
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAchieving an exceptionally high loading of isolated cobalt single atoms on a porous carbon matrix for efficient visible-light-driven photocatalytic hydrogen production†
Rui
Shi‡
a,
Chengcheng
Tian‡
b,
Xiang
Zhu
 *bcd,
Cheng-Yun
Peng
a,
Bingbao
Mei
e,
Lin
He
*c,
Xian-Long
Du
e,
Zheng
Jiang
e,
Yong
Chen
*a and
Sheng
Dai
*b
*bcd,
Cheng-Yun
Peng
a,
Bingbao
Mei
e,
Lin
He
*c,
Xian-Long
Du
e,
Zheng
Jiang
e,
Yong
Chen
*a and
Sheng
Dai
*b
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, HKU-CAS Joint Laboratory on New Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: chenyong@mail.ipc.ac.cn
bChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: dais@ornl.gov
cState Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute of Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, 730000, China. E-mail: helin@licp.cas.cn
dDepartment of Chemistry, Texas A&M University, College Station, Texas 77843, USA. E-mail: zhuxiang.ecust@gmail.com; xiang@licp.cas.cn
eShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201204, China
First published on 16th January 2019
Abstract
Single-atom catalysts (SACs) have shown great potential in a wide variety of chemical reactions and become the most active new frontier in catalysis due to the maximum efficiency of metal atom use. The key obstacle in preparing SAs lies in the development of appropriate supports that can avoid aggregation or sintering during synthetic procedures. As such, achieving high loadings of isolated SAs is nontrivial and challenging. Conventional methods usually afford the formation of SAs with extremely low loadings (less than 1.5 wt%). In this work, a new in situ preparation strategy that enables the synthesis of isolated cobalt (Co) SAs with an exceptionally high metal loading, up to 5.9 wt%, is developed. The approach is based on a simple one-step pyrolysis of a nitrogen-enriched molecular carbon precursor (1,4,5,8,9,12-hexaazatriphenylene hexacarbonitrile) and CoCl2. Furthermore, due to the successful electron transfer from carbon nitride to the isolated Co SAs, we demonstrate a high-performance photocatalytic H2 production using Co SAs as a co-catalyst, and the evolution rate is measured to be 1180 μmol g−1 h−1. We anticipate that this new study will inspire the discovery of more isolated SACs with high metal loadings, evidently advancing the development of this emerging type of advanced catalysts.
Introduction
Single-atom catalysts (SACs) have attracted extensive attention as a new scientific frontier, effectively bridging the fields of heterogeneous and homogeneous catalysis.1–5 Composed of isolated metal atoms dispersed on a support, SACs display distinctly different catalytic behavior to metal nanoparticles (NPs) while simultaneously maximizing the metal efficiency.6–8 As such, they offer great potential for achieving superior catalytic activity and selectivity, particularly for systems based on noble metals.9,10 However, it has been well documented that the surface free energy of metals increases significantly with decreasing particle size, promoting aggregation or sintering during synthetic procedures.11,12 As such, conventional synthetic approaches generally afford the formation of isolated SAs with extremely low metal loadings (less than 1.5 wt%) since small loadings can help to avoid aggregation and prevent the formation of metal nanocrystals. This has significantly impeded the development of SACs on account of the limited studies of their macroscopic properties.13–17 Therefore, while the synthesis of uniform and stable SACs with high metal loadings is highly desired, it nevertheless remains a great challenge in this field.Herein, we report a new in situ synthesis approach for the preparation of isolated cobalt (Co) SAs with a significantly large metal loading and further demonstrate their application as an efficient co-catalyst for visible-light-driven photocatalytic H2 evolution. Photocatalytic hydrogen (H2) evolution from water splitting represents one of the promising methods for effectively storing renewable solar energy in the chemical form and has attracted tremendous attention.18–21 Loading a co-catalyst is highly required in many systems, where the co-catalyst could promote efficient charge separation from the semiconductor to the co-catalyst's surface.22–26 Although great progress has been achieved, the synthesis of SACs, especially non-noble metal SACs, for photocatalytic H2 evolution remains rare.27–30 The key to our success lies in a simple one-step pyrolysis of a nitrogen-enriched molecular carbon precursor (1,4,5,8,9,12-hexaazatriphenylene hexacarbonitrile) and CoCl2, whereas isolated Co SAs were synthesized and immobilized on a porous nitrogen-doped carbon support. The loading of Co SAs was as high as 5.9 wt%. Based on the successful electron transfer from the photosensitizer g-C3N4 (ref. 31 and 32) to Co SAs, a high-performance photocatalytic H2 production was achieved, suggesting promising catalysis applications of this new material.
