
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDithioesters: simple, tunable, cysteine-selective H2S donors†
Matthew M.
Cerda
,
Turner D.
Newton
,
Yu
Zhao
 ,
Brylee K.
Collins
,
Christopher H.
Hendon
and
Michael D.
Pluth
*
,
Brylee K.
Collins
,
Christopher H.
Hendon
and
Michael D.
Pluth
*
Department of Chemistry and Biochemistry, Materials Science Institute, Institute of Molecular Biology, University of Oregon, Eugene, Oregon 97403, USA. E-mail: pluth@uoregon.edu
First published on 30th November 2018
Abstract
Dithioesters have a rich history in polymer chemistry for RAFT polymerizations and are readily accessible through different synthetic methods. Here we demonstrate that the dithioester functional group is a tunable motif that releases H2S upon reaction with cysteine and that structural and electronic modifications enable the rate of cysteine-mediated H2S release to be modified. In addition, we use (bis)phenyl dithioester to carry out kinetic and mechanistic investigations, which demonstrate that the initial attack by cysteine is the rate-limiting step of the reaction. These insights are further supported by complementary DFT calculations. We anticipate that the results from these investigations will allow for the further development of dithioesters as important chemical motifs for studying H2S chemical biology.
Introduction
Prevalent in the field of polymer chemistry, dithioesters are utilized as chain transfer agents in reversible addition-fragmentation chain transfer (RAFT) polymerizations.1 Arising from the ability to readily react with propagating radical monomers, the design of thiocarbonyl-containing motifs such as dithioesters, trithiocarbonates, and dithiocarbamates has been well studied to enhance the RAFT polymerization of various monomers.2,3 We recently demonstrated that related thionoesters, which are structural isomers of thioesters bearing a thiocarbonyl motif, undergo a chemoselective reaction with cysteine to generate a dihydrothiazole and hydrogen sulfide (H2S).4 Despite recent advancements in the synthesis of thionoesters,5 the ability to prepare a diverse library of thionoester-based H2S donors is limited by the availability of stable, readily-accessible starting materials. However, based on structural similarities between thionoesters and dithioesters, we hypothesized that dithioesters could provide similar reactivity in the presence of cysteine and allow for the development of tunable H2S donors with precise control over H2S release rates and efficiencies.Complementing carbon monoxide and nitric oxide, hydrogen sulfide (H2S) is now classified as an important biological signaling molecule.6 Commonly referred to as gasotransmitters, these small, gaseous molecules are produced endogenously, readily permeate cell membranes, and exert physiological responses upon binding to or reacting with cellular and/or molecular targets.7 The involvement of H2S-mediated signaling has been observed in a variety of physiological processes such as vasodilation,8 angiogenesis,9 and scavenging of reactive oxygen species.10 In recent years, researchers have become increasingly interested in utilizing H2S donors for the development of both important research tools and novel therapeutics.11,12 Towards this goal, researchers have relied heavily on the use of sodium hydrosulfide (NaSH), which is a water-soluble source of H2S, for preliminary studies. However, a comparison between H2S production from native enzymes including cystathionine-β-synthase and cystathionine-γ-lyase against exogenous administration of H2S (via NaSH) has revealed stark differences which should be taken into consideration.13 The dissolution of NaSH in buffered solutions leads to a rapid, almost spontaneous increase in local H2S concentration,14 whereas endogenous H2S production occurs at a slower, at a slower rate. To address this issue, researchers have developed synthetic H2S donors, which release H2S either passively via hydrolysis or in the presence of a specific analyte at rates comparable to enzymatic H2S production (Fig. 1).15–18
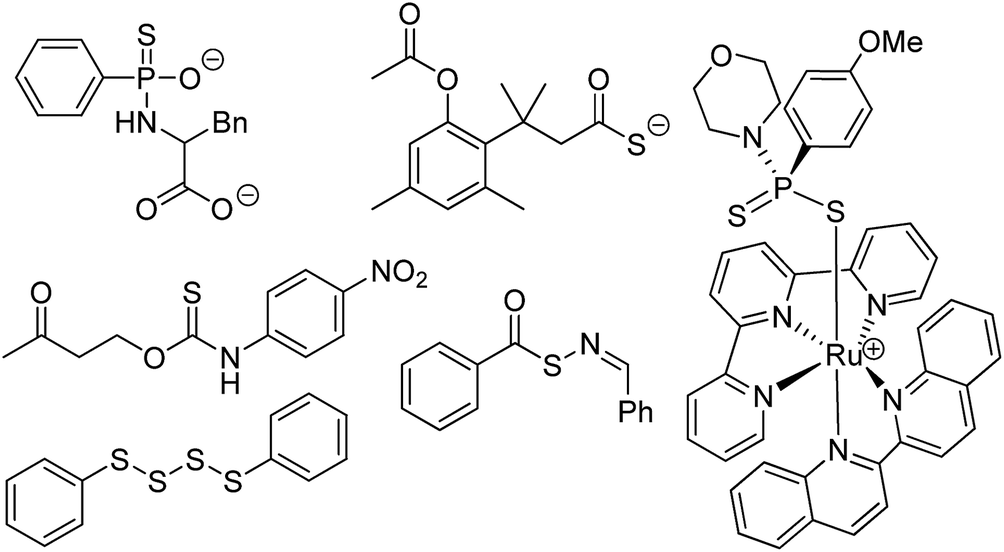 | ||
| Fig. 1 Selected examples of synthetic H2S donors. | ||
Drawing parallels to the dissolution of NaSH, small molecules derived from dithiole-3-thione19 and phosphorodithioate20,21 motifs respectively have been synthesized as hydrolysis-activated H2S donors, although we note the overall low H2S-releasing efficiencies from these donors. Towards improving releasing yields and tuning rates of H2S release, analyte-responsive small molecules have been prepared to release H2S as a function of pH,22 in the presence of native enzymes,23 and upon irradiation with light.24–26 Initially reported by our group,27 the conversion of carbonyl sulfide (COS) to H2S by carbonic anhydrase has been used to access a library of COS/H2S donors.28–30 Inspired by the endogenous production of H2S via enzymatic conversion of cysteine, a number of thiol-triggered H2S donors have been reported. Abundant in nature, organic polysulfides31 have been shown to release H2S in the presence of biological thiols including cysteine32 and reduced glutathione (GSH).33 The generation of persulfides, an important class of biologically-relevant reactive sulfur species,34via thiol-mediated reduction has been used to prepare a series of synthetic H2S donors with potent protective effects against myocardial-ischemia reperfusion injury.25 Similarly, the generation of N-mercaptan species in the presence of thiols has been demonstrated as a viable platform for H2S delivery with tunable releasing kinetics.35–37 Under physiological conditions, thionoesters react rapidly with cysteine via a native chemical ligation-like mechanism to afford an equivalent of phenol, H2S, and a cysteine-derived dihydrothiazole (Fig. 2a). Although dithioesters have been used to prepare polymeric H2S donors,38,39 direct use of this motif as an H2S donor scaffold has not been reported. Herein, we present our findings on the development of dithioesters as novel, cysteine-triggered H2S donors (Fig. 2b). We demonstrate the release of H2S in the presence of cysteine from a representative (bis)phenyl dithioester with a comprehensive kinetic analysis. Additionally, we demonstrate the ability to tune the rate of H2S release from dithioesters via simple alkyl functionalization.
