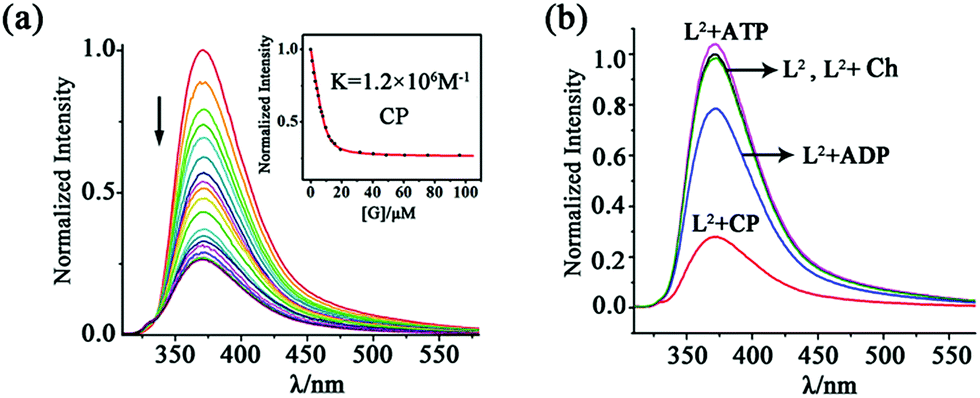 | ||
| Fig. 5 (a) Fluorescence titration of 10 μM L2 (excited at 300 nm) with CP in DMSO/2% H2O solution. Inset: fluorescence intensity at 372 nm as a function of the concentration of added CP. (b) Normalized spectra of 10 μM L2 upon addition of 2.0 equiv. of different guests in DMSO/2% H2O solution. | ||
To better understand the dual-site binding mechanism of CP by the tripodal ligands, fluorescence titrations of L2 with some related species were carried out for comparison with CP. The binding ability of the hexa-urea moiety was elucidated by using guests containing only the anionic phosphate tail, such as dibutylphosphate (DBP) and butylphosphate (BP) (Scheme 1b). It can be seen that the anion binding site shows a slightly smaller affinity to the dianionic fragment of BP (binding constant K = 6.4 × 105 M−1, Fig. S19†), which is about half of that (K = 1.2 × 106 M−1) for CP featuring the same monoester head. However, the binding to the monoanionic dibutylphosphate DBP (K = 9.2 × 103 M−1, Fig. S20†) is much weaker than that to CP. On the other hand, for butyl-trimethylammonium (BTA), which contains the quaternary ammonium head but not the phosphate tail, only a negligible fluorescence change of L2 was observed (Fig. S21†). Therefore, it appears that the coordination of the phosphate tail by the hexa-urea has more contribution to CP binding, which may in turn organize the three arms in a convergent fashion, thus providing a partial aromatic box for binding the cationic head.
Having established the strong binding of CP by L2, we then evaluated the selectivity over some related/competing guests (Scheme 1b). As mentioned above, CP is a key intermediate in the metabolic process and may be used to monitor the function of choline kinase. To this respect, the three species, i.e. choline (Ch), ADP and ATP, which are all involved in the process of ATP to ADP conversion, were tested.
The addition of ADP caused a fluorescence quenching of host L2 but the rate is much smaller than in the case of CP. While 2.0 equiv. of CP led to 80% quenching, only 20% decrease was generated by 2.0 equiv. of ADP (and 15 equivalents were needed to complete the change; Fig. 5b and S22†). Consequently, the binding constant of ADP (7.8 × 104 M−1) is about 15 times smaller than that of CP (1.2 × 106 M−1). This fluorescence quenching induced by the guest could be attributed to the promotion of the photo-induced electron transfer (PET) process from the tren nitrogen to the naphthalene fluorophores after guest binding.18 In contrast, 2.0 equiv. of ATP caused a slight fluorescence enhancement of L2 (Fig. 5b), which was further increased with large amounts of ATP (up to 50 equiv., Fig. S23†). This is possibly due to the protonation of the tertiary bridgehead amine of L2 by ATP (pKa = 4.06), which is a stronger acid than ADP (pKa = 6.40),19 leading to prohibition of the PET process and thus fluorescence turn-on.20 The protonation mechanism is further proved by the fact that HClO4 can also induce fluorescence enhancement of the ligand (Fig. S24†). In addition, Ch caused a negligible fluorescence change of L2 (Fig. S25†).