Experimental
Preparation of g-C3N4
The photosensitizer g-C3N4 was prepared through a typical thermal polymerization procedure.27 In a typical process, 5 g of urea was put into an alumina crucible and heated to 823 K for 4 h under an air atmosphere. The resultant yellow powder was washed with deionized water and ethanol, and then dried.Preparation of Co–N–C
A mixture of 1,4,5,8,9,12-hexaazatriphenylene hexacarbonitrile (HAT-6CN, 116 mg) and anhydrous CoCl2 (392 mg) (molar ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) was added into a quartz ampoule inside a glove box. The ampoule was then evacuated, vacuum-sealed, and then heated at 450 °C for 20 h and 600 °C for another 20 h. The heating rate is 5 °C min−1. The obtained black powder was subsequently ground and washed thoroughly with dilute 2 M HCl, water, acetone and dichloromethane. The desired product was dried in an oven at 80 °C.
:10) was added into a quartz ampoule inside a glove box. The ampoule was then evacuated, vacuum-sealed, and then heated at 450 °C for 20 h and 600 °C for another 20 h. The heating rate is 5 °C min−1. The obtained black powder was subsequently ground and washed thoroughly with dilute 2 M HCl, water, acetone and dichloromethane. The desired product was dried in an oven at 80 °C.
Different contents of CoCl2 (1:15, 1:20, 1:30, molar ratio) were used towards the synthesis of Co–N–C with a diverse Co loading.
Metal free N–C was prepared in the absence of CoCl2 using the above pyrolysis method.
Preparation of Co–N–C/g-C3N4 composites
The hybrid materials were prepared as follows: Co–N–C and g-C3N4 were mixed with a certain mass ratio and ground in an agate mortar for about 20 min.Photoelectrochemical analysis
The electrochemical properties were obtained on a CHI 660E electrochemical work station (Chenhua Instrument, Shanghai, China) with a conventional three-electrode system. A working electrode was prepared as follows: 1 mg of the sample and 50 uL of Nafion (5% solution) were dispersed in 1 mL CH3CH2OH by at least 2 h sonication to prepare a homogeneous catalyst colloid. Then, 100 μL of the above suspension was deposited onto 1 × 2 cm2 indium tin oxide (ITO) glass, and then dried at room temperature for 48 h to obtain working electrodes. The prepared electrodes, a platinum flake and an Ag/AgCl (saturated KCl) electrode were used as the working, counter and reference electrodes, respectively. A 0.1 M Na2SO4 solution was employed as the electrolyte. Mott–Schottky plots were obtained under direct current potential polarization at different frequencies (2000, 3000 Hz) and at the potential range from −0.6 V to 0.4 V. Electrochemical impedance spectroscopy (EIS) was performed at an applied potential of 0.01 V versus Ag/AgCl over the frequency range of 1 MHz to 0.1 Hz.Photocatalytic measurement
Photocatalytic H2 evolution experiments were carried out in a 15 mL quartz tube sealed with a silicone rubber septum. In a typical photocatalytic experiment, 2 mg of the samples were suspended in an aqueous solution containing sacrificial electron donors (5 mL, 10 vol%). The system was deoxygenated with Ar for 30 min to remove the air before irradiation. A LED light source (12 W, λ = 420 ± 10 nm) was used as the irradiation light source. The quantities of the H2 evolution were measured using a gas chromatograph (GC-2014C, Shimadzu, with Ar as the carrier gas), which was equipped with a 5 Å molecular sieve column (3 mm × 2 mm) and a thermal-conductivity detector. The detection of isotope D2 is exactly the same as that of H2, except that the carrier gas is replaced by H2. The apparent quantum efficiency (AQE) was calculated according to the equation below:| AQE = 2 × NH2/NP × 100%, |
Results and discussion
To illustrate this approach, the preparation route is shown in Scheme 1. 1,4,5,8,9,12-hexaazatriphenylene hexacarbonitrile (HAT-6CN), a nitrogen (N)-enriched molecular carbon monomer, was rationally synthesized and employed for the construction of our desired porous carbon matrix. The metal–support interaction has been proven to play a crucial role in stabilizing metal SACs. Doping rich N-based sites into carbon architectures provides a facile means to enhance this interaction, as a way to stabilize isolated metal SAs, such as Co–N4 and Ni–N4.33–37 Previously, we reported the successful synthesis of a N-enriched porous triazine-linked framework based on the ZnCl2-promoted ionothermal polymerization of an analogue motif, 3,7,11-trimethoxy-2,6,10-tricyano-1,4,5,8,9,12-hexaazatriphenylene (HAT-3CN, Scheme S1†).38 We hypothesized that this new type of N-doped material derived from the HAT ring could be employed as a novel support for the immobilization of SAs, for example, Co SAs. In this regard, CoCl2 was used as a surrogate for ZnCl2. It has been well documented that pyrolyzing aromatic ortho dinitriles at a high temperature in the presence of metal salts, like ZnCl2 and CuCl2, affords the formation of phthalocyanine-linked frameworks based on the condensation reaction of nitrile groups.39,40 In this context, HAT-6CN and CoCl2 (1:10, molar ratio) were first sealed in a quartz ampoule and then pyrolyzed at 450 °C for 20 h and 600 °C for another 20 h, as a way to create abundant N-doped sites inside the carbon matrix. The obtained crude material was thoroughly washed with a diluted acid solution to remove impurities. Synthetic details are presented in the experimental section.