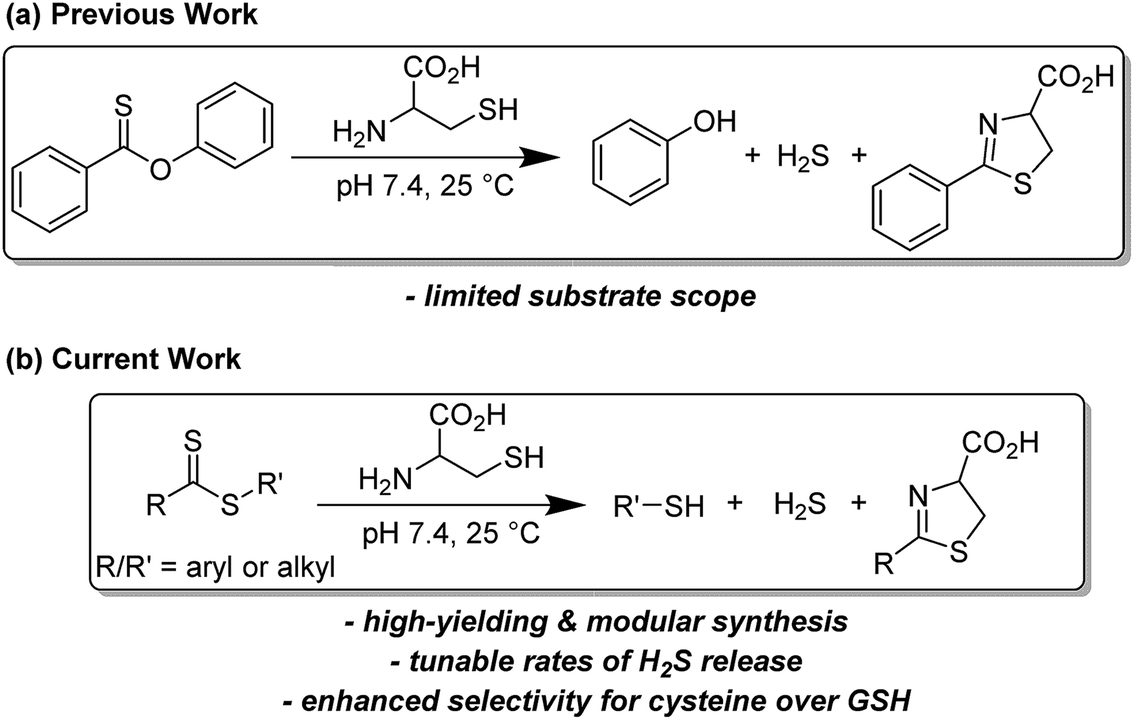 | ||
| Fig. 2 (a) Cysteine-triggered H2S release from thionoesters. (b) Cysteine-triggered H2S release from dithioesters. | ||
Results and discussion
Building upon our previous work, we hypothesized that the intermediate dithioester formed during the chemoselective reaction of cysteine with thionoesters could provide a platform for future investigations leading to the development of novel, cysteine-triggered H2S donors. Unfortunately, the functionalization of thionoester-based H2S donors is hindered by a limited number of stable, isolable alkyl chlorothionoformates. By contrast, the enhanced reactivity of Lawesson's reagent toward thioesters over esters allows for convenient functionalization and synthetic accessibility of various dithioesters.40 To prepare different dithioesters, we treated the desired thiol with the desired acid chloride in the presence of triethylamine followed by thionation with Lawesson's reagent to access to a library of dithioesters with good to excellent yields (Scheme 1). The modularity of this synthetic route readily allows for the introduction of various functional groups and can be expanded upon to include payloads of interest including fluorophores and known therapeutics for the development of H2S-releasing prodrugs. In the interest of accessing tunable H2S donors, we hypothesized modulation of thiocarbonyl electrophilicity and thiolate leaving group ability would directly alter the rates of cysteine-triggered H2S release from dithioesters.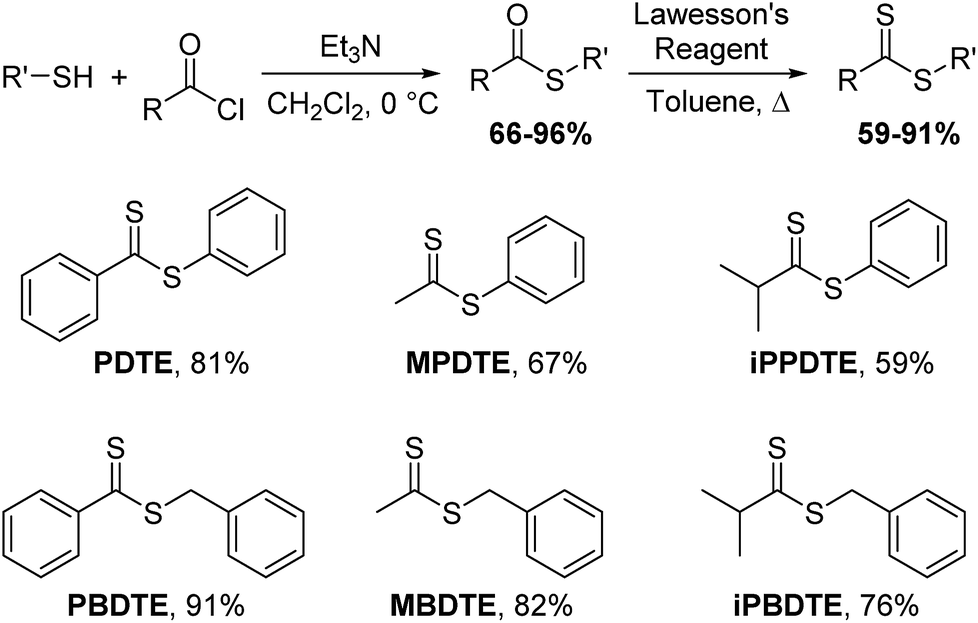 | ||
| Scheme 1 General synthesis of dithioesters. | ||
With synthesized dithioesters in hand, we began our studies by examining the reactivity of PDTE (25 μM) as a representative dithioester towards various concentrations of cysteine (250, 500, 1000, and 1250 μM) and monitoring the release of H2S via the spectrophotometric methylene blue assay, which allows for H2S quantification (Fig. 3a).41 Consistent with our hypothesis, we observed increasing amounts of H2S released with increasing cysteine concentrations from PDTE. To quantify the H2S-releasing efficiency, we used a methylene blue calibration curve generated with NaSH (see ESI†) and we found 25 μM PDTE released approximately 17 μM H2S after 1 h, which corresponds to a releasing efficiency of ∼68%. Under identical conditions, we note a slight decrease in H2S-releasing efficiency between dithioesters and thionoesters (80%), although both of these constructs are efficient H2S-releasing motifs. To quantify the rate of H2S release, we were able to fit these releasing curves and obtain pseudo-first order rate constants (kobs). A plot of log[Cys] versus log(kobs) provided a linear plot with slope near one, which suggests the overall reaction is first order in cysteine and proceeds via a mechanism similar to the reaction of thionoesters with cysteine (Fig. 3b). A plot of [Cys] versus kobs yielded a second-order rate constant (k2) of 1.8 ± 0.1 M−1 s−1 (Fig. 3c), which is approximately five times slower relative to cysteine-triggered H2S from thionoesters (9.1 ± 0.3 M−1 s−1). This difference in H2S-releasing kinetics prompted us to further investigate the kinetics of cysteine-triggered H2S release from dithioesters and elucidate potential mechanistic differences between both classes of H2S donors.
