
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogenoborane mediated allene cyclooligomerization†
Xin
Tao
,
Christian
Wölke
,
Constantin G.
Daniliuc
 ,
Gerald
Kehr
and
Gerhard
Erker
*
,
Gerald
Kehr
and
Gerhard
Erker
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: erker@uni-muenster.de
First published on 2nd January 2019
Abstract
The halogenoboranes XB(C6F5)2 (X: Cl, Br) react with allene to give a mixture of the cyclotrimer 1,3,5-trimethylenecyclohexane (1) and the related halogenoborylated cyclotetramerization products 3a and 3b. Alkyl-substituted allenes were catalytically cyclotrimerized metal-free by XB(C6F5)2 (X: Cl, Br) to give cis,trans-2,4,6-trialkyl-1,3,5-trimethylenecyclohexanes under mild conditions.
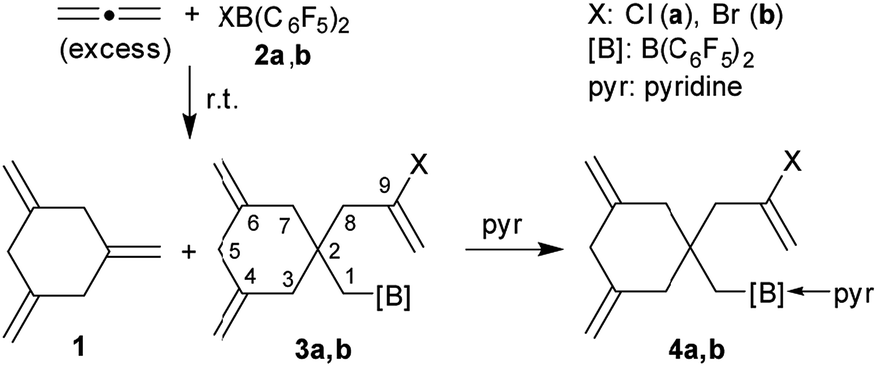
Allenes serve as important organic building blocks.1 The parent allene, propadiene, has been cyclotrimerized at a variety of transition metal catalysts, but this usually gave predominately 1,2,4-trimethylenecyclohexane and only minor amounts of the 1,3,5-trimethylenecyclohexane isomer 1.2,3 Compound 1 had been synthesized in stoichiometric reaction sequences.4 We recently developed the metal-free, HB(C6F5)2 catalysed formation of isomerically pure 1 by allene cyclotrimerization under mild conditions.5 We also had recently shown that a small series of 1-alkynes could be converted to linear oligomers by treatment with either of the halogenoboranes XB(C6F5)22a (X: Cl) or 2b (X: Br). The resulting products contained a halide substituent at one end of the oligoacetylene chain and the B(C6F5)2 functional group at the other.6 This prompted us to investigate which type of a reaction allene would undergo with either of the reagents 2a and b. We have performed these reactions and here report about their surprising outcome.
We first carried out the reaction of excess allene with ClB(C6F5)2 (2a) in d8-toluene in a Young NMR tube at room temperature. After 7 h reaction time we monitored the NMR features of a mixture that contained the products 1,3,5-trimethylenecyclohexane (1) and the chloroborylated allene tetramer 3a in a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (both present in ca. 8 mol% in the mixture) plus unreacted 2a (ca. 10 mol%) and allene (ca. 75 mol%). After 24 h the amount of the pair of allene cyclooligomerization products had almost doubled and there remained only ca. 3 mol% of the chloroborane 2a. That was almost completely consumed after 48 h reaction time at room temperature. The products 1 and 3a were identified spectroscopically from the mixture [1: δ 4.53, 2.70 (1H)]. Compound 3a shows the typical olefinic exo-methylene pairs of 1H NMR resonances at δ 5.08, 4.84 (9-CH2
:1 ratio (both present in ca. 8 mol% in the mixture) plus unreacted 2a (ca. 10 mol%) and allene (ca. 75 mol%). After 24 h the amount of the pair of allene cyclooligomerization products had almost doubled and there remained only ca. 3 mol% of the chloroborane 2a. That was almost completely consumed after 48 h reaction time at room temperature. The products 1 and 3a were identified spectroscopically from the mixture [1: δ 4.53, 2.70 (1H)]. Compound 3a shows the typical olefinic exo-methylene pairs of 1H NMR resonances at δ 5.08, 4.84 (9-CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) and δ 4.63, 4.48 (4,6-CH2) as well as the AX-spin system of the diastereotopic 3,7-CH2 pairs (δ 2.24, 2.00, 2JHH = 14.0 Hz) and the AB pattern of the 5-CH2 at δ 2.58, 2.54. The 1-CH2 group at boron and the 8-CH2 unit give rise to 1H NMR signals at δ 2.18 and δ 2.26, respectively (atom numbering scheme analogous to Fig. 1). Compound 3a shows a broad 11B NMR resonance at δ 71.5, which is typical for a planar tri-coordinate Lewis acidic borane in this situation (Δ19Fm,p = 13.2 ppm, for details see the ESI†).
