
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface radio-mineralisation mediates chelate-free radiolabelling of iron oxide nanoparticles†
P. Stephen
Patrick
 *a,
Lara K.
Bogart
d,
Thomas J.
Macdonald
c,
Paul
Southern
d,
Michael J.
Powell
c,
May
Zaw-Thin
a,
Nicolas H.
Voelcker
ef,
Ivan P.
Parkin
c,
Quentin A.
Pankhurst
d,
Mark F.
Lythgoe
a,
Tammy L.
Kalber
a and
Joseph C.
Bear
*b
*a,
Lara K.
Bogart
d,
Thomas J.
Macdonald
c,
Paul
Southern
d,
Michael J.
Powell
c,
May
Zaw-Thin
a,
Nicolas H.
Voelcker
ef,
Ivan P.
Parkin
c,
Quentin A.
Pankhurst
d,
Mark F.
Lythgoe
a,
Tammy L.
Kalber
a and
Joseph C.
Bear
*b
aCentre for Advanced Biomedical Imaging (CABI), Department of Medicine, University College London, London WC1E 6DD, UK. E-mail: peter.patrick@ucl.ac.uk
bSchool of Life Science, Pharmacy & Chemistry, Kingston University, Penrhyn Road, Kingston upon Thames, KT1 2EE, UK. E-mail: J.Bear@kingston.ac.uk
cMaterials Chemistry Centre, Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
dUCL Healthcare Biomagnetics Laboratory, 21 Albemarle Street, London, W1S 4BS, UK
eMonash Institute of Pharmaceutical Sciences, Monash University, Parkville, Australia
fCommonwealth Scientific and Industrial Research Organisation (CSIRO), Clayton, Australia
First published on 9th January 2019
Abstract
We introduce the concept of surface radio-mineralisation (SRM) to describe the chelate-free radiolabelling of iron-oxide and ferrite nanoparticles. We demonstrate the effectiveness of SRM with both 111In and 89Zr for bare, polymer-matrix multicore, and surface-functionalised magnetite/maghemite nanoparticles; and for bare Y3Fe5O12 nanoparticles. By analogy with geological mineralisation (the hydrothermal deposition of metals as minerals in ore bodies or lodes) we demonstrate that the heat-induced and aqueous SRM process deposits radiometal-oxides onto the nanoparticle or core surfaces, passing through the matrix or coating if present, without changing the size, structure, or magnetic properties of the nanoparticle or core. We show in a mouse model followed over 7 days that the SRM is sufficient to allow quantitative, non-invasive, prolonged, whole-body localisation of injected nanoparticles with nuclear imaging.
Introduction
The tuneable properties of iron oxide nanoparticles (IONPs), primarily magnetite, maghemite, or mixtures thereof, and their capacity for functionalisation suit them for numerous biomedical applications: hyperthermia therapy for cancer; drug, cell and gene delivery, and as diagnostic or cell tracking agents.1–7 The ability of superparamagnetic nanoparticles to de-phase the MRI-detectable water proton (1H) signal enables their detection in tissues as hypo-intensities, confirming delivery and retention with high resolution and sensitivity. Yet, in practice, identification of IONPs with magnetic resonance imaging (MRI) is often ambiguous due to endogenous signal hypo-intensities including the lungs, bone, and gut (Fig. 1). Particle quantification is also hampered by saturation of signal loss at high concentrations, insufficient sensitivity to low concentrations, and aggregation effects on relaxivity.8,9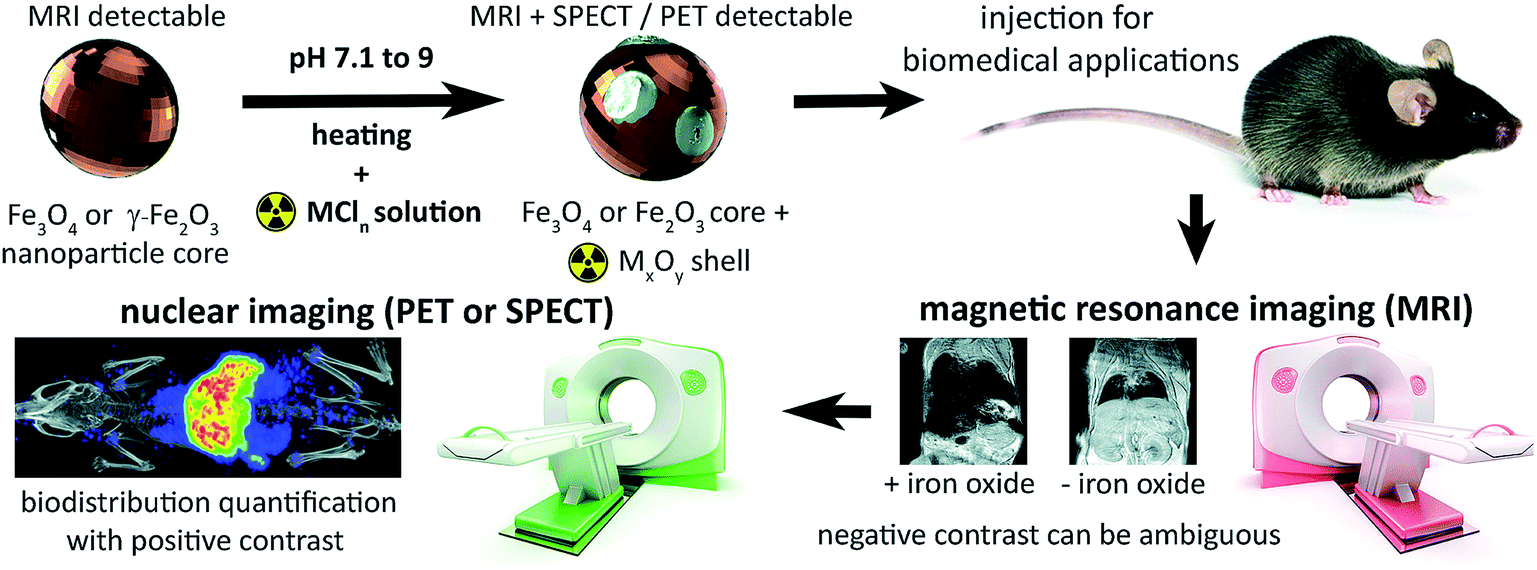 | ||
| Fig. 1 Synthesis of radiolabelled IONPs using radiometal chloride salts (MCln) to form an oxidised radiometal coating allows whole-body non-invasive quantitative imaging using PET or SPECT, in addition to high-resolution detection using MRI. | ||