On the other hand, 1H NMR spectroscopy revealed that the guests ADP (Δδav = 0.90 ppm) and ATP (Δδav = 0.95 ppm) led to some downfield shifts of the urea NH signals of L2 (Fig. S24, Table S5†) which are smaller than those with CP. The proton He of ATP and He’ of ADP show about 0.2 ppm upfield shifts, demonstrating the shielding effect imposed by L2. The signal of H12 proton on the naphthyl unit of L2 at 7.52 ppm shifted to 7.38 ppm (for the ADP adduct) and 7.46 ppm (for the ATP adduct), respectively, indicating a stronger π–π interaction between ADP and the naphthyl groups than ATP. This may explain why the binding constant of L2·ADP is higher than that of L2·ATP, although some protons of L2 experience very similar shifts in the presence of ADP or ATP (Fig. S26†). Competitive experiments with these guests were also carried out. Significant fluorescence quenching (about 50%) of L2 at 372 nm was still observed in the presence of 1.0 equiv. of CP, ADP and ATP (Fig. S27†), although the decrease rate is smaller than that (about 65%) with CP alone. The results demonstrate that L2 shows considerable selectivity to CP over some competitive guests (ADP, ATP and Ch).
Next, two related compounds, i.e. the zwitterionic choline derivatives taurine and betaine that contain different anionic tails (Scheme 1b), were also examined. These two guests gave rise to no obvious fluorescence quenching of L2 in DMSO/2% water (Fig. S28 and S29†), highlighting again the good selectivity of L2 for CP and the crucial role of the phosphate moiety in this recognition process. Moreover, consistent with the fluorescence studies, 1H NMR spectroscopy indicated no complexation between taurine or betaine and L2 (Fig. S30†).
Conclusions
Two tripodal hexa-urea receptors with aromatic substituents are able to encapsulate choline phosphate (CP) even in a competitive aqueous medium. The exact binding mode is revealed by the crystal structures, which show the coordination of phosphate by the urea groups and binding of the quaternary ammonium head by a ‘composite aromatic box’ mimicking the key interactions found in biological systems. NMR and fluorescence experiments indicate that the receptors show certain selectivity to choline phosphate over other competitive species. This work presents a nice example of both X-ray evidence and solution studies on the binding of CP and should open a new avenue for the mimicking of anionic or zwitterionic guest binding. Further improvement of the selectivity of the receptor for such guests is underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21771144, 21772154 and 21701132).Notes and references
- (a) T. Pawson and J. D. Scott, Trends Biochem. Sci., 2005, 30, 286 CrossRef CAS PubMed; (b) S. Chakraborti, S. Das, P. Kar, B. Ghosh, K. Samanta, S. Kolley, S. Ghosh, S. Roy and T. Chakraborti, Mol. Cell. Biochem., 2007, 298, 1 CrossRef CAS PubMed; (c) A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603 CrossRef CAS PubMed.
- (a) C. Sohlenkamp, I. M. López-Lara and O. Geiger, Prog. Lipid Res., 2003, 42, 115 CrossRef CAS PubMed; (b) G. J. Doherty and H. T. McMahon, Annu. Rev. Biophys., 2008, 37, 65 CrossRef CAS PubMed; (c) K. Glunde, Z. M. Bhujwalla and S. M. Ronen, Nat. Rev. Cancer, 2011, 11, 835 CrossRef CAS PubMed; (d) B. Rubio-Ruíz, A. Conejo-García, P. Ríos-Marco, M. P. Carrasco-Jiménez, J. Segovia, C. Marco, M. A. Gallo, A. Espinosa and A. Entrena, Eur. J. Med. Chem., 2012, 50, 154 CrossRef PubMed.