) and δ 4.63, 4.48 (4,6-CH2) as well as the AX-spin system of the diastereotopic 3,7-CH2 pairs (δ 2.24, 2.00, 2JHH = 14.0 Hz) and the AB pattern of the 5-CH2 at δ 2.58, 2.54. The 1-CH2 group at boron and the 8-CH2 unit give rise to 1H NMR signals at δ 2.18 and δ 2.26, respectively (atom numbering scheme analogous to Fig. 1). Compound 3a shows a broad 11B NMR resonance at δ 71.5, which is typical for a planar tri-coordinate Lewis acidic borane in this situation (Δ19Fm,p = 13.2 ppm, for details see the ESI†).
 | ||
| Fig. 1 A view of the molecular structure of compound 4b. Selected bond lengths (Å) and angles (°): B1–C1 1.634(2) B1–N21 1.661(2) C4–C11 1.323 (2) C6–C12 1.327(2) C9–C10 1.351(9) C9–Br1 1.910(5) B1–C1–C2 124.4(1) C1–C2–C8 108.5(1) C2–C8–C9 116.3(3) C8–C9–C10 124.5(6) C8–C9–Br1 118.4(4). | ||
The reaction of allene with BrB(C6F5)2 (2b) was carried out analogously. The reaction was directly followed by NMR spectroscopy. It resulted in the formation of the products 1 and 3b in a ca. 1:2 ratio (4 h, r.t.). Compound 3b was characterized from the mixture by NMR spectroscopy. It shows similar spectra as its chloro-substituted analogue 3a (see the ESI† for details and the depicted spectra).
We prepared compound 3a on a preparative scale. After direct treatment with pyridine we isolated the Lewis adduct 4a as a white solid in 53% yield (Scheme 1). It was characterized by C, H, N elemental analysis, by spectroscopy and by X-ray diffraction (see the ESI† for details). It showed very similar parameters as the analogous bromine containing compound 4b (see below).
 | ||
| Scheme 1 Reaction of allene with the halogenoboranes 2a and 2b. | ||
Treatment of in situ prepared 3b with a slight excess of pyridine gave the adduct 4b, which we isolated in 72% yield as a white solid on a 100 mg scale. The X-ray crystal structure analysis (Fig. 1) showed the central six-membered ring that was constructed by connecting three allene units. It features the exo-methylene CC double bonds annulated at carbon atoms C4 and C6 and it has the CH2–CBrCH2 unit, derived from the fourth connected allene unit, attached at the ring carbon atom C2. This carbon atom also bears the CH2–B(C6F5)2 substituent, which has the pyridine donor added to its boron atom. The central six-membered carbocycle of compound 4b features a distorted chair-like ring conformation.
In solution (CD2Cl2) compound 4b shows the 1H NMR CH2 signals of the symmetry equivalent pair of exo-methylene groups at the ring carbons C4 and C6 and the pair of signals of the C9CH2 moiety. The C3/C7 CH2 hydrogen atoms are pairwise diastereotopic and the C8 and C1 CH2 groups both show 1H NMR singlets. Compound 4b shows a 11B NMR resonance in the typical tetra-coordinated borane range at δ −1.4 (Δ19Fm,p = 5.6 ppm).