These limitations have spurred research into conjugation and chelation chemistry for radiolabelling IONPs, to enable their detection and quantification with nuclear imaging.2,10,11 Single photon emission computed tomography (SPECT), and positron emission tomography (PET) detect γ-radiation respectively produced directly or indirectly (after positron annihilation) following radioisotope decay. This offers accurate and sensitive quantification of imaging isotopes across the body, without endogenous background signals from tissue. Traditionally, nanoparticle radiolabelling requires surface functionalisation with organic chelators10 – increasing the complexity, time and cost of synthesis. A standard method of radiolabelling IONPs has proven elusive as radiometals differ in co-ordination numbers and atomic radii, therefore requiring different chelating agents and conjugation strategies.12
Radiochemical doping provides one alternative to the use of chelators, whereby radiometals such as 64Cu and 111In are incorporated in the iron oxide core during its synthesis.13,14 This has the advantage of stable radiolabel retention. However its practicality is reduced by the necessity of synthesising (and possibly functionalising) particles on-demand before every use due to the constraints of isotope half-lives. A more user friendly and clinically-translatable approach would allow last-minute labelling of off-the-shelf iron oxide nanoparticles with the chosen isotope prior to injection. With this in mind, Chen et al. demonstrated a post-synthesis, chelate-free method for radiolabelling uncoated iron oxides using radioarsenic (71As, 72As, 74As, 76As),15 which was followed by a similar demonstration using 69Ge, also for PET imaging.16 To expand upon this, Boros et al. labelled the FDA-approved, carbohydrate coated IONP ferumoxytol (Feraheme®) with a range of more commonly available radiometal isotopes, including 89Zr and 64Cu for PET and 111In for SPECT.17 Particles were heated in an aqueous solution with the radiometal chloride (≥80 °C was optimal for most metals tested, including Zr, Cu, and In) at a pH between 7–9. However, despite ongoing interest in this method,2,7 the nature of the chemical interaction between the metal isotopes and iron oxide nanoparticles remained unidentified.18
Here, we address this problem by applying a combination of energy-dispersive X-ray spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), time of flight mass spectrometry (ToF-SIMS) and room temperature 57Fe Mössbauer spectroscopy, to elucidate the mechanistics of heat-induced iron oxide and ferrite radiolabelling as described by Boros et al. We establish that it operates primarily through mineralisation of the radiometal onto the particle surface as a radiometal oxide (see Fig. 1). We show that this surface radiomineralisation (SRM) has no effect on the structural and chemical properties of commercially available maghemite and magnetite/maghemite-based IONPs, and that as the key magnetic properties of the particles remain unchanged, their utility for MRI and other biomedical applications is retained. Finally, we demonstrate tracking of 111In radiolabelled IONPs using whole-body, non-invasive SPECT imaging, thereby illustrating key advantages over the use of MRI alone.