- (a) A. Peschel, R. W. Jack, M. Otto, L. V. Collins, P. Staubitz, G. Nicholson, H. Kalbacher, W. F. Nieuwenhuizen, G. Jung, A. Tarkowski, K. P. M. van Kessel and J. A. G. van Strijp, J. Exp. Med., 2001, 193, 1067 CrossRef CAS PubMed; (b) S. L. Hazen and G. M. Chisolm, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12515 CrossRef CAS PubMed; (c) A. Yalcin, B. Clem, S. Makoni, A. Clem, K. Nelson, J. Thornburg, D. Siow, A. N. Lane, S. E. Brock, U. Goswami, J. W. Eaton, S. Telang and J. Chesney, Oncogene, 2010, 29, 139 CrossRef CAS PubMed; (d) M. Sahun-Roncero, B. Rubio-Ruiz, G. Saladino, A. Conejo-Garcia, A. Espinosa, A. Velazquez-Campoy, F. L. Gervasio, A. Entrena and R. Hurtado-Guerrero, Angew. Chem., Int. Ed., 2013, 52, 4582 CrossRef CAS PubMed.
- (a) J. O. Magrans, A. R. Ortiz, M. A. Molins, P. H. P. Lebouille, J. Sanchez-Quesada, P. Prados, M. Pons, F. Gago and J. de Mendoza, Angew. Chem., Int. Ed., 1996, 35, 1712 CrossRef CAS; (b) F. Cuevas, S. D. Stefano, J. O. Magrans, P. Prados, L. Mandolini and J. de Mendoza, Chem.–Eur. J., 2000, 6, 3228 CrossRef CAS PubMed.
- (a) S. Moerkerke, J. Wouters and I. Jabin, J. Org. Chem., 2015, 80, 8720 CrossRef CAS PubMed; (b) E. Brunetti, S. Moerkerke, J. Wouters, K. Bartik and I. Jabin, Org. Biomol. Chem., 2016, 14, 10201 RSC; (c) P. J. Costa, I. Marques and V. Félix, Biochim. Biophys. Acta, 2014, 1838, 890 CrossRef CAS PubMed.
- (a) F. H. Zelde, R. Salvio and J. Rebek Jr, Chem. Commun., 2006, 1280 RSC; (b) L. Trembleau and J. Rebek Jr, Science, 2003, 301, 1219 CrossRef CAS PubMed; (c) P. Ballester and M. A. Sarmentero, Org. Lett., 2006, 8, 3477 CrossRef CAS PubMed.
- D. Zhang, G. Gao, L. Guy, V. Robert, J.-P. Dutasta and A. Martinez, Chem. Commun., 2015, 51, 2679 RSC.
- (a) Y. Satow, G. H. Cohen, E. A. Padlan and D. R. Davies, J. Mol. Biol., 1986, 190, 593 CrossRef CAS PubMed; (b) R. Glockshuber, J. Stadlmüller and A. Pliickthun, Biochemistry, 1991, 30, 3049 CrossRef CAS PubMed; (c) S. Rudikoff, M. Potter, D. M. Segal, E. A. Padlan and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 3689 CrossRef CAS PubMed.
- (a) S. F. Martin, B. C. Follows, P. J. Hergenrother and B. K. Trotter, Biochemistry, 2000, 39, 3410 CrossRef CAS PubMed; (b) E. Hough, L. K. Hansen, B. Birknes, K. Jynge, S. Hansen, A. Hordvik, C. Little, E. Dodson and Z. Derewenda, Nature, 1989, 338, 357 CrossRef CAS PubMed; (c) J. H. Exton, J. Biol. Chem., 1990, 265, 1 CAS.
- I. Fujisawa, Y. Kitamura, R. Okamoto, K. Murayam, R. Kato and K. Aoki, J. Mol. Struct., 2013, 1038, 188 CrossRef CAS.
- (a) K. Bowman-James, A. Bianchi and E. García-España, Anion Coordination Chemistry, Wiley-VCH, Weinheim, 2011 CrossRef; (b) P. A. Gale, E. N. W. Howe, X. Wu and M. J. Spooner, Coord. Chem. Rev., 2018, 375, 333 CrossRef CAS; (c) A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603 CrossRef CAS PubMed; (d) G. I. Vargas-Zúñiga and J. L. Sessler, Coord. Chem. Rev., 2017, 345, 281 CrossRef PubMed; (e) C. Guo, S. Sun, Q. He, V. M. Lynch and J. L. Sessler, Org. Lett., 2018, 20, 5414 CrossRef CAS PubMed; (f) A. Caballero, F. Zapata and P. D. Beer, Coord. Chem. Rev., 2013, 257, 2434 CrossRef CAS; (g) J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2018, 57, 584 CrossRef CAS PubMed.