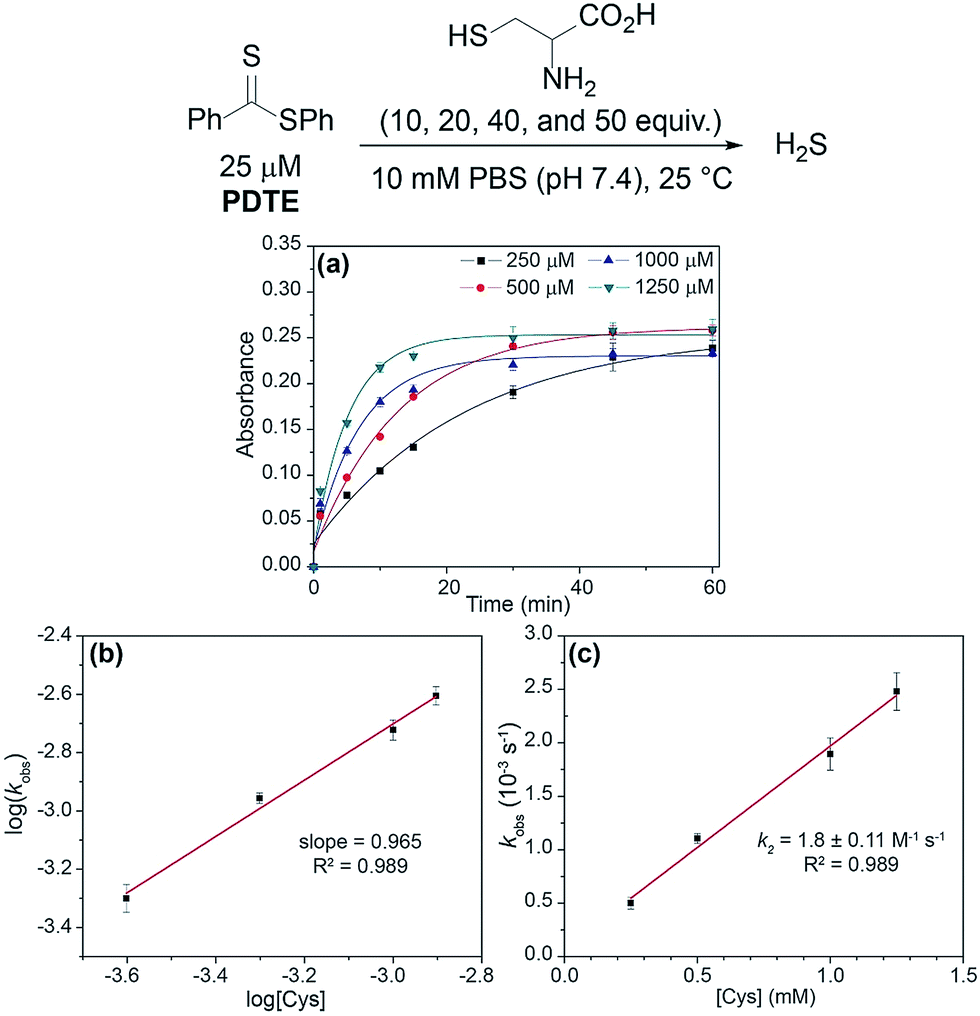 | ||
| Fig. 3 (a) Release of H2S from PDTE (25 μM) in the presence of increasing cysteine concentrations (250, 500, 1000, and 1250 μM). (b) Plot of log(kobs) vs. log([Cys]). (c) Plot of kobsvs. [Cys]. | ||
To further probe the reactivity of dithioesters with respect to H2S release, we next investigated the effect of cysteine derivatives and related thiol-based nucleophiles on H2S release from PDTE (Fig. 4). In the absence of nucleophiles, we did not observe H2S release under hydrolytic conditions. Although the conversion of a thiocarbonyl to the corresponding carbonyl is thermodynamically favorable with an enthalpic gain of ∼43 kcal mol−1 when comparing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S versus CO bond strengths, the hydrolysis of dithioesters is a slow process and can be considered negligible when considering the rate of cysteine-triggered H2S release.42,43 Further supporting a similar mechanism of cysteine-triggered H2S release between thionoesters and dithioesters, masking of either the thiol or amine moieties in cysteine completely abolished H2S release from PDTE. Additionally, the use of cysteine methyl ester did not affect H2S release when compared to H2S release in the presence of cysteine. To assess the effect of cysteine analogues on H2S release, we measured H2S release in the presence of homocysteine and penicillamine. Interestingly, we observed a significant reduction in the H2S release rate in the presence of homocysteine relative to cysteine-triggered H2S release. In the presence of penicillamine, we failed to observe significant H2S release, which we attribute to a reduction in nucleophilicity due to the presence of geminal methyl groups.
S versus CO bond strengths, the hydrolysis of dithioesters is a slow process and can be considered negligible when considering the rate of cysteine-triggered H2S release.42,43 Further supporting a similar mechanism of cysteine-triggered H2S release between thionoesters and dithioesters, masking of either the thiol or amine moieties in cysteine completely abolished H2S release from PDTE. Additionally, the use of cysteine methyl ester did not affect H2S release when compared to H2S release in the presence of cysteine. To assess the effect of cysteine analogues on H2S release, we measured H2S release in the presence of homocysteine and penicillamine. Interestingly, we observed a significant reduction in the H2S release rate in the presence of homocysteine relative to cysteine-triggered H2S release. In the presence of penicillamine, we failed to observe significant H2S release, which we attribute to a reduction in nucleophilicity due to the presence of geminal methyl groups.
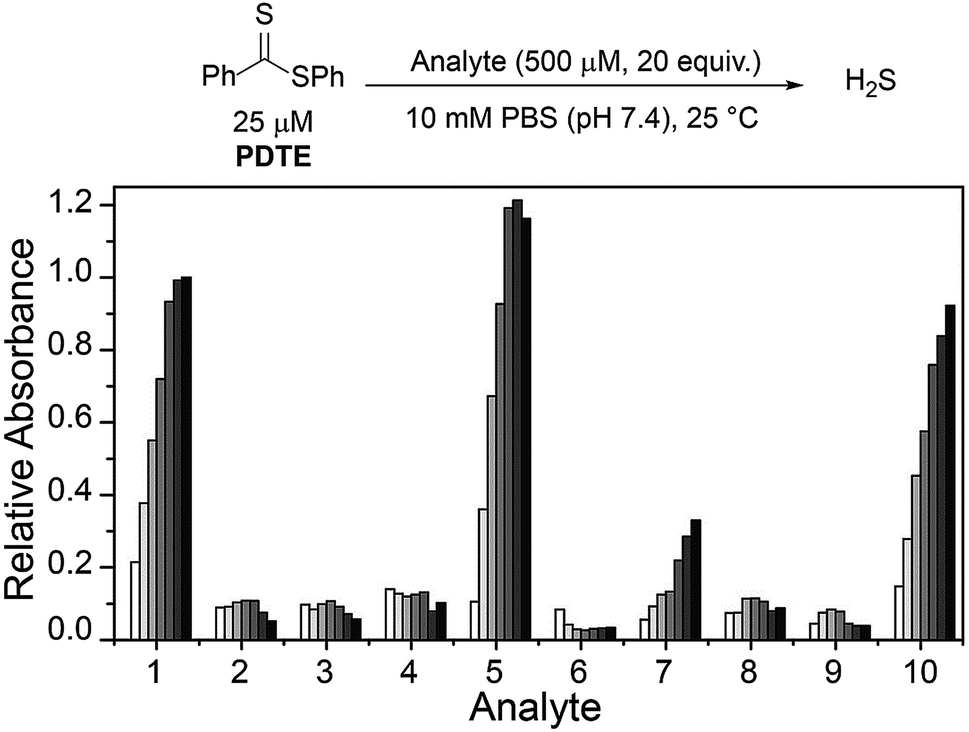 | ||
| Fig. 4 Selectivity of H2S release from PDTE in the presence of different analytes. Data were acquired at 1, 5, 10, 15, 30, 45, and 60 min. Methylene blue absorbance values are relative to the maximum absorbance value obtained from H2S release in the presence of cysteine (1). Analytes: H2O (2), N-acetyl-L-cysteine (3), S-methyl-L-cysteine (4), L-cysteine methyl ester hydrochloride (5), N-acetyl-L-cysteine methyl ester (6), DL-homocysteine (7), DL-penicillamine (8), GSH (9), cysteine + 1 mM GSH (10). | ||
We next examined the release of H2S from PDTE in the presence of reduced glutathione (GSH), which is the most abundant biological thiol to determine the effect of competitive thiols on H2S release. In the presence of 500 μM GSH, we did not observe significant H2S release, but also could not rule out that transthioesterification by GSH, which would result in consumption of the dithioester moiety with a lack of H2S release. Considering the nucleophilicity of the departing thiophenol, we hypothesized that the reversibility of transthioesterification is likely to be more efficient for dithioesters than for thionoesters, which should result in enhanced selectivity for cysteine over GSH. To test this hypothesis, we measured H2S release from PDTE in the presence of 500 μM cysteine and 1 mM GSH. In support of the expected enhanced reversibility, we observed minimal change on the cysteine-triggered H2S release from PDTE even in the presence of excess GSH. This presents a considerable advancement over the thionoester scaffold, which yielded a decreased H2S-releasing efficiency in the presence of GSH and cysteine. Taken together, these results demonstrate the selectivity of the dithioester scaffold for cysteine over other thiol-based nucleophiles including GSH and we proposed the following mechanism of H2S release (Scheme 2).
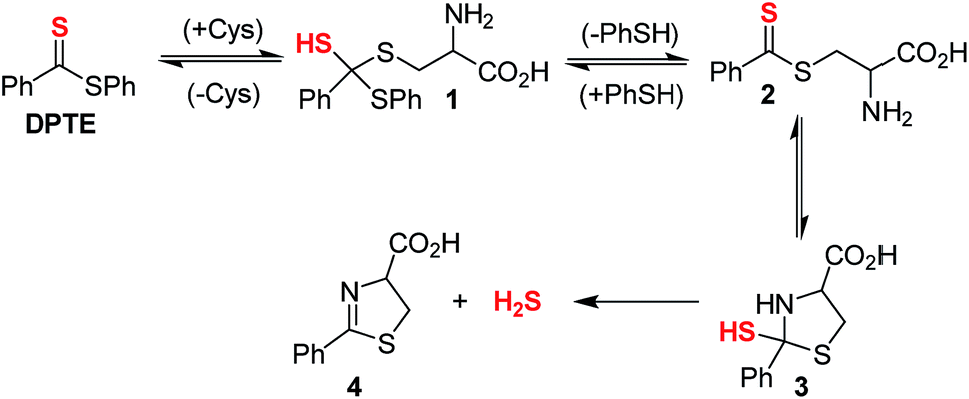 | ||
| Scheme 2 Proposed mechanism for release of H2S from PDTE in the presence of cysteine. | ||
Initial nucleophilic attack by cysteine on PDTE generates tetrahedral intermediate 1, which collapses upon thiocarbonyl formation and extrusion of thiophenol to yield intermediate 2. Nucleophilic attack by the pendant amine generates intermediate 3, which extrudes H2S upon formation of dihydrothiazole 4. Based on the negligible loss in H2S-releasing efficiency in the presence of excess GSH, the generation of 1 and 2 is likely highly reversible and could provide enhanced selectivity of the dithioester moiety for cysteine. In the mechanism of cysteine-triggered release of H2S from thionoesters, the poor nucleophilicity of the departing alcohol likely impedes formation of the initial thionoester. In our proposed mechanism of H2S release from dithioesters, the departing thiolate bears enhanced nucleophilicity and likely reacts with the intermediate dithioester to regenerate the initial dithioester effectively reversing cysteine addition. Additionally, the enhanced reversibility of transthioesterification supports the considerably slower kinetics between dithioesters and thionoesters. Taken together, we hypothesized this enhanced reversibility would result in the initial nucleophilic attack by cysteine being the rate-determining step of cysteine-triggered H2S release from dithioesters.