Results
We initially investigated the ability of the reported method17 to induce radiolabelling of commercially-available nanoparticles representing the two magnetic materials most commonly used in biomedical applications: maghemite (γ-Fe2O3), and magnetite (Fe3O4).4 As we have shown previously, commercially available magnetite is typically a magnetite/maghemite mixture despite being labelled as >98% magnetite, which we confirmed here using a model-independent fitting of the 57Fe Mössbauer spectrum of the “magnetite” sample, as per Fock and Bogart et al.,19 which indicated a mixture comprising ≈63 wt% magnetite and 37 wt% maghemite. The maghemite and the magnetite/maghemite particles were then labelled via heating to 90 °C at pH 9 in the presence of 100 to 200 kBq of either 111In or 89Zr, for 90 min (Table 1). This resulted in 79% to 94% retention of the radioactivity on the particles; assessed by either thin layer chromatography (TLC) or magnetic separation.| Nominal chemical composition | Nominal particle diameter | Supplier | Matrix | 111In RCY (%) | 89Zr RCY (%) | ||
|---|---|---|---|---|---|---|---|
| TLC, n = 11 (SEM) | Magnetic separation, n = 4 (SEM) | TLC, n = 10 (SEM) | Magnetic separation, n = 4 (SEM) | ||||
| a Magnetic separation of Y3Fe5O12 was not completely successful as indicated by visual inspection. | |||||||
| N/A control | — | — | — | 0.7 (0.5) | 0.4 (0.2) | 2.46 (2.9) | 0.4 (0.2) |
| Maghemite (γ-Fe2O3) | 20–40 nm | Alfa Aesar | Bare | 79.1 (4.9) | 79.3 (6.8) | 94.2 (0.7) | 94.7 (0.4) |
| Magnetite (Fe3O4) | 50–100 nm | Sigma Aldrich | Bare | 85.2 (3.1) | 78.6 (6.4) | 94.9 (1.1) | 94.2 (0.7) |
| Y3Fe5O12 | <100 nm | Sigma Aldrich | Bare | 88.2 (3.7) | 66.6 (3.5)a | 91.9 (1.3) | 71.9 (3.5)a |
The radiolabelling efficiency was slightly higher using 89Zr than with 111In, consistent with the previous report on ferumoxytol.17 Labelling was repeated using non-radioactive (natural abundance) metal isotopes of InCl3 and ZrCl4 (10 μmol metal chloride additive per 100 mg of IONPs). Metal additives were successfully incorporated into IONPs as assessed by inductively coupled plasma mass spectrometry, ICP-MS (17 to 51%), giving the same trend of higher labelling efficiencies for Zr compared to In (Table S1†). To show the broader applicability of this process with alternative iron oxides, yttrium iron oxide nanoparticles (Y3Fe5O12) were also successfully radiolabelled (Table 1).
In order to establish the effect of In and Zr labelling on the physical properties of magnetite/maghemite and Y3Fe5O12 nanoparticles, we have used both transmission electron microscopy (TEM) and powder X-ray diffraction (pXRD) and observed no discernible change in the physical structure of the nanoparticles following the labelling treatment (Fig. 2). TEM imaging clearly showed that there was no change in the physical structure of the nanoparticles followed the labelling treatment (Fig. 2). The core sizes of each sample displayed the familiar log-normal size distribution, which remained unchanged regardless of reaction conditions. Indeed, due to the highly polydisperse nature of the nanoparticles (as seen by the large standard deviation), any change in size was impossible to see: 35.3 ± 20.5 nm for the maghemite, 19.1 ± 15.8 nm for the Y3Fe5O12 and 116.6 ± 79.0 nm for the magnetite/maghemite. High resolution TEM analysis (Fig. 2A–C and S14–S22†) clearly shows the lack of core/shell structure and no change in the lattice planes of the nanoparticles, consistent with pXRD observations (Fig. S1–S4†). The inability to detect either In or Zr via pXRD suggests an amorphous and non-crystalline incorporation, as would be expected based on the relatively low reaction temperature. The presence of small amounts of In and Zr within the samples was confirmed with energy dispersive X-ray spectroscopy (EDS) (Fig. 2F and S14–S22†).
 | ||
| Fig. 2 Characterisation of maghemite (Alfa Aesar) γ-Fe2O3 following 111In labelling. High resolution TEM images (A–C), show no discernible change in lattice structure from bulk iron oxide. (C) Lattice d-spacing of 2.87 Å assigned as the (220) plane of maghemite. (D) SQUID measurement of maghemite (γ-Fe2O3) before and after modification shows comparable magnetisation curves. (E) Mössbauer spectra acquired for maghemite (Fe2O3) particles before and after heat induced labelling with non-radioactive In and Zr additives. No change in the proportion of Fe atoms in a maghemite environment was seen following the labelling reaction. (F) EDS spectrum shows the presence of added In. (G) ToF-SIMS demonstrates a sharp decrease in In concentration with increasing sputter time, indicating its surface bound nature. | ||
To quantify any change in composition following heating and radiolabelling, the maghemite and magnetite/maghemite particles were analysed with room temperature 57Fe Mössbauer spectroscopy19 before and after In and Zr labelling (Fig. S23†). For the labelled maghemite samples there was no change in the measured spectra for all of the treated samples; we confirmed this quantitatively by observing no change to the value of the α parameter (the numerical proportion of Fe atoms in the magnetite environment), with α = 0 ± 0.04, consistent with pure maghemite.19,20 Similarly, for the treated Sigma Aldrich “magnetite” samples there was no change to the spectra, with the best fit isomer shift indicating α = 0.52 ± 0.02, corresponding to a magnetite content of ca. 56 wt%. Such observations strongly suggest that there is no incorporation of either the In or Zr within the sub-lattice structures of the iron oxide, indicating a surface location.
We confirmed this using X-ray photoelectron spectroscopy (XPS; a surface (<10 nm) sensitive technique) to probe the oxidation state and quantities of the In and Zr additives (Fig. S5–S13†). All samples showed the presence and absence of the In and/or Zr additives where appropriate. Fe environments were largely unchanged after In/Zr treatment, with only 0.1 eV variation across the three samples in Fe 2p scans, in good agreement with the Mössbauer spectra. The Fe 2p3/2 values of 710.6 (AA maghemite), 710.7 (SA magnetite), and 710.3 eV (Y3Fe5O12) are indicative of γ-Fe2O3 (710.6, 710.7 eV) and Y3Fe5O12 (710.3 eV) respectively.21,22 There was also little variation within the Y3Fe5O12 samples, with a single environment, and a variation in the Y 3d5/2 range of 157.0–157.3 eV displayed, similar to that observed in Y2O3.23
Both the In and Zr additives were clearly seen in single chemical environments, for all treated samples. Scans of In 3d showed In with a 3+ oxidation state, assigned as In2O3 at 444.3 eV and Zr 3d scans showed a Zr 3d5/2 peak at 182.0 eV assigned as ZrO2, with Zr in the 4+ oxidation state. The high oxidation states of the additive elements and the absence of any observable change or indeed new chemical environments in either the O 1s or Fe 2p high resolution scans, leads us to conclude that the additives are surface bound and not fully integrated (doped) into the iron oxide structure, which is supported by pXRD and Mössbauer spectroscopy (Fig. S1–S4†). Based on the amount of In detected in the samples by ICP-MS, this gives a ratio of 1 In atom for every 3 to 20 surface Fe atoms per nanoparticle for the magnetite/maghemite and maghemite particles respectively – consistent with the absence of shell detection with TEM.
The surface location of additives was further established with time of flight-secondary ion mass spectroscopy (ToF-SIMS), which was used to remove atomic monolayers of metal ions from the surface of the IONPs. Fig. 2G presents the ToF-SIMS depth profile for the In doped maghemite (γ-Fe2O3-In), in which the In concentration shows a sharp drop with etching time. The slight increase in iron concentration can be attributed to its dominance within the core of the NPs, which becomes more clear on removal of the In. These measurements were complemented by the ToF-SIMS depth profile measurements of the Y3Fe5O12 NPs (Fig. S28, ESI†). From this, we propose that the In atoms are surface bound, consistent with the mild nature of the radiolabelling reaction and its negligible effects on the structural (by TEM) and physical (vide infra) properties of the particles. ToF-SIMS of the Zr-doped maghemite and Y3Fe5O12 NPs was complicated by the ionisation efficiency of Zr and the overlapping mass with the Y fragment. Furthermore, ToF-SIMS is more surface sensitive than XPS; which means the ion beam penetration was considerably lower than that of the XPS measurements (sampling depth is considerably less).24 Despite this, Zr was successfully detected using XPS (Fig. S5–S13†).
Following the demonstration of In and Zr surface mineralisation using this method, we next sought to demonstrate wider utility with a range of maghemite and magnetite-based nano- and microparticles coated for biomedical application. A small selection of commercially-available magnetic nanoparticles was chosen for variation in size and coating, and labelled with either 111In or 89Zr according to the protocol described above. As with the uncoated maghemite and magnetite/maghemite particles (Table 1), this resulted in efficient radiochemical yields (RCYs) between 68 and 95% as assessed with TLC and magnetic separation (Table 2).
| Particle type | Nominal particle diameter | Matrix | 111In % RCY | 89Zr % RCY | ||
|---|---|---|---|---|---|---|
| TLC (SEM), n = 11 | Magnetic separation (SEM), n = 4 | TLC (SEM), n = 10 | Magnetic separation (SEM), n = 8 | |||
| FluidMag | 50 nm | Citrate | 68.5 (3.1) | — | 86.3 (1.8) | — |
| FluidMag | 100 nm | Citrate | 70.3 (3.8) | 81.1 (7.7) | 87.9 (2.2) | 83.4 (5.9) |
| FluidMag | 200 nm | Citrate | 69.1 (3.5) | 84.9 (6.7) | 93.3 (0.9) | 94.8 (0.5) |
| Biomag Maxi | 3–12 μm | Carboxyl functionalised alkoxysilane | 71.4 (3.1) | 86.1 (6.0) | 82.8 (1.6) | 93.3 (1.5) |
| SiMag | 500 nm | Silanol | 80.4 (3.1) | 83.7 (1.5) | 71.5 (2.8) | 64.3 (5.0) |
To monitor the effects of the radiolabelling procedure on the magnetic properties of a selection of these particles, super-conducting quantum interference device (SQUID) measurements were taken after labelling with non-radioactive ZrCl4 and InCl3 additives at a ratio 10 μmol per 100 mg particle (see Table S2, Fig. S24–S27†). For each particle type except the Y3Fe5O12 (which showed altered coercivity but not saturation magnetisation or remanence), magnetisation curves were comparable between unmodified particles and those labelled with Zr and In (Fig. 2D).
We lastly demonstrate that particles labelled with this method are suitable for in vivo imaging with MRI and SPECT. FluidMag CT was chosen as a representative particle for biomedical application as it is commercially available and has previously been evaluated for stem cell labelling and hyperthermia.25,26 MRI was done prior to and 2.5 h following intravenous injection to monitor the distribution of the labelled particles in wild type mice (C57BL/6 strain). Signal hypo-intensity was present in the lungs only pre-injection (Fig. 3A), and post-injection in lungs, liver, and kidneys (Fig. 3B) – consistent with previous reports of excretory organ nanoparticle uptake.27 An equivalent dose of unmodified stock particles were injected into a separate animal, and gave comparable distribution of MRI contrast as the radiolabelled particles, predominantly in the liver (Fig. 3B and C).
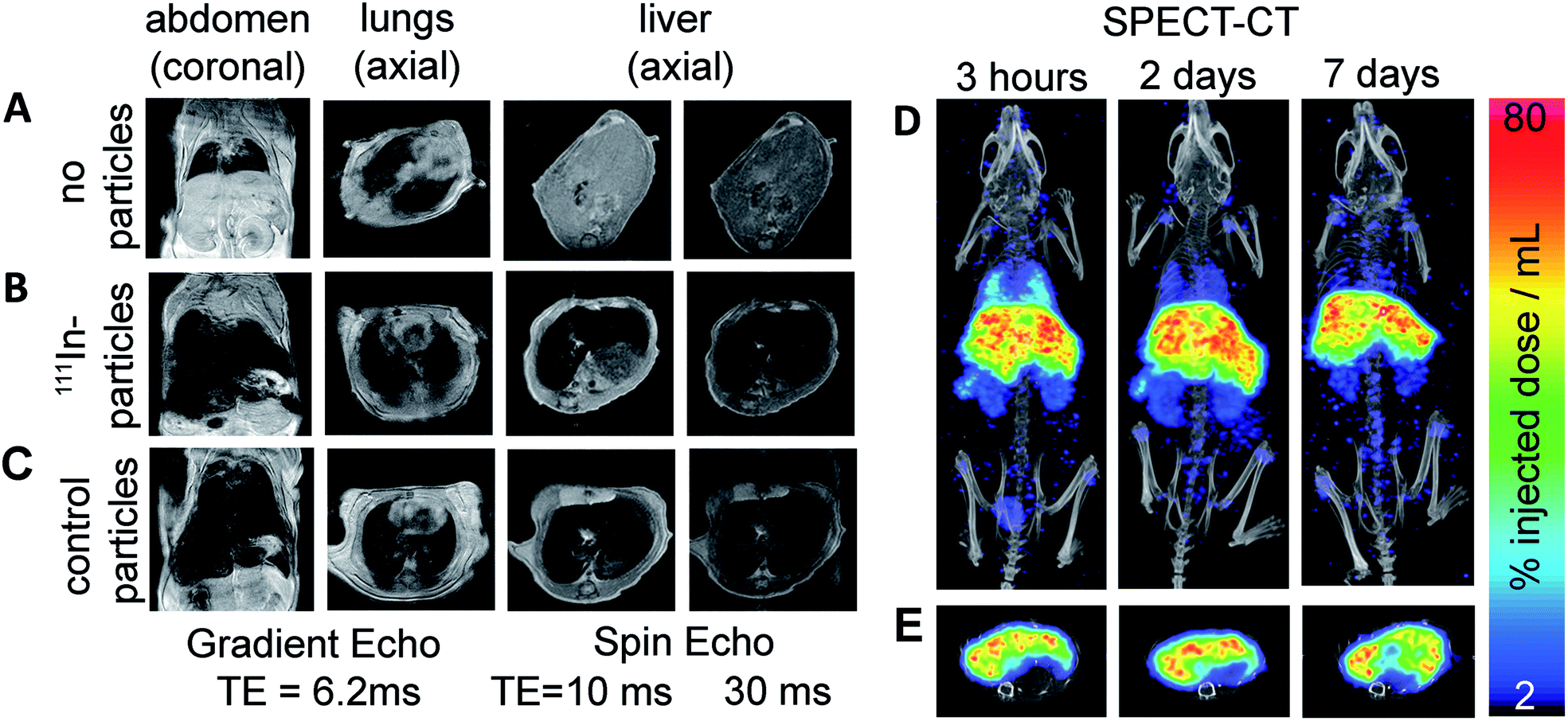 | ||
| Fig. 3 MRI pre (A) and post (B) injection of the 111In-FluidMagCT, or (C) post-FluidMagCT (control). (D) Maximum intensity projection 111In SPECT-CT at 3 h, 2 and 7 d post-injection confirms presence of labelled iron oxides in the liver, lung, kidneys, and spleen of C57BL/6 mice. (E) Corresponding axial slices show co-localisation of the radiolabelled IONPs and the liver. | ||
SPECT-CT imaging at 3 h, 2 d, and 7 d post injection confirmed the location of the 111In-labelled IONPs within the liver and kidneys (see Fig. 3), showing additional retention within the lungs which was difficult to identify on the MR images due to endogenous contrast. At 2 and 7 d SPECT-CT showed a clearing of the particles from the lungs – again not detectable using MRI. Quantification of total activity with SPECT ROI analysis showed the majority of activity (54.5%) retained in the liver after 7 days.
Conclusion
In conclusion, using a combination of TEM, 57Fe Mössbauer spectroscopy, XPS, ToF-SIMS and SQUID we have demonstrated that heat-mediated chelate-free radiolabelling method as described by Boros et al.17 operates by mineralisation of the radiometal (111In or 89Zr) as an oxide on the surface of the IONP that does not alter the magnetic and physical properties of the particle core. Further, we have shown that this surface radio-mineralisation (SRM) is compatible with a range of nano- and microscale iron oxide particles (maghemite and magnetite/maghemite), as well as yttrium iron oxide (Y3Fe5O12) as a representative non-typical iron oxide. ToF-SIMS analysis shows radiometal incorporation is limited to the surface of the particles, consistent with the lack of change in the magnetite–maghemite content of the starting material. With the exception of a slight effect on the Y3Fe5O12 particles this was confirmed by means of SQUID analysis, which showed that their original magnetisation curves were retained following labelling.This report affords a better understanding of the heat-induced chelate-free radiolabelling method. We anticipate that this will encourage its use in investigating the bio-distribution of the IONP-based biomedical therapeutics and diagnostics, thus combining the quantitative high-sensitivity of PET/SPECT imaging with the high-resolution detail of MRI.
Experimental section
Detailed descriptions of the instrumentation, materials and experimental protocols can be found in the ESI.†Ethical statement
All animal studies were approved by the University College London Biological Services Ethical Review Committee and licensed under the UK Home Office regulations and the Guidance for the Operation of Animals (Scientific Procedures) Act 1986 (Home Office, London, United Kingdom). All animal methods were performed in accordance to institutional ethical guidelines and regulations. During all in vivo imaging, mice were maintained at 37 °C under isofluorane breathable anaesthesia (1–2%) in oxygen. A small animal physiological monitoring system (SA Instruments, Stony Brook, NY) was used to maintain respiration rate. Mice (C57BL/6; male) were obtained from Charles River at 4 months old.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Dr Stanislav Strekopytov for the ICP-MS analysis at the Imaging and Analysis Centre, Natural History Museum, London. P. S. P. acknowledges funding from the UK Regenerative Medicine Platform (MRC: MR/K026739/1) and MRC grant MR/R026416/1. T. L. K. is funded by an EPSRC Early Career Fellowship (EP/L006472/1). J. B. and T. J. M. acknowledge the Ramsay Memorial Trust for their financial assistance through the Ramsay Memorial Fellowship. T. J. M. and N. H. V. would like to thank Dr John Denman from the University of South Australia for performing the ToF-SIMS measurements and assisting with the analysis.References
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi and A. Shaw, et al., Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues, Nat. Nanotechnol., 2016, 11(11), 986–994 CrossRef CAS PubMed.
- H. S. Yuan, M. Q. Wilks, M. D. Normandin, G. El Fakhri, C. Kaittanis and L. Josephson, Heat-induced radiolabeling and fluorescence labeling of Feraheme nanoparticles for PET/SPECT imaging and flow cytometry, Nat. Protoc., 2018, 13(2), 392–412 CrossRef CAS PubMed.
- A. H. Lu, E. L. Salabas and F. Schuth, Magnetic nanoparticles: Synthesis, protection, functionalization, and application, Angew. Chem., Int. Ed., 2007, 46(8), 1222–1244 CrossRef CAS PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, Applications of magnetic nanoparticles in biomedicine, J. Phys. D: Appl. Phys., 2003, 36(13), R167–R181 CrossRef CAS.
- J. J. Connell, P. S. Patrick, Y. Yu, M. F. Lythgoe and T. L. Kalber, Advanced cell therapies: targeting, tracking and actuation of cells with magnetic particles, Regener. Med., 2015, 10(6), 757–772 CrossRef CAS PubMed.
- P. G. Kyrtatos, P. Lehtolainen, M. Junemann-Ramirez, A. Garcia-Prieto, A. N. Price and J. F. Martin, et al., Magnetic Tagging Increases Delivery of Circulating Progenitors in Vascular Injury, JACC Cardiovasc. Interv., 2009, 2(8), 794–802 CrossRef PubMed.
- M. D. Normandin, H. Yuan, M. Q. Wilks, H. H. Chen, J. M. Kinsella and H. Cho, et al., Heat-Induced Radiolabeling of Nanoparticles for Monocyte Tracking by PET, Angew. Chem., 2015, 54(44), 13002–13006 CrossRef CAS PubMed.
- J. C. Bear, P. S. Patrick, A. Casson, P. Southern, F. Y. Lin and M. J. Powell, et al., Magnetic hyperthermia controlled drug release in the GI tract: solving the problem of detection, Sci. Rep., 2016, 6, 34271 CrossRef CAS PubMed.
- K. M. Bennett, E. M. Shapiro, C. H. Sotak and A. P. Koretsky, Controlled aggregation of ferritin to modulate MRI relaxivity, Biophys. J., 2008, 95(1), 342–351 CrossRef CAS PubMed.
- D. Ni, E. B. Ehlerding and W. Cai, Multimodality Imaging Agents with PET as the Fundamental Pillar, Angew. Chem., 2018 DOI:10.1002/anie.201806853.
- R. T. M. de Rosales, R. Tavare, R. L. Paul, M. Jauregui-Osoro, A. Protti and A. Glaria, et al., Synthesis of Cu-64(II)-Bis(dithiocarbamatebisphosphonate) and Its Conjugation with Superparamagnetic Iron Oxide Nanoparticles: In Vivo Evaluation as Dual-Modality PET-MRI Agent, Angew. Chem., Int. Ed., 2011, 50(24), 5509–5513 CrossRef PubMed.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Coordinating Radiometals of Copper, Gallium, Indium, Yttrium, and Zirconium for PET and SPECT Imaging of Disease, Chem. Rev., 2010, 110(5), 2858–2902 CrossRef CAS.
- R. M. Wong, D. A. Gilbert, K. Liu and A. Y. Louie, Rapid size-controlled synthesis of dextran-coated, 64Cu-doped iron oxide nanoparticles, ACS Nano, 2012, 6(4), 3461–3467 CrossRef CAS PubMed.
- J. Zeng, B. Jia, R. Qiao, C. Wang, L. Jing and F. Wang, et al., In situ111In-doping for achieving biocompatible and non-leachable 111In-labeled Fe3O4 nanoparticles, Chem. Commun., 2014, 50(17), 2170–2172 RSC.
- F. Chen, P. A. Ellison, C. M. Lewis, H. Hong, Y. Zhang and S. Shi, et al., Chelator-free synthesis of a dual-modality PET/MRI agent, Angew. Chem., 2013, 52(50), 13319–13323 CrossRef CAS PubMed.
- R. Chakravarty, H. F. Valdovinos, F. Chen, C. M. Lewis, P. A. Ellison and H. Luo, et al., Intrinsically Germanium-69-Labeled Iron Oxide Nanoparticles: Synthesis and In-Vivo Dual-Modality PET/MR Imaging, Adv. Mater., 2014, 26(30), 5119–5123 CrossRef CAS.
- E. Boros, A. M. Bowen, L. Josephson, N. Vasdev and J. P. Holland, Chelate-free metal ion binding and heat-induced radiolabeling of iron oxide nanoparticles, Chem. Sci., 2015, 6(1), 225–236 RSC.
- J. Lamb and J. P. Holland, Advanced Methods for Radiolabeling Multimodality Nanomedicines for SPECT/MRI and PET/MRI, J. Nucl. Med., 2018, 59(3), 382–389 CrossRef CAS PubMed.
- J. Fock, L. K. Bogart, D. Gonzalez-Alonso, J. I. Espeso, M. F. Hansen and M. Varon, et al., On the ‘centre of gravity’ method for measuring the composition of magnetite/maghemite mixtures, or the stoichiometry of magnetite-maghemite solid solutions, via Fe-57 Mossbauer spectroscopy, J. Phys. D: Appl. Phys., 2017, 50(26), 65005 CrossRef.
- G. M. da Costa, C. Blanco-Andujar, E. De Grave and Q. A. Pankhurst, Magnetic Nanoparticles for In Vivo Use: A Critical Assessment of Their Composition, J. Phys. Chem. B, 2014, 118(40), 11738–11746 CrossRef CAS.
- S. L. T. Andersson and R. F. Howe, An X-ray photoelectron study of metal clusters in zeolites, J. Phys. Chem., 1989, 93(12), 4913–4920 CrossRef CAS.
- M.-S. Jang, I.-J. Roh, J. Park, C.-Y. Kang, W. J. Choi and S.-H. Baek, et al., Dramatic enhancement of the saturation magnetization of a sol-gel synthesized Y3Fe5O12 by a mechanical pressing process, J. Alloys Compd., 2017, 711, 693–697 CrossRef CAS.
- Y. Uwamino, T. Ishizuka and H. Yamatera, X-ray photoelectron spectroscopy of rare-earth compounds, J. Electron Spectrosc. Relat. Phenom., 1984, 34(1), 67–78 CrossRef CAS.
- T. J. Macdonald, K. Wu, S. K. Sehmi, S. Noimark, W. J. Peveler and H. du Toit, et al., Thiol-Capped Gold Nanoparticles Swell-Encapsulated into Polyurethane as Powerful Antibacterial Surfaces Under Dark and Light Conditions, Sci. Rep., 2016, 6, 39272 CrossRef CAS PubMed.
- C. Blanco-Andujar, D. Ortega, P. Southern, Q. A. Pankhurst and N. T. Thanh, High performance multi-core iron oxide nanoparticles for magnetic hyperthermia: microwave synthesis, and the role of core-to-core interactions, Nanoscale, 2015, 7(5), 1768–1775 RSC.
- T. L. Kalber, K. L. Ordidge, P. Southern, M. R. Loebinger, P. G. Kyrtatos and Q. A. Pankhurst, et al., Hyperthermia treatment of tumors by mesenchymal stem cell-delivered superparamagnetic iron oxide nanoparticles, Int. J. Nanomed., 2016, 11, 1973–1983 CrossRef CAS.
- P. S. Patrick, Q. A. Pankhurst, C. Payne, T. Kalber and M. Lythgoe, Magnet-Targeted Delivery and Imaging, in Design and Applications of Nanoparticles in Biomedical Imaging, ed. J. W. M. Bulte and M. J. Modo, Springer, 2017, pp. 123–152 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04895a |
| This journal is © The Royal Society of Chemistry 2019 |