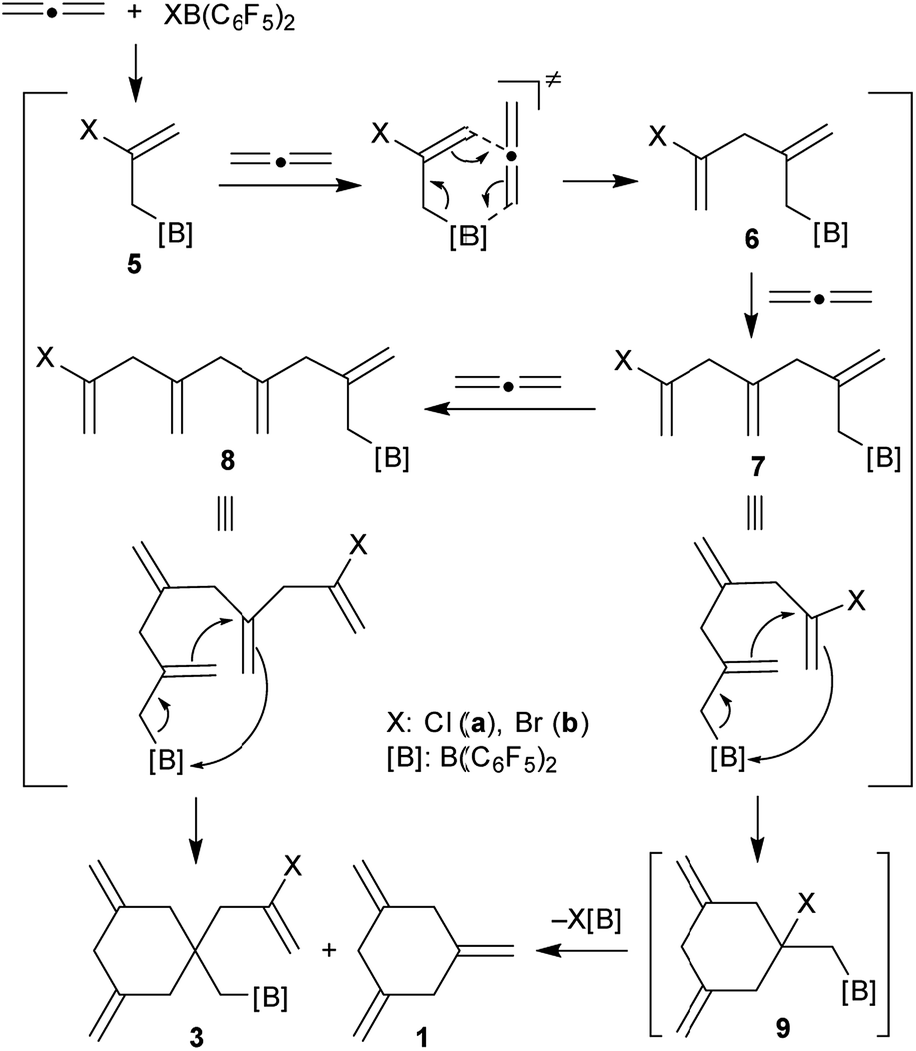
It is probably reasonable to assume that our reaction sequence starts with a halogenoboration reaction7 of allene (Scheme 2). This would generate the allylborane 5 that probably undergoes an allylboration8 reaction with a second equivalent of allene to form the intermediate 6. Since this contains an allylborane subunit it can undergo another allylboration reaction to give 7. This sequence could in principle be propagated to form a respective series of linear borylated allene oligomers 7, 8etc., were there not the attractive possibility of these systems to undergo alternative intramolecular allylboration.9 In the case of 7 this would lead to the cyclization product 9. Subsequent XB(C6F5)2 elimination provides an attractive pathway to the observed product 1,3,5-trimethylenecyclohexane (1). This would in principle constitute a cyclotrimerization of allene catalysed by the XB(C6F5)2 reagents. However, the intramolecular allylboration of 8, giving the other experimentally observed products 3, represents a competing stoichiometric reaction branch that eventually removes the XB(C6F5)2 reagent from the system (Scheme 2) and thus terminates the catalytic sequence of the formation of 1.
 | ||
| Scheme 2 Reaction pathway for the XB(C6F5)2 mediated allene cyclooligomerization. | ||
Compounds 3a,b are reactive boron Lewis acids. With the bulky tBu3P Lewis base (10) they react by ring closure10,11 to eventually give the zwitterionic product 12 (Scheme 3). As a typical example, the reaction of the in situ generated chloride containing derivative 3a was carried out with a ca. two molar equivalents of tBu3P in toluene (24 h, 60 °C). Workup with pentane and dichloromethane in this case gave a ca. 79:21 mixture of compound 12 (characterization see below) and the [tBu3PH+]Cl− phosphonium salt 11a (31P NMR: δ 47.9, 1JPH ∼463 Hz, some of the stoichiometric by-product 11a was probably lost during the workup procedure).
 | ||
| Scheme 3 Reaction of the products 3 with two molar equiv. of tBu3P. | ||
The reaction between the more reactive borane 3b and tBu3P was carried out similarly (toluene, 24 h, r.t.). Workup in this case gave the pure compound 12 as a white solid, which we isolated in 65% yield. It was characterized by C, H, N elemental analysis, by spectroscopy and by X-ray diffraction (single crystals of compound 12 were obtained from dichloromethane/pentane by the diffusion method). The X-ray crystal structure analysis shows the newly formed borataspiro[5,5]undecene framework (Fig. 2) which was selectively formed by boron induced allene tetramerization followed by phosphane induced ring closure reaction. The zwitterionic compound has the bulky tBu3P-substituent attached at the C(9)C(10) carbon–carbon double bond. The adjacent six-membered ring shows carbon atoms C4 and C6 which serve both as the ring sp2-carbons of the pair of exo-methylene CCH2 groups.
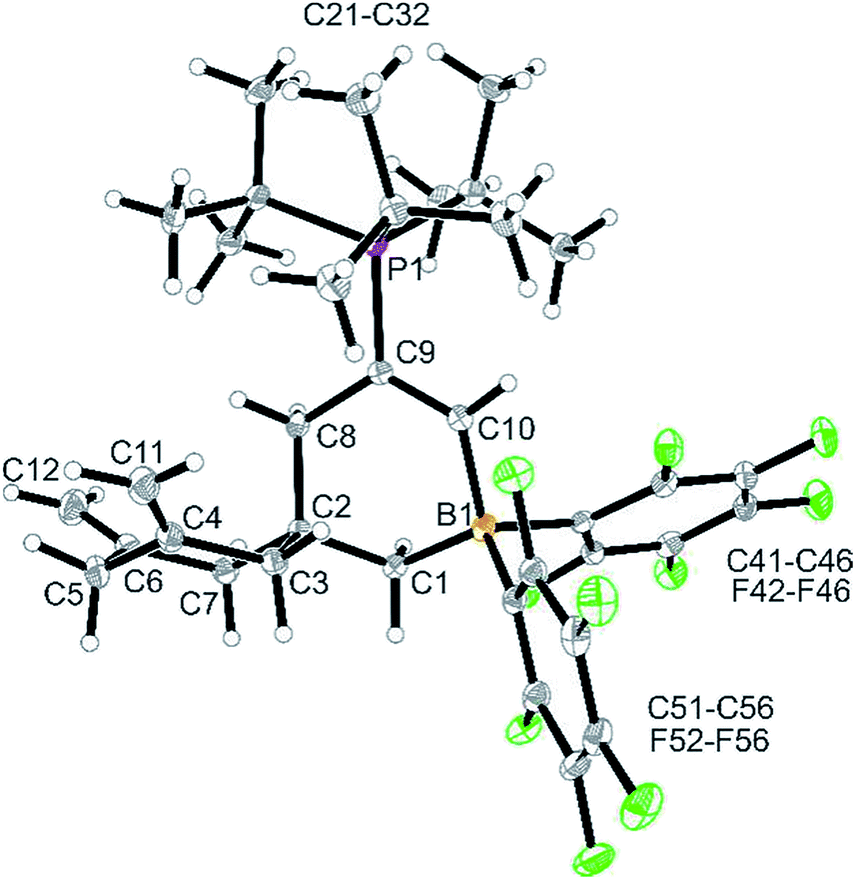 | ||
| Fig. 2 A view of the molecular structure of the zwitterionic FLP product 12. Selected bond lengths (Å) and angles (°): B1–C1 1.630(4) B1–C10 1.624(4) C4–C11 1.328(4) C6–C12 1.323(5) C9–C10 1.343(4) C9–P1 1.834(3) B1–C1–C2 113.5(2) C1–C2–C8 108.5(2) C1–B1–C10 107.0(2) C2–C2–C7 108.0(2) C2–C8–C9 114.0(2) C8–C9–C10 121.4(2) C9–C10–B1 125.7(3). | ||
In solution (CD2Cl2) compound 12 shows the typical 1H NMR doublet of the tBu3P-substituent. It features the CH signals of the newly formed internal olefinic moiety at δ 8.21 [1H: d, 3JPH = 27.8 Hz; 13C: δ 177.0 (broad)]. The BCH2 group shows up as a 1H NMR signal at δ 1.20 and the C(8)H2 methylene group as a doublet at δ 2.06 (3JPH = 4.5 Hz). The pair of olefinic exomethylene groups of the adjacent carbocyclic six-membered ring shows typical 1H NMR signals at δ 4.61/4.41 and the resonances of the pairwise diastereotopic methylene hydrogen atoms at the C3/C7 pair and at C5 (see the ESI† for details).
We also reacted a pair of alkyl-substituted allenes with the halogenoboranes 2 and found a marked dependence of the overall reaction pathway on the substitution pattern. In the two cases investigated the competition between the two previously observed pathways, namely catalytic cyclotrimerization and stoichiometric formation of a tetramer derivative, was shifted to the catalytic side.
We treated n-octylallene (13c), which we prepared according to a procedure reported by Ma et al.12 with 10 mol% of ClB(C6F5)2 (2a) in d8-toluene solution at 60 °C. Workup after 48 h reaction time involving purification by chromatography gave the cyclotrimer 14c as the major product (Scheme 4). Compound 14c was isolated in 44% yield as a colorless oil. The NMR spectra showed the symmetry features of the cis,trans-2,4,6-trialkyl-substituted 1,3,5-trimethylenecyclohexane diastereomer. It features the 1H NMR triplets of the 2,6-CH and the 4-CH ring hydrogens in a 2:1 intensity ratio. The 1-H2C unit shows one 1H NMR resonance, whereas the 3,5-H2C moieties showed two due to their stereochemically unsymmetrical environment (see the ESI† for further details). The BrB(C6F5)2 borane is an equally efficient metal-free cyclotrimerization catalyst for the n-octylallene 13c. The catalytic reaction was carried out under analogous conditions and gave the product 14c in 46% yield after workup (some minor byproduct was isolated (ca. 20%) but not positively identified as yet). We performed the XB(C6F5)2 catalysed alkylallene cyclotrimerization reaction for a second example: the reaction of n-dodecylallene (13d) with either of the Cl/BrB(C6F5)2 borane (10 mol%, 60 °C, 48 h in d8-toluene) gave the tri-substituted cyclotrimer 14d with cis,trans-attachment of the long chained alkyl groups as the major product. We isolated it as a colorless oil in 40% yield from the ClB(C6F5)2 catalysed reaction [54% with BrB(C6F5)2] (see the ESI† for the characterization of 14d).
 | ||
| Scheme 4 XB(C6F5)2 (X: Cl, Br) catalyzed cyclotrimerization of alkyl substituted allenes. | ||
We had previously shown that HB(C6F5)2 serves as an efficient metal-free catalyst for the cyclotrimerization of allene to 1,3,5-trimethylenecyclohexane and of cyclohexylallene to cis,trans-2,4,6-tricyclohexyl-1,3,5-trimethylenecyclohexane.5 Our present study shows that the strongly electrophilic halogenoboranes XB(C6F5)2 (X: Cl, Br) in principle can induce the same catalytic reaction. Both these strongly electrophilic halogeno-boranes serve as catalysts for the cyclotrimerization of long-chain n-alkylallenes 13c,d to give the respective cyclotrimers 14c,d as the major products. Ring-closure and elimination is sufficiently effective to close the catalytic cycle with liberation of the respective XB(C6F5)2 (X: Cl, Br) catalyst (Scheme 2). However, the reaction seems to be sensitive to sterics in the competition between ring-closure and further allene incorporation. In the case of the unsubstituted parent allene substrate we found a substantially competing further oligomerization step which eventually leads to the formation of the stoichiometric products 3a and 3b, thereby consuming the borane catalyst of the allene cyclotrimerization reaction and, thus, leading to termination of the catalytic reaction in this specific case. Nevertheless, our study has shown that allene oligomerization beyond trimerization is possible with such borane systems. This makes us hopeful that it might be possible to open novel ways of utilization of allenes by a further development of metal-free reactions along the characteristic lines that have shown up in this study.
We have started to use the allene cyclotrimerization products 1 and 14 as the starting materials for the conversion to the respective arene isomers. It is known that 1,3,5-trimethylene-cyclohexane is resistant to thermal isomerization, but it was reported that it could be isomerized by treatment with acid.2a,13 We repeated this reaction: treatment of 1 with 5 mol% of p-toluene sulfonic acid in toluene solution at r.t. for 1.5 h cleanly converted 1 to mesitylene (Scheme 5). We also treated the n-octyl- and cyclohexylallene cyclotrimers 14c14 and 14e5 with p-toluene sulfonic acid. Compound 14c was fully converted on a preparative scale with 10 mol% of the acid catalyst during 48 h at r.t. The hexa-substituted arene 15c was isolated as an oil in 78% after workup. It was characterized by 1H and 13C NMR spectroscopy (see the ESI† for details). Compound 14e needed slightly more vigorous conditions. It required treatment with 10 mol% of the p-toluene sulfonic acid catalyst at 80 °C for 5 h to become converted. This reaction is not overly selective, but we isolated the aromatic isomer 15e in 37% yield from the reaction mixture. The compound was characterized by spectroscopy and by an X-ray crystal structure analysis (see the ESI† for details). In principle, these reactions have shown that the metal-free X–B(C6F5)2 catalyzed allene cyclotrimerization opens attractive pathways to the synthesis of interesting highly substituted arene products.
 | ||
| Scheme 5 Acid catalyzed isomerization reactions of allene-cyclotrimers to mesitylene derivatives. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for the European Research Council is gratefully acknowledged. We thank Dr Atsushi Ueno and Jennifer Möricke for providing us with samples of the halogenoboranes 2a,b.Notes and references
- (a) A. S. K. Hashmi, Angew. Chem., Int. Ed. Engl., 2000, 39, 3590 CrossRef CAS; (b) N. Krause and A. S. K. Hashmi, Modern Allene Chemistry, Wiley-VCH, Weinheim, Germany, 2004 CrossRef; (c) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef PubMed; (d) N. Krause, Cumulenes and Allenes, Science of Synthesis, Thieme, Stuttgart, Germany, 2007, vol. 44 Search PubMed; (e) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (f) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed; (g) B. Alcaide and P. Almendros, Chem. Soc. Rev., 2014, 43, 2886 RSC; (h) W. Yang and A. S. K. Hashmi, Chem. Soc. Rev., 2014, 43, 2941 RSC.
- (a) R. E. Benson and R. V. Lindsey Jr., J. Am. Chem. Soc., 1959, 81, 4247 CrossRef CAS; (b) R. J. De Pasquale, J. Organomet. Chem., 1971, 32, 381 CrossRef CAS; (c) J. Furukawa, J. Kiji and K. Ueo, Makromol. Chem., 1973, 170, 247 CrossRef CAS.
- (a) D. J. Pasto, N.-Z. Huang and C. W. Eigenbrot, J. Am. Chem. Soc., 1985, 107, 3160 CrossRef CAS; (b) C. Hernández-Díaz, E. Rubio and J. González, Eur. J. Org. Chem., 2016, 265 CrossRef.
- (a) B. M. Mikhailov, V. N. Smirnov, O. D. Smirnova, E. P. Prokofev and A. S. Shashkov, Izv. Akad. Nauk SSSR, Ser. Khim., 1979, 2340 CAS; (b) M. E. Gurskii, S. V. Baranin, A. S. Shashkov, A. I. Lutsenko and B. M. Mikhailov, J. Organomet. Chem., 1983, 246, 129 CrossRef CAS; (c) Y. N. Bubnov, M. E. Gurskii, S. Y. Erdyakov, O. A. Kizas, G. D. Kolomnikova, N. Y. Kuznetsov, T. V. Potapova, O. A. Varzatskii and Y. Z. Voloshin, J. Organomet. Chem., 2009, 694, 1754 CrossRef CAS.
- X. Tao, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2017, 56, 1376 CrossRef CAS PubMed.
- A. Ueno, J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Chem.–Eur. J., 2018, 24, 10044 CrossRef CAS PubMed.
- (a) M. F. Lappert and B. Prokai, J. Organomet. Chem., 1964, 1, 384 CrossRef CAS; (b) S. Hara, H. Dojo, S. Takinami and A. Suzuki, Tetrahedron Lett., 1983, 24, 731 CrossRef CAS; (c) Y. Satoh, H. Serizawa, S. Hara and A. Suzuki, J. Am. Chem. Soc., 1985, 107, 5225 CrossRef CAS; (d) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (e) N. Matsumi and Y. Chujo, Polym. Bull., 1997, 39, 2952 CrossRef; (f) C. Wang, T. Tobrman, Z. Xu and E. Negishi, Org. Lett., 2009, 6, 4092 CrossRef PubMed; (g) A. Suzuki, Heterocycles, 2010, 80, 15 CrossRef CAS; (h) J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon and M. J. Ingleson, Angew. Chem., Int. Ed., 2013, 52, 7518 CrossRef CAS PubMed; (i) J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627 CrossRef CAS PubMed.
- (a) B. M. Mikhailov and Y. N. Bubnov, Izv. Akad. Nauk SSSR, Ser. Khim., 1964, 1874 CAS; (b) B. M. Mikhailov and Y. N. BubnovOrganoboron Compounds in Organic Synthesis, Harvood, Chur, 1984 Search PubMed; (c) Y. N. Bubnov, Pure Appl. Chem., 1987, 59, 895 CAS; (d) R. W. Hoffmann, Pure Appl. Chem., 1988, 60, 123 CAS; (e) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207 CrossRef CAS.
- (a) J. Krüger and R. W. Hoffmann, J. Am. Chem. Soc., 1997, 119, 7499 CrossRef; (b) Y. Yamamoto, K. Kurihara, A. Yamada, M. Takahashi, Y. Takahashi and N. Miyaura, Tetrahedron, 2003, 59, 537 CrossRef CAS; (c) N. G. Bandur, D. Brückner, R. W. Hoffmann and U. Koert, Org. Lett., 2006, 8, 3829 CrossRef CAS PubMed.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed.
- (a) J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968 CrossRef CAS PubMed; (b) J. B. Sortais, T. Voss, G. Kehr, R. Fröhlich and G. Erker, Chem. Commun., 2009, 7417 RSC; (c) T. Voss, J.-B. Sortais, R. Fröhlich, G. Kehr and G. Erker, Organometallics, 2011, 30, 584 CrossRef CAS; (d) X. X. Zhao and D. W. Stephan, J. Am. Chem. Soc., 2011, 133, 12448 CrossRef CAS PubMed; (e) G. Ménard, L. Tran, J. S. J. McCahill, A. J. Lough and D. W. Stephan, Organometallics, 2013, 32, 6759 CrossRef.
- J. Huang and S. Ma, J. Org. Chem., 2009, 74, 1763 CrossRef PubMed.
- (a) M. E. Gurskii, S. Yu. Erdyakov, T. V. Potapova and Yu. N. Bubnov, Russ. Chem. Bull., 2008, 57, 802 CrossRef CAS; (b) Y. N. Bubnov, M. E. Gurskii, S. Y. Erdyakov, O. A. Kizas, G. D. Kolomnikova, N. Y. Kuznetsov, T. V. Potapova, O. A. Varzatskii and Y. Z. Voloshin, J. Organomet. Chem., 2009, 694, 1754 CrossRef CAS.
- X. Tao, C. G. Daniliuc, D. Dittrich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2018, 57, 13922 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, further spectral and crystallographic data. CCDC 1862533–1862535 and 1881527. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04790a |
| This journal is © The Royal Society of Chemistry 2019 |