To confirm the formation of a cysteine-based dihydrothiazole, PDTE (100 μM) was treated with L-cysteine methyl ester (2 mM, 20 equiv.) and a reaction aliquot after 1 h was subjected to HPLC analysis (see ESI†). In agreement with our proposed mechanism, the HPLC analysis revealed the formation of the expected dihydrothiazole in ∼61% yield, which is consistent with the H2S-releasing efficiency of PDTE as measured via the methylene blue assay. To gain insights on the rate-determining step, we measured the effect of temperature on the rate of H2S release from PDTE (25 μM) in the presence of cysteine (500 μM, 20 equiv.) (Fig. 5). If nucleophilic attack by cysteine on PDTE is the rate-determining step of the reaction, we would expect to observe an entropy of activation (ΔS‡) of approximately −20 eu, which would be characteristic of a bimolecular reaction.44,45
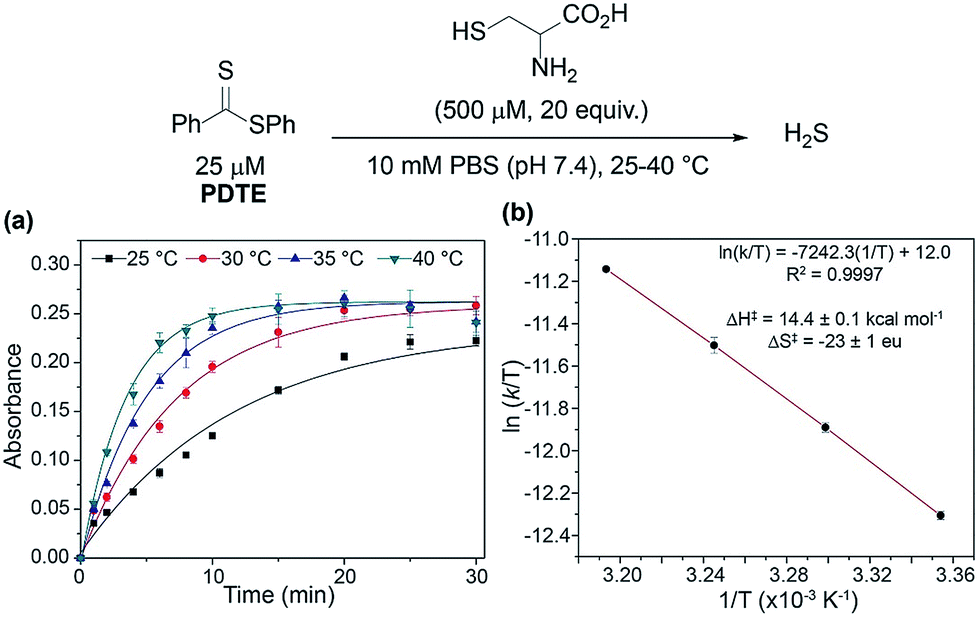 | ||
| Fig. 5 (a) Effect of temperature on rate of H2S release from PDTE (25 μM) in the presence of cysteine (500 μM, 20 equiv.). (b) Eyring analysis of cysteine-triggered H2S release from PDTE. | ||
Upon measuring the rates of H2S release at different temperatures, we constructed an Eyring plot using the obtained kobs values, which afforded ΔS‡ = −23 ± 1 eu. The observed ΔS‡ supports our mechanistic hypothesis and is consistent with the initial addition of cysteine to the dithioester to generate 1 being the rate-determining step of cysteine-triggered H2S release from dithioesters. In the reaction of thionoesters with cysteine, we interpreted an experimentally-determined ΔS‡ = −38 ± 3 eu to suggest the intramolecular cyclization as the rate determining step. In comparison between both mechanisms, by simply altering the nucleophilicity of the leaving group (i.e. alcohol vs. thiol) we have shunted the rate-determining step of the reaction. Additionally, our results suggest that cysteine-triggered H2S release from dithioesters and native chemical ligation share a similar rate-determining step.
To complement our experimental results, we used density functional theory (DFT) to examine the potential energy surface for H2S release from dithioesters. Because PDTE was used for our experimental mechanistic investigations, we chose to investigate the reaction of PDTE with cysteine thiolate using Gaussian 09 at the B3LYP/6-311++G(d,p) level of theory applying the IEF-PCM water solvation model. We found that the initial nucleophilic attack by cysteine thiolate on PDTE resulted in an activation barrier of 19.1 kcal mol−1, which was the highest barrier on the reaction coordinate and qualitatively agrees with the experimentally-observed ΔH‡ of 14.4 kcal mol−1. The resultant transthioesterified cysteine adduct is 3.7 kcal mol−1 more stable than the PDTE starting material, and subsequently undergoes an intramolecular S to N thioacyl transfer reaction with an associated barrier of 8.9 kcal mol−1, resulting in the final and more thermodynamically-favorable dihydrothiazole product (Fig. 6).
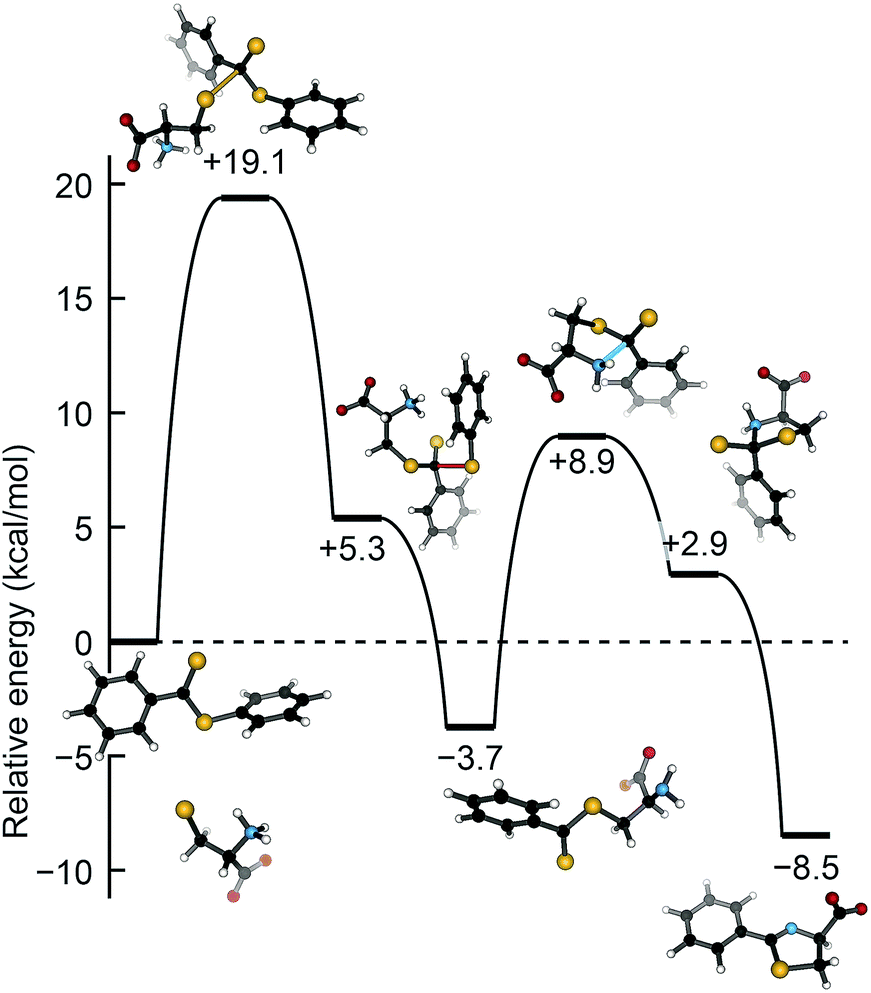 | ||
| Fig. 6 Potential energy surface for the attack of cysteine thiolate on PDTE. Calculations were performed in Gaussian 09 at the B3LYP/6-311++G(d,p) level of theory applying the IEF-PCM water solvation model. | ||
The ease of synthesis of dithioester compounds also enabled further investigation into the effect of alkyl functionalization on H2S release. We hypothesized that the electrophilicity at the thiocarbonyl position could be modified directly by introducing various alkyl, and thus alter the rate of H2S release. Additionally, we hypothesized that modification of the departing thiolate nucleophilicity would also alter the rate of H2S release from dithioesters. We anticipated the introduction of inductively donating alkyl groups, such as methyl and isopropyl, would decrease the thiocarbonyl electrophilicity and resulting in decreased rates of H2S release. Additionally, we anticipated that replacement of thiophenol with benzyl mercaptan would result in decreased rates of H2S release due to the enhanced nucleophilicity of benzyl mercaptan over thiophenol. To test our hypotheses, we measured H2S release from our library of alkyl functionalized dithioesters (25 μM) in the presence of cysteine (500 μM, 20 equiv.) (Fig. 7).
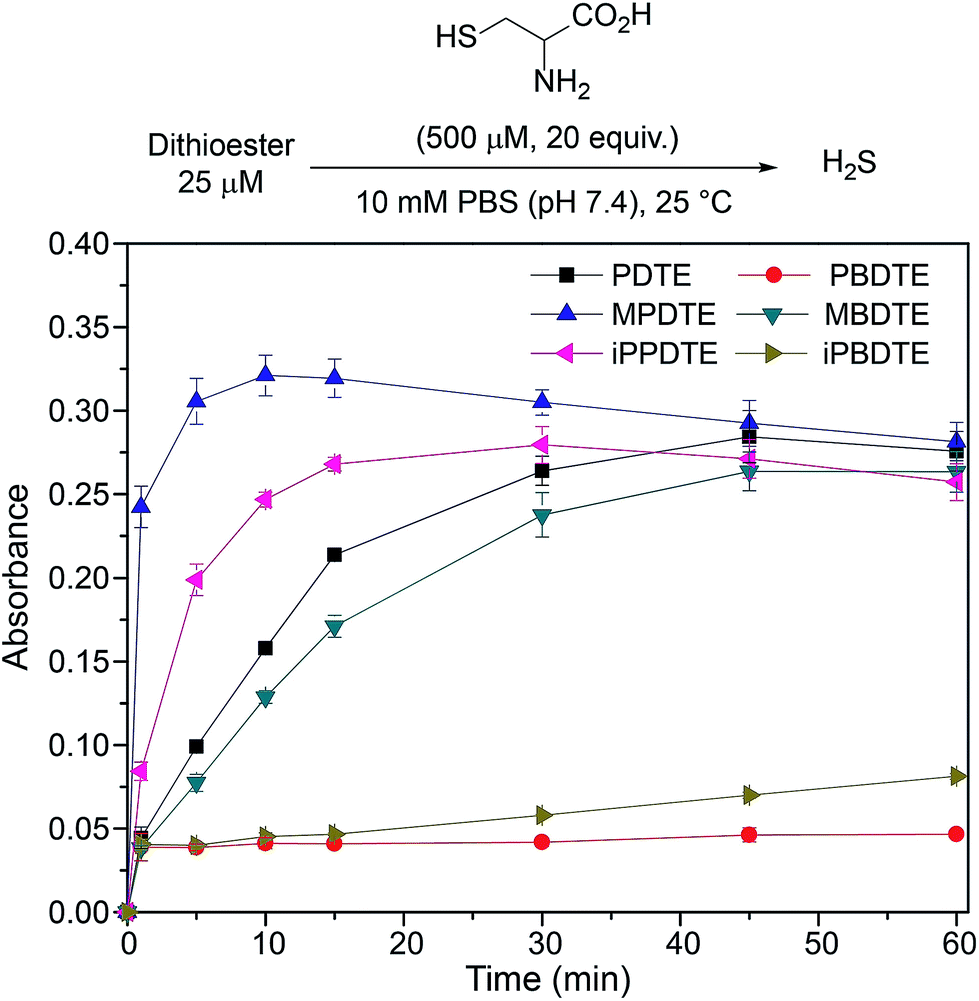 | ||
| Fig. 7 Effect of alkyl functionalization on the rate of H2S release from dithioesters. | ||
Contrary to our initial hypothesis, the incorporation of inductively-donating alkyl groups led to enhanced rates of H2S release relative to PDTE. To better understand the apparent releasing trends, we considered the stability of the charge-separated thiocarbonyl motif and the effect of different alkyl groups (Fig. 8). In the case of PDTE, a charge-separated thiocarbonyl yields a benzylic carbocation which can readily delocalize via resonance effectively altering the electrophilicity of the thiocarbonyl via delocalization of the carbocation. In the presence of inductively donating groups such as methyl and isopropyl in MPDTE and iPPDTE respectively, the resulting carbocation is localized to the thiocarbonyl position, which would result in enhanced rates of H2S release in comparison to PDTE. Incorporation of an isopropyl group in iPPDTE, however, also introduces the potential for an intermediate 1,2-methyl shift which would partially delocalize the carbocation and hinder H2S release relative to MPDTE. Considering these contributions, we interpret the enhanced release of H2S from alkyl-functionalized dithioesters as a reflection of altered thiocarbonyl electrophilicity via carbocation delocalization. Although this outcome was contrary to our initial hypothesis, these findings demonstrate the ability to tune the rate of H2S release from dithioesters by simple alkyl substitutions.
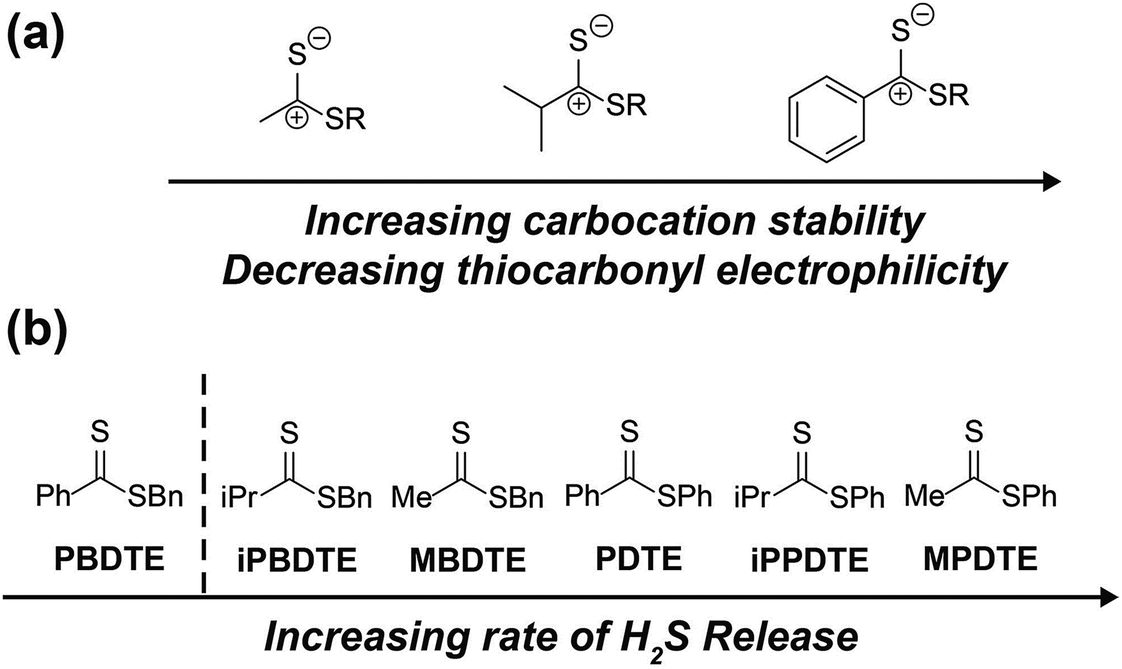 | ||
| Fig. 8 (a) Effect of pendant groups on thiocarbonyl electrophilicity and carbocation stability. (b) Scale of increasing H2S releasing rates for reported dithioesters. | ||
Furthering our studies, we also examined the effect of benzyl mercaptan as a leaving group on H2S release from MBDTE, PBDTE, and iPBDTE respectively. In comparison to thiophenol, benzyl mercaptan is a considerably better nucleophile and likely perturbs the equilibrium of transthioesterification to disfavor the addition of cysteine. In concert with the effect of alkyl groups on thiocarbonyl electrophilicity, we anticipated H2S release from dithioesters bearing benzyl mercaptan to be dramatically altered relatively to their thiophenol analogue. In agreement with our hypothesis, H2S release was observed exclusively from MBDTE. Considering the lack of carbocation delocalization by the pendant methyl group, this result suggests the thiocarbonyl moiety in MBDTE is sufficiently electrophilic to promote H2S release in the presence of cysteine. Alternatively, we observed effectively no H2S release from PBDTE and relatively slow H2S release from iPBDTE. Considering the effect of alkyl groups on thiocarbonyl electrophilicity via carbocation delocalization in concert with the enhanced nucleophilicity of benzyl mercaptan, a significantly hindered rate of release from iPBDTE and complete shutdown of H2S release from PBDTE is also in agreement with our hypothesis. Overall, we found the use of benzyl mercaptan decreased overall H2S-releasing efficiency and yielded hindered rates of release in comparison to thiophenol-based dithioesters. Taken together, these findings demonstrate the rate of H2S release from dithioesters can be enhanced or decreased as a function of alkyl substitution.
Conclusions
Advancing our previous work on synthetic H2S donors via the chemoselective condensation of thionoesters and cysteine, we have identified that dithioesters are a viable and tunable platform for developing cysteine-triggered H2S donors. Using PDTE as a representative dithioester, we have demonstrated H2S release in the presence of cysteine proceeds with good efficiency, and that dithioesters provide significant advancements over thionoesters, including ease of functionalization and enhanced selectivity for cysteine over other thiol-based nucleophiles including GSH. Mechanistic investigations, including rate-order analysis and activation parameters support a mechanism in which the initial nucleophilic attack by cysteine on the dithioesters is the rate-determining step of the reaction, which is also supported by computation. Furthermore, we demonstrated that the rate of H2S release from a small library of dithioesters can be controlled by alkyl substitution. The findings presented herein provide several advancements over thionoesters as cysteine-triggered H2S donors and provides a foundation for further development of dithioesters for expanding upon the chemical biology of H2S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported here was supported by the Dreyfus Foundation and the NIH (R01GM113030). NMR and MS capabilities in the UO CAMCOR facility are supported by the NSF (CHE-1427987, CHE-1625529).References
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- J. Chiefari, R. T. A. Mayadunne, C. L. Moad, G. Moad, E. Rizzardo, A. Postma, M. A. Skidmore and S. H. Thang, Macromolecules, 2003, 36, 2273–2283 CrossRef CAS.
- Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- M. M. Cerda, Y. Zhao and M. D. Pluth, J. Am. Chem. Soc., 2018, 140, 12574–12579 CrossRef CAS PubMed.
- J. J. Newton, R. Britton and C. M. Friesen, J. Org. Chem., 2018, 83, 12784–12792 CrossRef CAS PubMed.
- A. K. Mustafa, M. M. Gadalla and S. H. Snyder, Sci. Signaling, 2009, 2, re2 Search PubMed.
- R. Wang, FASEB J., 2002, 16, 1792–1798 CrossRef CAS PubMed.
- W. Zhao, J. Zhang, Y. Lu and R. Wang, EMBO J., 2001, 20, 6008–6016 CrossRef CAS PubMed.
- Z. Altaany, G. Yang and R. Wang, J. Cell. Mol. Med., 2013, 17, 879–888 CrossRef CAS PubMed.
- G. Yang, K. Zhao, Y. Ju, S. Mani, Q. Cao, S. Puukila, N. Khaper, L. Wu and R. Wang, Antioxid. Redox Signaling, 2013, 18, 1906–1919 CrossRef CAS PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, L. K. De La Cruz, M. Roy Choudhury, A. Anifowose and B. Wang, Med. Res. Rev., 2018, 38, 57–100 CrossRef CAS PubMed.
- M. Whiteman, L. Li, P. Rose, C. H. Tan, D. B. Parkinson and P. K. Moore, Antioxid. Redox Signaling, 2010, 12, 1147–1154 CrossRef CAS PubMed.
- E. R. DeLeon, G. F. Stoy and K. R. Olson, Anal. Biochem., 2012, 421, 203–207 CrossRef CAS PubMed.
- C. R. Powell, K. M. Dillon and J. B. Matson, Biochem. Pharmacol., 2018, 149, 110–123 CrossRef CAS PubMed.
- C. Szabo and A. Papapetropoulos, Pharmacol. Rev., 2017, 69, 497–564 CrossRef PubMed.
- P. Bora, P. Chauhan, K. A. Pardeshi and H. Chakrapani, RSC Adv., 2018, 8, 27359–27374 RSC.
- Y. Zhao, T. D. Biggs and M. Xian, Chem. Commun., 2014, 50, 11788–11805 RSC.
- L. Li, G. Rossoni, A. Sparatore, L. C. Lee, P. Del Soldato and P. K. Moore, Free Radical Biol. Med., 2007, 42, 706–719 CrossRef CAS PubMed.
- L. Li, M. Whiteman, Y. Y. Guan, K. L. Neo, Y. Cheng, S. W. Lee, Y. Zhao, R. Baskar, C. H. Tan and P. K. Moore, Circulation, 2008, 117, 2351–2360 CrossRef CAS PubMed.
- C. M. Park, Y. Zhao, Z. Zhu, A. Pacheco, B. Peng, N. O. Devarie-Baez, P. Bagdon, H. Zhang and M. Xian, Mol. BioSyst., 2013, 9, 2430–2434 RSC.
- J. Kang, Z. Li, C. L. Organ, C. M. Park, C. T. Yang, A. Pacheco, D. Wang, D. J. Lefer and M. Xian, J. Am. Chem. Soc., 2016, 138, 6336–6339 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, K. Ji, Z. Pan, V. Chittavong and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 4514–4518 CrossRef CAS PubMed.
- Z. Xiao, T. Bonnard, A. Shakouri-Motlagh, R. A. L. Wylie, J. Collins, J. White, D. E. Heath, C. E. Hagemeyer and L. A. Connal, Chemistry, 2017, 23, 11294–11300 CrossRef CAS PubMed.
- N. O. Devarie-Baez, P. E. Bagdon, B. Peng, Y. Zhao, C. M. Park and M. Xian, Org. Lett., 2013, 15, 2786–2789 CrossRef CAS PubMed.
- J. J. Woods, J. Cao, A. R. Lippert and J. J. Wilson, J. Am. Chem. Soc., 2018, 140, 12383–12387 CrossRef CAS PubMed.
- A. K. Steiger, S. Pardue, C. G. Kevil and M. D. Pluth, J. Am. Chem. Soc., 2016, 138, 7256–7259 CrossRef CAS PubMed.
- Y. Zhao, A. K. Steiger and M. D. Pluth, Angew. Chem., Int. Ed., 2018, 57, 13101–13105 CrossRef CAS PubMed.
- A. K. Sharma, M. Nair, P. Chauhan, K. Gupta, D. K. Saini and H. Chakrapani, Org. Lett., 2017, 19, 4822–4825 CrossRef CAS PubMed.
- C. R. Powell, J. C. Foster, B. Okyere, M. H. Theus and J. B. Matson, J. Am. Chem. Soc., 2016, 138, 13477–13480 CrossRef CAS PubMed.
- M. Pluth, T. Bailey, M. Hammers, M. Hartle, H. Henthorn and A. Steiger, Synlett, 2015, 26, 2633–2643 CrossRef CAS.
- F. Ercole, M. R. Whittaker, M. L. Halls, B. J. Boyd, T. P. Davis and J. F. Quinn, Chem. Commun., 2017, 53, 8030–8033 RSC.
- M. M. Cerda, M. D. Hammers, M. S. Earp, L. N. Zakharov and M. D. Pluth, Org. Lett., 2017, 19, 2314–2317 CrossRef CAS PubMed.
- M. R. Filipovic, J. Zivanovic, B. Alvarez and R. Banerjee, Chem. Rev., 2018, 118, 1253–1337 CrossRef CAS PubMed.
- Y. Zhao, H. Wang and M. Xian, J. Am. Chem. Soc., 2011, 133, 15–17 CrossRef CAS PubMed.
- Y. Zhao, C. Yang, C. Organ, Z. Li, S. Bhushan, H. Otsuka, A. Pacheco, J. Kang, H. C. Aguilar, D. J. Lefer and M. Xian, J. Med. Chem., 2015, 58, 7501–7511 CrossRef CAS PubMed.
- J. C. Foster, C. R. Powell, S. C. Radzinski and J. B. Matson, Org. Lett., 2014, 16, 1558–1561 CrossRef CAS PubMed.
- J. C. Foster, S. C. Radzinski, X. Zou, C. V. Finkielstein and J. B. Matson, Mol. Pharm., 2017, 14, 1300–1306 CrossRef CAS PubMed.
- F. Ercole, F. M. Mansfeld, M. Kavallaris, M. R. Whittaker, J. F. Quinn, M. L. Halls and T. P. Davis, Biomacromolecules, 2016, 17, 371–383 CrossRef CAS PubMed.
- T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2007, 107, 5210–5278 CrossRef CAS PubMed.
- L. M. Siegel, Anal. Biochem., 1965, 11, 126–132 CrossRef CAS PubMed.
- G. Levesque, P. Arsène, V. Fanneau-Bellenger and T.-N. Pham, Biomacromolecules, 2000, 1, 400–406 CrossRef CAS PubMed.
- D. B. Thomas, A. J. Convertine, R. D. Hester, A. B. Lowe and C. L. McCormick, Macromolecules, 2004, 37, 1735–1741 CrossRef CAS.
- Organic Reaction Mechanisms 1967, ed. B. Capon, M. J. Perkins and C. W. Rees, Wiley-Interscience, London, 1967, ch. 12 Search PubMed.
- E. V. Anslyn and D. A. Dougherty, in Modern Physical Organic Chemistry, University Science, Sausalito, CA, 2006, ch. 7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectra, computational details, tabulated geometries. See DOI: 10.1039/c8sc04683b |
| This journal is © The Royal Society of Chemistry 2019 |