- (a) C. Jia, B. Wu, S. Li, Z. Yang, Q. Zhao, J. Liang, Q.-S. Li and X.-J. Yang, Chem. Commun., 2010, 46, 5376 RSC; (b) J. Zhao, D. Yang, Y. Zhao, X.-J. Yang, Y.-Y. Wang and B. Wu, Angew. Chem., Int. Ed., 2014, 53, 6632 CrossRef CAS PubMed; (c) B. Wu, J. Liang, J. Yang, C. Jia, X.-J. Yang, H. Zhang, N. Tang and C. Janiak, Chem. Commun., 2008, 1762 RSC; (d) S. Li, C. Jia, B. Wu, Q. Luo, X. Huang, Z. Yang, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 5721 CrossRef CAS PubMed; (e) B. Wu, F. Cui, Y. Lei, S. Li, N. S. Amadeu, C. Janiak, Y.-J. Lin, L.-H. Weng, Y.-Y. Wang and X.-J. Yang, Angew. Chem., Int. Ed., 2013, 52, 5096 CrossRef CAS PubMed; (f) D. Yang, J. Zhao, Y. Zhao, Y. Lei, L. Cao, X.-J. Yang, M. Davi, N. S. Amadeu, C. Janiak, Z. Zhang, Y.-Y. Wang and B. Wu, Angew. Chem., Int. Ed., 2015, 54, 8658 CrossRef CAS PubMed.
- (a) C. Jia, B. Wu, S. Li, X. Huang, Q. Zhao, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 486 CrossRef CAS PubMed; (b) X. Huang, B. Wu, C. Jia, B. P. Hay, M. Li and X.-J. Yang, Chem.–Eur. J., 2013, 19, 9034 CrossRef CAS PubMed.
- (a) A. Pramanik, M. E. Khansari, D. R. Powell, F. R. Fronczek and M. A. Hossain, Org. Lett., 2014, 16, 366 CrossRef CAS PubMed; (b) M. Emami Khansari, A. Mirchi, A. Pramanik, C. R. Johnson, J. Leszczynski and M. A. Hossain, Sci. Rep., 2017, 7, 6032 CrossRef PubMed.
- (a) C. Jia, W. Zuo, D. Yang, Y. Chen, L. Cao, R. Custelcean, J. Hostaš, P. Hobza, R. Glaser, Y.-Y. Wang, X.-J. Yang and B. Wu, Nat. Commun., 2017, 8, 938 CrossRef PubMed; (b) W. Zuo, Z. Huang, Y. Zhao, W. Xu, Z. Liu, X.-J. Yang, C. Jia and B. Wu, Chem. Commun., 2018, 54, 7378 RSC.
- G. N. Nagy, L. Marton, A. Contet, O. Ozohanics, L.-M. Ardelean, Á. Révész, K. Vékey, F. D. Irimie, H. Vial, R. Cerdan and B. G. Vértessy, Angew. Chem., Int. Ed., 2014, 53, 13471 CrossRef CAS PubMed.
- P. Kuzmic, Anal. Biochem., 1996, 237, 260 CrossRef CAS PubMed.
- (a) T. Gunnlaugsson, P. E. Kruger, T. C. Lee, R. Parkesh and G. M. Hussey, Tetrahedron Lett., 2003, 44, 6575 CrossRef CAS; (b) W.-X. Liu and Y.-B. Jiang, Org. Biomol. Chem., 2007, 5, 1771 RSC; (c) T. Gunnlaugsson, A. P. Davis, J. E. O'Brien and M. Glynn, Org. Lett., 2002, 4, 2449 CrossRef CAS PubMed.
- R. M. Smith and A. E. Martell, Critical Stability Constants, Springer, Boston, MA, 1975 Search PubMed.
- H. Xie, S. Yi, X. Yang and S. Wu, New J. Chem., 1999, 23, 1105 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization data and corresponding CIF files. CCDC 1859609–1859611. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04338h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |