
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd-catalyzed asymmetric allylic substitution cascade using α-(pyridin-1-yl)-acetamides formed in situ as nucleophiles†
Kun
Yao
a,
Qianjia
Yuan
b,
Xingxin
Qu
a,
Yangang
Liu
a,
Delong
Liu
 *a and
Wanbin
Zhang
*ab
*a and
Wanbin
Zhang
*ab
aShanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Pharmacy, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, P. R. China. E-mail: dlliu@sjtu.edu.cn; wanbin@sjtu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, P. R. China
First published on 4th December 2018
Abstract
A Pd-catalyzed asymmetric allylic substitution cascade reaction, using α-(pyridin-1-yl)-acetamides (formed in situ) as nucleophiles, has been developed, generating chiral piperidine-containing amino acid derivatives via a one-pot procedure in high yields and with up to 96% ee. The products can be easily converted into potential bioactive compounds, unnatural chiral amino acids and dipeptides.
Piperidines are among the most prevalent nitrogen ring systems found in numerous bioactive compounds and natural products.1 They are also prevalent in many well-selling pharmaceuticals, such as donepezil, methylphenidate, raloxifene, risperidone and paliperidone.2 Although several strategies have been developed to obtain piperidines,3 the activation of easily accessible pyridine ring systems followed by functionalization is considered to be the most efficient methodology.4
Quaternary pyridinium salts A (Fig. 1), generally used as activated pyridines, are very attractive synthons for synthetic chemists because of their ease of preparation and unique reactivity.5–8 They have received significant interest for use in many types of transformation, for example, Michael additions,5a 1,3-dipole additions,5b Kröhnke reaction,5cetc. Therefore, quaternary pyridinium salts A are widely applied in the synthesis of heterocyclic compounds. From the viewpoint of functionalization, A can roughly be divided into three activated types: ketones A1,6 esters A27 and amides A3 (Fig. 1),8 which have been applied in the synthesis of anti-mycobacterial compounds,6a NF-κB inhibitors7a and 5-HT2c modulators,8a respectively. The main method to prepare these heterocyclic compounds is via functionalization of the α-positions of A1, A2 and A3. To the best of our knowledge, however, alkylation at the α-position of the aforementioned quaternary pyridinium salts via a substitution reaction, has not yet been developed.
 | ||
| Fig. 1 Generally used pyridine quaternary salts. | ||
Pd-catalyzed allylic substitution is a powerful synthetic tool for the formation of C–C and C–X bonds (X = N, O, S, etc.).9 Our group has developed several novel Pd-catalyzed asymmetric alkylations for the construction of biologically-active chiral molecules with excellent catalytic behavior,10 particularly using several activated nucleophiles formed in situ by either an organic10a–d or transition-metallic catalyst.10i–m Herein, we disclose the use of pyridine quaternary salts A, formed in situ from pyridines and haloacetamides, as novel activated nucleophiles, allowing for the preparation of piperidine-containing amino acid derivatives easily via a cascade process (Scheme 1). Differing from previously reported methodologies in which the pyridine ring of the activated species A is generally removed or transferred to a fused aromatic ring moiety in the final products,5–8 in our methodology the pyridine rings were reduced to piperidine groups in the products. The obtained N-azacyclic (e.g. piperidyl) substituted α-amino acid structural motif is present in numerous bioactive compounds, natural products and medicines.3a,3c,11
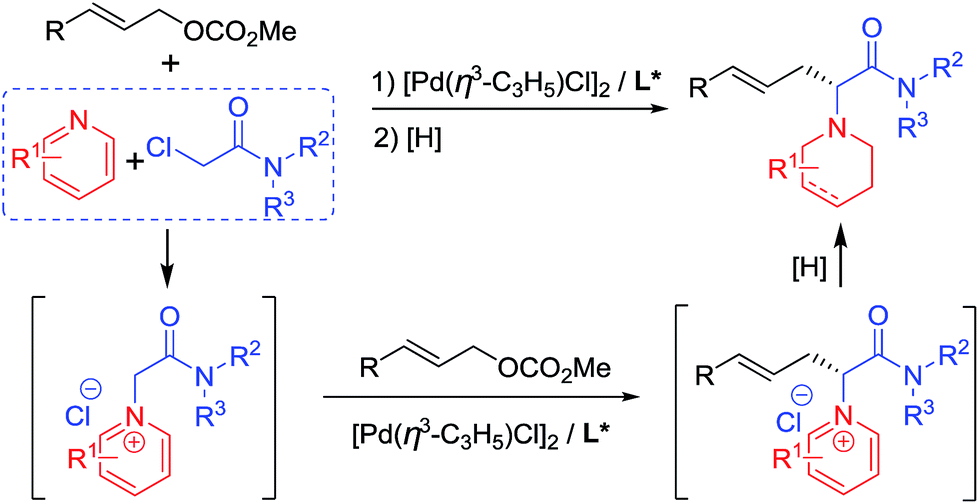 | ||
| Scheme 1 Pd-catalyzed asymmetric allylic substitution cascade. | ||
Initially, we carried out the reaction by using both ketones A1 and esters A2 as nucleophiles. However, only racemic products were obtained albeit in high yields, probably due to the strong acidity of the α-hydrogen.12 Therefore, pyridine quaternary salt A3, bearing a less acidic α-hydrogen than that of A1 and A2, was used, providing the desired products with excellent catalytic behaviour. After a dearomatizing reduction, a series of piperidine-containing amino acid derivatives can be obtained in high yields and with excellent enantioselectivities (Scheme 1).
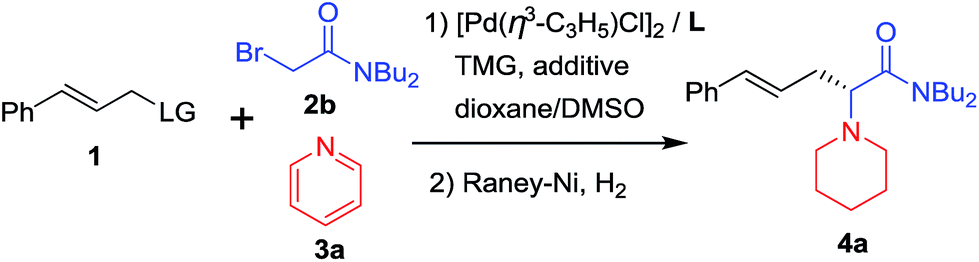
Our study began with the reaction of cinnamyl acetate (1a), 2-chloro-N,N-dibutylacetamide (2a) and pyridine (3a) using a catalytic system consisting of [Pd(η3-C3H5)Cl]2 and planar chiral phosphino-oxazoline ligands L113 under a nitrogen atmosphere at 20 °C over 12 h. Unfortunately, no reaction occurred because chloroacetamide is too inert to form quaternary pyridinium salts. Next, the more reactive 2-bromo-N,N-dibutylacetamide (2b) was employed. To our delight, the reaction proceeded smoothly and the desired product 4a was formed in 92% yield but with low enantioselectivity (Table 1, entry 1).14 Next, planar chiral ligands L2 (entry 2) and L3 (entry 3) were also employed, and somewhat lower yields and enantioselectivities were obtained compared with that of L1 (entry 1). However, no reaction occurred when t-Bu-PHOX was used in place of the above planar chiral ligands (entry 4). After screening the effects of base and solvent on the reaction, it was found that the use of ferrocenyl ligand L1 and 1,1,3,3-tetramethylguanidine (TMG) in a mixed solvent system of dioxane/DMSO (10/1, v/v) provided the desired product 4a in 92% yield and 77% ee (Table 1, entry 1). Then, several additives, such as CsF, LiCl, LiBr and LiI were employed to improve the yield and enantioselectivity of the alkylated product (entries 5–8).10f,15 The reaction with LiI as an additive gave 4a in 94% yield and 91% ee (entry 8). Next, allyl substrates bearing OCO2Me (1b) and OBoc (1c) as leaving groups were examined (entries 9 and 10). It was found that 1b bearing OCO2Me as a leaving group provided better results (92% yield and 94% ee) than that of 1a and 1c. Finally, we lowered the reaction temperature in order to further improve the enantioselectivity. To our delight, the product 4a was obtained in up to 96% yield and 95% ee when the reaction was carried out at 5 °C (entry 11). Further lowering the reaction temperature to 0 °C led to a sharp decline in yield because the solvent turned viscous at this temperature. Different substituents on the nitrogen atom of 2 were also examined and 2b with two n-Bu groups gave the best results.14 In addition, since the use of LiI improved the enantioselectivitiy as mentioned above (entry 8) and because the use of LiI would most likely increase the activity of the chloroacetamide 2a, we employed 2a as a substrate again in place of 2b. As expected, the desired product 4a was obtained in excellent yield and enantioselectivity (entry 12). Therefore, 2a was chosen for use in the subsequent reactions because it is less expensive and easy to access than 2b.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | L | LG of 1 | Temp. (°C) | Additive | Yieldb (%) | eec (%) |
a Reaction conditions: 1a (0.1 mmol) with 2b (0.2 mmol) and 3a (0.5 mmol) in a mixed solvent of dioxane/DMSO (10/1, v/v) under nitrogen atmosphere with a catalytic system of [Pd(η3-C3H5)Cl]2 (2.5 mol%) and L (6.0 mol%) with TMG as a base (0.16 mmol) in the presence of an additive (0.1 mmol) at indicated temperature for 12 h; The pyridine quaternary salt was reduced by RANEY®Ni under hydrogen atmosphere.
b Isolated yields.
c Determined by HPLC using a chiral Daicel IC-3 column.
d Chloroacetamide 2a was used.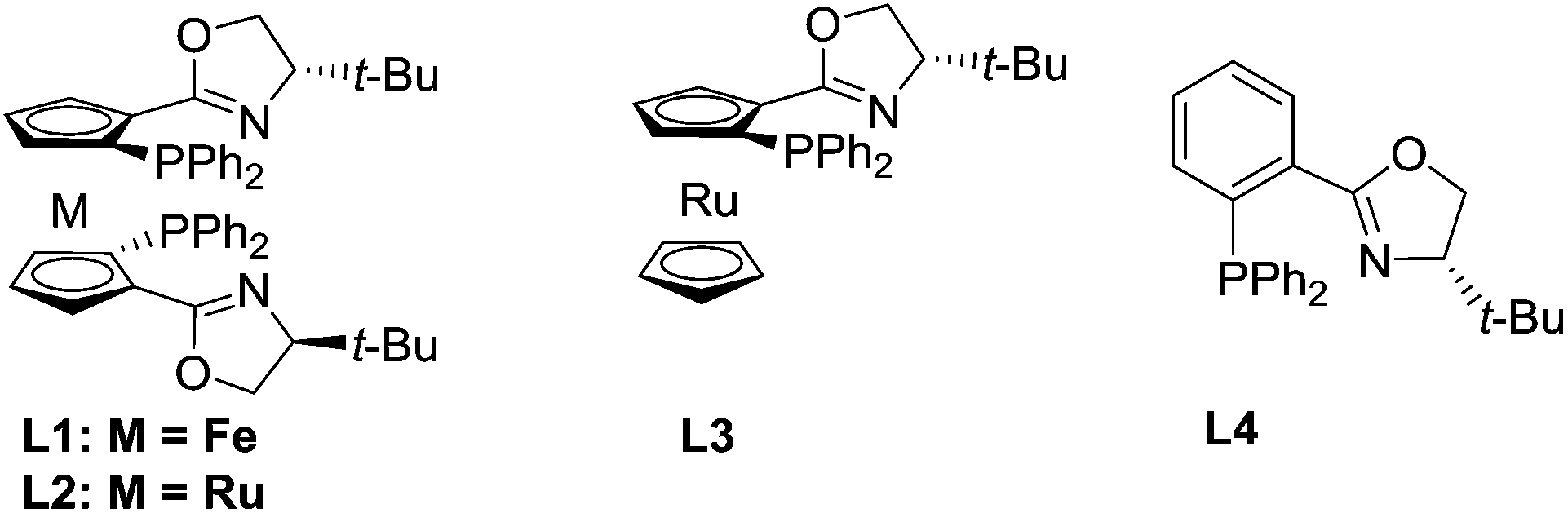
|
||||||
| 1 | L1 | OAc | 20 | — | 92 | 77 |
| 2 | L2 | OAc | 20 | — | 90 | 41 |
| 3 | L3 | OAc | 20 | — | 66 | 31 |
| 4 | L4 | OAc | 20 | — | NR | — |
| 5 | L1 | OAc | 20 | CsF | 95 | 41 |
| 6 | L1 | OAc | 20 | LiCl | 93 | 55 |
| 7 | L1 | OAc | 20 | LiBr | 93 | 71 |
| 8 | L1 | OAc | 20 | LiI | 94 | 91 |
| 9 | L1 | OCO2Me | 20 | LiI | 92 | 94 |
| 10 | L1 | OBoc | 20 | LiI | 91 | 91 |
| 11 | L1 | OCO2Me | 5 | LiI | 96 | 95 |
| 12d | L1 | OCO2Me | 5 | LiI | 96 | 95 |
With the optimized reaction conditions in hand (Table 1, entry 9), substrates with different aryl substituents were explored (Scheme 2). First, cinnamyl carbonates bearing Me groups at the 2-, 3- and 4-positions of the phenyl ring were examined, and ees of approximately 90% were obtained for all substrates (4b, 4c and 4d). 4d with an Me group at the 4-position of the phenyl ring provided the best enantioselectivity. When Me groups were replaced by OMe groups, nearly identical results were obtained and the substrate with an OMe group at the 4-position of the phenyl ring gave the best result (4e, 4f and 4g). Therefore, substrates with Et, i-Pr and t-Bu at 4-position of the phenyl ring were examined and all afforded satisfactory results (4h, 4i and 4j). Substrates bearing electron-withdrawing groups on the phenyl ring were next considered. When substrates with an F atom at the 2, 3- or 4-positions of the phenyl ring were used, the desired products were obtained in high yields and around 90% ees (4k and 4lvs.4m). Similarly, 4m with an F atom at the 4-position of the phenyl ring gave the best enantioselectivity. Therefore, substrates with electron-withdrawing groups, such as Cl, Br and CF3 at the 4-position of the phenyl ring, have been examined and all afforded promising results (4n, 4o and 4p). To our delight, 4p bearing a CF3 group gave its corresponding product with 96% ee. When a Ph group was introduced, the reaction proceeded smoothly providing the corresponding product in 91% yield and 88% ee (4q). When the phenyl ring was replaced by a 2-naphthyl or 2-furyl group, the desired products were obtained in more than 90% yields and enantioselectivities for both substrates (4r and 4s). An allylic substrate bearing 1,2-disubstituted groups was tested with the corresponding product 4t being formed in 94% yield and 57% ee. Next, allyl carbonates without an Ar functional group or with alkyl groups instead of Ar groups were also subjected to the reaction conditions and the reductant was changed to NaBH4. The desired products bearing a tetrahydropyridine group (5a–c) were obtained in good yields and enantioselectivities. Two 1,1′-disubstituted allylic substrates bearing alkyl groups were also subjected to the reaction conditions, giving the desired products in moderate yields but with low ee (5d and 5e). Finally, the n-Bu groups on the nitrogen atom of 2a were replaced by two 4-pentenyl groups, with the desired product 5f being obtained in excellent yield and enantioselectivity.
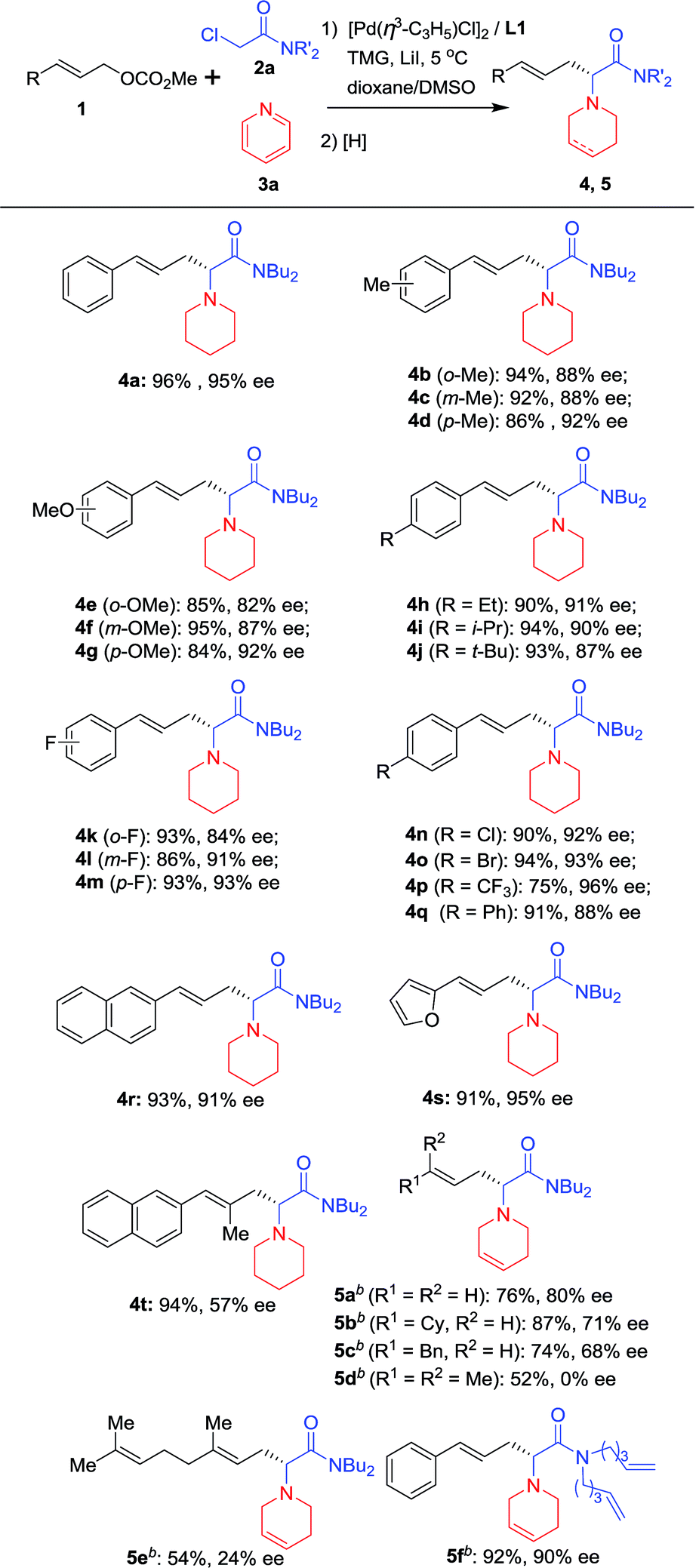 | ||
| Scheme 2 Scope of allylic substratesa. aUsing the optimal reaction conditions shown in Table 1. Isolated yields; ees were determined by HPLC using a chiral Daicel column. bThe pyridine quaternary salts were reduced with NaBH4. | ||
To further explore the substrate scope, pyridine heterocycles 3 bearing different substituents were examined using NaBH4 as a reductant (Scheme 3). First, we found that when 3 possesses a Me group at the 4-position of the pyridine ring, the corresponding product is obtained in higher yield and enantioselectivity than when the Me group is present at the 3-position of the pyridine (5g and 5h). Next, 3 bearing bulky alkyl or aryl groups substituted at the 4-position of the pyridine ring was used. The desired products were prepared in excellent yields and with around 90% ees (5i–5l). Finally, isoquinoline was used instead of pyridine and the corresponding dihydroisoquinoline derivative 5m was obtained in good yield and enantioselectivity. 5m contains a dihydroisoquinoline skeleton which is an important structural motif in alkaloid and biological molecules.16
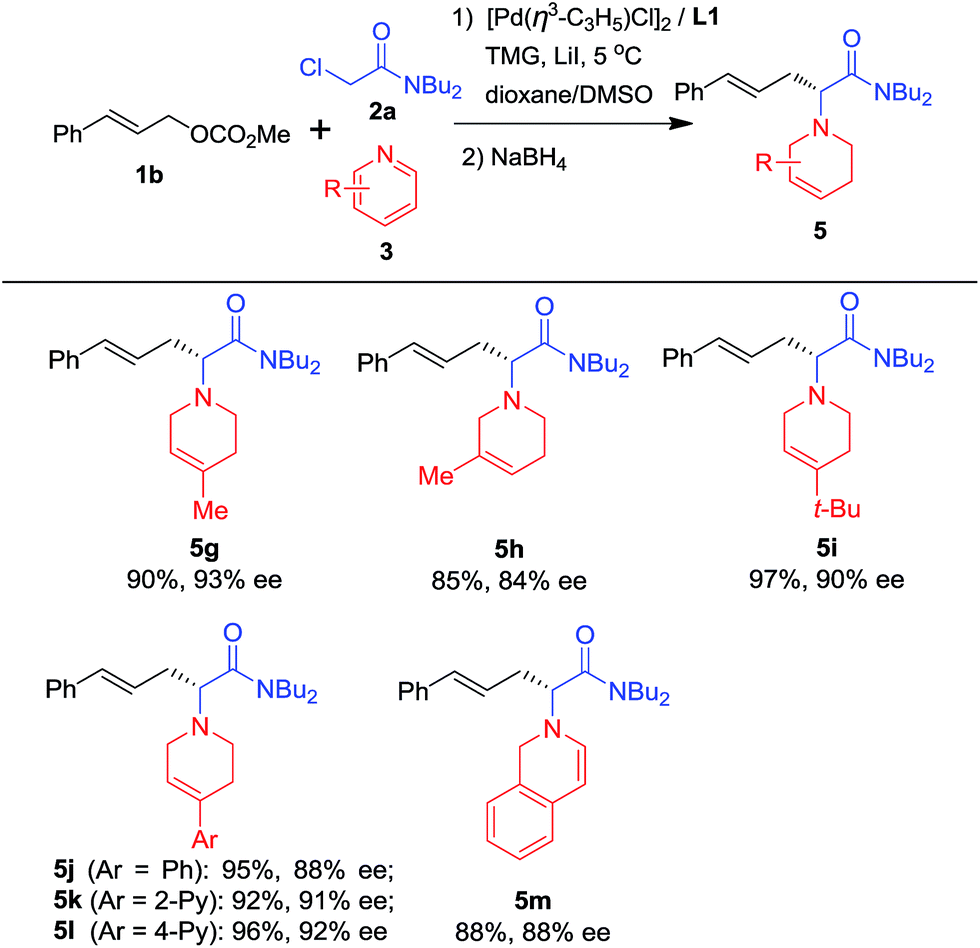 | ||
| Scheme 3 Scope of pyridinesa. aUsing the optimal reaction conditions shown in Table 1. The pyridine quaternary salt was reduced with NaBH4. Isolated yields; ees were determined by HPLC using a chiral Daicel column. | ||
To prove the utility of this new synthetic methodology, product 4a was reduced with LiAlH4 to provide chiral vicinal diamine 6 in excellent yield (Scheme 4, eqn (1)). The hydrogenation of the double bond of the allylic motif in 4q gave the product 7 in 83% yield without any loss in enantioselectivity (Scheme 4, eqn (2)). Derivatizations of 5fvia ring closing metathesis were conducted, forming the corresponding macrocyclic amino acetamide 8 in excellent yield without any loss in ee (Scheme 4, eqn (3)).17 Product 5j, with a phenyl group at the tetrahydropyridine ring, was oxidized to give the lactam 9 in excellent yield and with complete retention of configuration (Scheme 4, eqn (4)).18
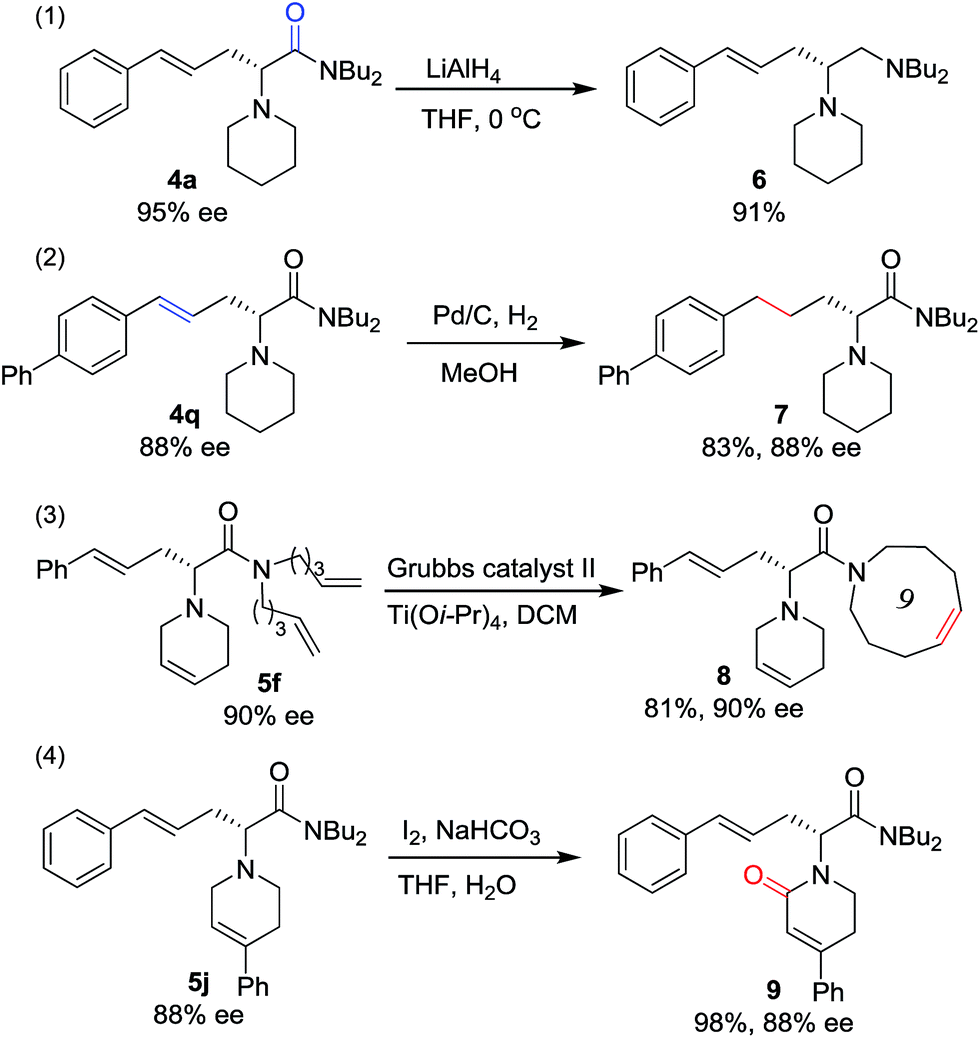 | ||
| Scheme 4 The transformation of 4 and 5. | ||
Reaction with chloroacetamide 2c under standard conditions was also conducted. After removal of the DMB group, product 10 was obtained in 92% yield and 90% ee. Further hydrolysis of 10 under mild conditions19 gave the unnatural chiral amino acid (R)-1120 in good overall yield (Scheme 5, top). 10 was also treated with Lawesson's reagent and the corresponding amino thioacetamide derivative 12 was formed in 31% yield and 88% ee (Scheme 5, top). Unnatural amino acids are often used for polypeptide drug candidate optimization since the peptide bonds consisting of unnatural amino acids are more protease-resistant, which increases stability under physiological conditions and changes the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D value.21 Thus, bromoacetyl protected L-proline allyl ester 13 was prepared and applied in the asymmetric intramolecular allylic substitution cascade, affording the desired dipeptide 14 in high yield and diastereoselectivity (Scheme 5, bottom).
D value.21 Thus, bromoacetyl protected L-proline allyl ester 13 was prepared and applied in the asymmetric intramolecular allylic substitution cascade, affording the desired dipeptide 14 in high yield and diastereoselectivity (Scheme 5, bottom).
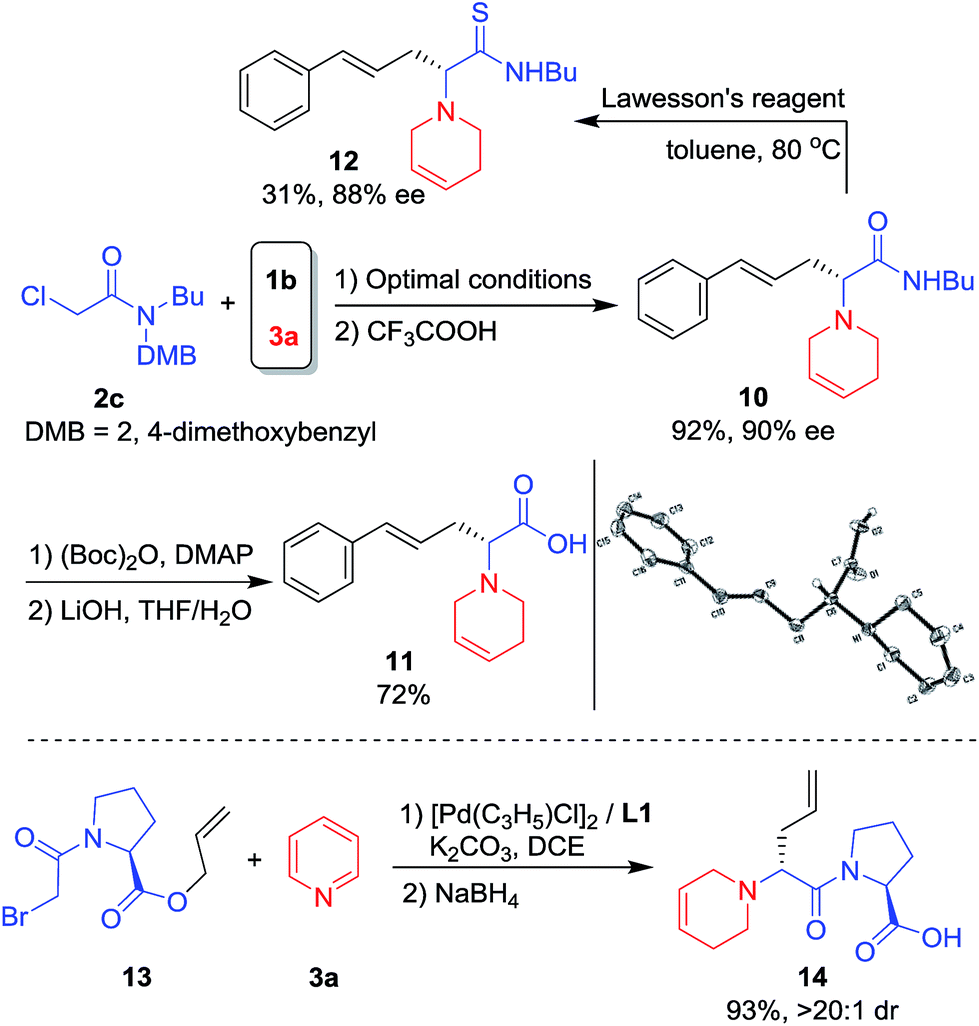 | ||
| Scheme 5 The synthesis of an unnatural amino acid and dipeptide. | ||
Conclusions
In summary, α-(pyridin-1-yl)-acetamides formed in situ have been used as nucleophiles in a Pd-catalyzed asymmetric allylic substitution cascade for the one-pot construction of chiral piperidine-based amino amides in high yields and with up to 96% ee. Using our methodology, several types of unnatural chiral amino acids and dipeptides containing piperidine or tetrahydropyridine substituents, which play an important role in natural products and medicines, have been prepared with excellent asymmetric behaviour.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (No. 21472123, 21672142, 21620102003 and 21831005), Shanghai Municipal Education Commission (No. 201701070002E00030). We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University.Notes and references
- For selected reviews, see: (a) S. Källström and R. Leino, Bioorg. Med. Chem., 2008, 16, 601 CrossRef PubMed; (b) J. Bolleddula, K. DeMent, J. P. Driscoll, P. Worboys, P. J. Brassil and D. L. Bourdet, Drug Metab. Rev., 2014, 46, 379 CrossRef CAS PubMed; (c) F. I. Carroll and R. E. Dolle, ChemMedChem, 2014, 9, 1638 CAS.
- M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265 CrossRef PubMed.
- For selected recent examples, see: (a) S. S. M. Spoehrle, T. H. West, J. E. Taylor, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 11895 CrossRef CAS PubMed; (b) N. Münster, N. A. Parker, L. van Dijk, R. S. Paton and M. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 9468 CrossRef PubMed; (c) V. Arredondo, S. C. Hiew, E. S. Gutman, I. D. U. A. Premachandra and D. L. Van Vranken, Angew. Chem., Int. Ed., 2017, 56, 4156 CrossRef CAS PubMed.
- For selected reviews, see: (a) W. Sliwa, Curr. Org. Chem., 2003, 7, 995 CrossRef CAS; (b) P. Madaan and V. K. Tyagi, J. Oleo Sci., 2008, 57, 197 CrossRef CAS PubMed; (c) S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453 RSC. For selected examples, see: (d) Z.-S. Ye, M.-W. Chen, Q.-A. Chen, L. Shi, Y. Duan and Y.-G. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10181 CrossRef CAS PubMed; (e) M. Chang, Y. Huang, S. Liu, Y. Chen, S. W. Krska, I. W. Davies and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 12761 CrossRef CAS PubMed; (f) Z. Yang, Q. Wu, W. Shao and S.-L. You, J. Am. Chem. Soc., 2015, 137, 15899 CrossRef CAS PubMed; (g) J.-H. Xu, S.-C. Zheng, J.-W. Zhang, X.-Y. Liu and B. Tan, Angew. Chem., Int. Ed., 2016, 55, 11834 CrossRef CAS PubMed; (h) Y. Wang, Y. Liu, D. Zhang, H. Wei, M. Shi and F. Wang, Angew. Chem., Int. Ed., 2016, 55, 3776 CrossRef CAS PubMed.
- For selected examples, see: (a) T. M. Kadayat, C. Song, Y. Kwon and E.-S. Lee, Bioorg. Chem., 2015, 62, 30 CrossRef CAS PubMed; (b) R.-B. Hu, S. Sun and Y. Su, Angew. Chem., Int. Ed., 2017, 56, 10877 CrossRef CAS PubMed; (c) D. Lebedev, Y. Pineda-Galvan, Y. Tokimaru, A. Fedorov, N. Kaeffer, C. Copéret and Y. Pushkar, J. Am. Chem. Soc., 2018, 140, 451 CrossRef CAS PubMed.
- For selected examples, see: (a) R. Danac and I. I. Mangalagiu, Eur. J. Med. Chem., 2014, 74, 664 CrossRef CAS PubMed; (b) P. Thapa, K.-Y. Jun, T. M. Kadayat, C. Park, Z. Zheng, T. B. T. Magar, G. Bist, A. Shrestha, Y. Na, Y. Kwon and E.-S. Lee, Bioorg. Med. Chem., 2015, 23, 6454 CrossRef CAS PubMed; (c) P. S. O'Hora, C. A. Incerti-Pradillos, M. A. Kabeshov, S. A. Shipilovskikh, A. E. Rubtsov, M. R. J. Elsegood and A. V. Malkov, Chem.–Eur. J., 2015, 21, 4551 CrossRef PubMed.
- For selected examples, see: (a) Y. Fu, J. Ma, X. Shi, X.-Y. Song, Y. Yang, S. Xiao, J. Li, W.-J. Gu, Z. Huang, J. Zhang and J. Chen, Biochem. Pharmacol., 2017, 135, 126 CrossRef CAS PubMed; (b) N. B. Chernysheva, A. S. Maksimenko, F. A. Andreyanov, V. P. Kislyi, Y. A. Strelenko, V. N. Khrustalev, M. N. Semenova and V. V. Semenov, Tetrahedron, 2017, 73, 6728 CrossRef CAS; (c) A. Szymanska-Michalak, D. Wawrzyniak, G. Framski, J. Stawinski, J. Barciszewski and A. Kraszewski, Eur. J. Med. Chem., 2018, 144, 682 CrossRef CAS PubMed; (d) K. Pandey, K. Rangan and A. Kumar, J. Org. Chem., 2018, 83, 8026 CrossRef CAS PubMed.
- For selected examples, see: (a) B. Gisela, B. Margaretha, H. Maria, B. Guenter, B. Wilfried, D. Karla, E. Thomas, H. Andreas, K. Hannes, L. Viktor, L. Anna, M. Helmut, P. Raimund and R. A. Lucia, Hexahydrodiazepinoquinolines as 5-HT2c modulators and their preparation, WO 2015136090, 2015; (b) A. J. Frey, Q. Wang, C. Busch, D. Feldman, L. Bottalico, C. A. Mesaros, I. A. Blair, A. Vachani and N. W. Snyder, Steroids, 2016, 116, 60 CrossRef CAS PubMed; (c) W. Zhang, Y.-M. Zhang, S.-H. Li, Y.-L. Cui, J. Yu and Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 11452 CrossRef CAS PubMed; (d) T. Anajafi, M. D. Scott, S. You, X. Yang, Y. Choi, S. Y. Qian and S. Mallik, Bioconjugate Chem., 2016, 27, 762 CrossRef CAS PubMed; (e) F. W. W. Hartrampf, T. Furukawa and D. Trauner, Angew. Chem., Int. Ed., 2017, 56, 893 CrossRef CAS PubMed.
- Selected reviews of Pd-catalyzed allylic substitutions: (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed; (b) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336 CrossRef CAS PubMed; (c) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (d) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS PubMed; (e) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427 RSC; (f) P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147 RSC; (g) B. M. Trost, Org. Process Res. Dev., 2012, 16, 185 CrossRef CAS PubMed; (h) A. Lumbroso, M. L. Cooke and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 1890 CrossRef CAS PubMed; (i) N. A. Butt, D. Liu and W. Zhang, Synlett, 2014, 25, 615 CrossRef CAS; (j) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558 CrossRef CAS PubMed; (k) N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929 RSC; (l) N. Butt, G. Yang and W. Zhang, Chem. Rec., 2016, 16, 2687 CrossRef PubMed; (m) J. Fu, X. Huo, B. Li and W. Zhang, Org. Biomol. Chem., 2017, 15, 9747 RSC.
- Selected recent papers: (a) X. Zhao, D. Liu, H. Guo, Y. Liu and W. Zhang, J. Am. Chem. Soc., 2011, 133, 19354 CrossRef CAS PubMed; (b) X. Zhao, D. Liu, F. Xie, Y. Liu and W. Zhang, Org. Biomol. Chem., 2011, 9, 1871 RSC; (c) X. Huo, M. Quan, G. Yang, X. Zhao, D. Liu, Y. Liu and W. Zhang, Org. Lett., 2014, 16, 1570 CrossRef CAS PubMed; (d) X. Huo, G. Yang, D. Liu, Y. Liu, I. D. Gridnev and W. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6776 CrossRef CAS PubMed; (e) X. Wei, D. Liu, Q. An and W. Zhang, Org. Lett., 2015, 17, 5768 CrossRef CAS PubMed; (f) K. Yao, D. Liu, Q. Yuan, T. Imamoto, Y. Liu and W. Zhang, Org. Lett., 2016, 18, 6296 CrossRef CAS PubMed; (g) Q. An, D. Liu, J. Shen, Y. Liu and W. Zhang, Org. Lett., 2017, 19, 238 CrossRef CAS PubMed; (h) C. Xia, J. Shen, D. Liu and W. Zhang, Org. Lett., 2017, 19, 4251 CrossRef CAS PubMed; (i) X. Huo, R. He, J. Fu, J. Zhang, G. Yang and W. Zhang, J. Am. Chem. Soc., 2017, 139, 9819 CrossRef CAS PubMed; (j) X. Huo, J. Fu, X. He, J. Chen, F. Xie and W. Zhang, Chem. Commun., 2018, 54, 599 RSC. We also developed several Ir-catalyzed asymmetric allylic substitution reactions, see: (k) X. Huo, R. He, X. Zhang and W. Zhang, J. Am. Chem. Soc., 2016, 138, 11093 CrossRef CAS PubMed; (l) R. He, P. Liu, X. Huo and W. Zhang, Org. Lett., 2017, 19, 5513 CrossRef CAS PubMed; (m) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080 CrossRef CAS PubMed.
- For selected examples, see: (a) D. Gnecco, J. Juárez, A. Galindo and R. G. Enríquez, ARKIVOC, 2003, 11, 56 Search PubMed; (b) M. Liniger, Y. Liu and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 13944 CrossRef CAS PubMed; (c) M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shevlin and P. J. Chirik, Science, 2018, 360, 888 CrossRef CAS PubMed.
- X.-M. Zhang, F. G. Bordwell, M. Van Der Puy and H. E. Fried, J. Org. Chem., 1993, 58, 3060 CrossRef CAS.
- (a) W. Zhang, Y. Adachi, T. Hirao and I. Ikeda, Tetrahedron: Asymmetry, 1996, 7, 451 CrossRef CAS; (b) D. Liu, F. Xie and W. Zhang, Tetrahedron Lett., 2007, 48, 7591 CrossRef CAS.
- See details in ESI.†.
- Halide additives might provide different catalytic behaviors in Pd-catalyzed asymmetric allylic substitution: (a) U. Burckhardt, M. Baumann and A. Togni, Tetrahedron: Asymmetry, 1997, 8, 155 CrossRef CAS; (b) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1999, 121, 4545 CrossRef CAS.
- M. Chrzanowska, A. Grajewska and M. D. Rozwadowska, Chem. Rev., 2016, 116, 12369 CrossRef CAS PubMed.
- Q. Yang, W.-J. Xiao and Z. Yu, Org. Lett., 2005, 7, 871 CrossRef CAS PubMed.
- R. J. Griffiths, G. A. Burley and E. P. A. Talbot, Org. Lett., 2017, 19, 870 CrossRef CAS PubMed.
- (a) J. R. Del Valle and M. Goodman, J. Org. Chem., 2004, 69, 8946 CrossRef CAS PubMed; (b) Y. Li, D.-M. Ji and M.-H. Xu, Org. Biomol. Chem., 2011, 9, 8452 RSC.
- CCDC 1860880 (11) contains the supplementary crystallographic data for this paper.†.
- (a) V. E. Cox and E. A. Gaucher, Curr. Protoc. Chem. Biol., 2015, 7, 223 Search PubMed; (b) M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807 CrossRef CAS PubMed; (c) N. Qvit, S. J. S. Rubin, T. J. Urban, D. Mochly-Rosen and E. R. Gross, Drug Discovery Today, 2017, 22, 454 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. CCDC 1860880. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04626c |
| This journal is © The Royal Society of Chemistry 2019 |