 | ||
| Scheme 1 Synthesis route and proposed structure for isolated Co single atoms. | ||
As expected, elemental analysis indicates an extremely high N content of 21.8 wt% within the resulting material Co–N–C. Pyridinic N (398.6 eV), Co–N (399.1 eV) and pyrrolic N (400.2 eV) were observed (Fig. S1†), respectively.41 These attractive N-doped sites may serve as anchors to stabilize Co species. To confirm this, the structure of Co–N–C was first examined by X-ray diffraction (XRD), and we did not obtain any diffraction peaks associated with large Co species (Fig. S2†), suggesting that the sizes of Co species within the architecture of Co–N–C are extremely small, potentially in terms of small nanoclusters or even single atoms. The observed broad peak originates from the partially graphitized structure of Co–N–C. Both the D band and graphitic G band were clearly observed in its Raman spectra (Fig. S3†). Additionally, as shown in the Co 2p XPS spectrum (Fig. S1†), the binding energies at 781.0 and 796.2 eV are assigned to Co 3p1/2 (Co2+) and Co 3p3/2 (Co2+), respectively, indicating the existence of Co–N moieties.42 The atomic ratio of C and N was estimated to be 1:0.24 by energy dispersive X-ray spectroscopy analysis (Fig. S4†), which is basically consistent with the results of elemental analysis. Elemental mapping results, as depicted in Fig. S5,† clearly indicate the existence of Co, N and C elements in the sample.
To get a better understanding of these Co species, we then performed aberration corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). As anticipated, abundant Co SAs were clearly observed (Fig. 1b), further supporting the XRD results. X-ray absorption spectra (XAS) measurements were then carried out, aiming to identify their structures. The difference in the intensity and the position in the Co K-edge X-ray absorption near-edge structure (XANES) spectra of Co–N–C and standard Co foil implies that the Co species are in different environments (Fig. 1d).42 The Co extended XAFS (EXAFS) spectra (Fig. 1e and f) were therefore collected to determine their local structures. In comparison with those of Co foil, the absence of a peak at ca. 2.1 Å, ascribed to the Co–Co bond (Fig. 1e), suggests that Co species within Co–N–C are not metallic Co. Instead, the new peak at ca. 1.4 Å (Fig. 1f) indicates that the Co species are coordinated with nitrogen atoms, thus implying that the Co–N coordination may account for the successful stabilization of these Co SAs.43 The coordination number for isolated Co centers is also quantified by least-squares EXAFS curve-fitting analysis (Fig. S6 and Table S1†). The fitting results demonstrated that the coordination number of Co centers in the first coordination sphere of Co–N–C is close to 4 at a distance of 1.46 Å based on the absorption-backscattering pair of Co–N. For standard Co foil, the coordination number of the Co atom in the first coordination sphere is 12 at a distance of 2.21 Å based on the absorption-backscattering pair of Co–Co. The difference in coordination environments of the Co atom between Co–N–C and Co foil further excludes the formation of Co–Co bands and confirms the presence of the CoN4 configuration. Taken together, HAADF-STEM and XANES as well as XRD results clearly confirm the successful synthesis of isolated Co SAs on this new HAT-derived nitrogen-rich carbon support.
 | ||
| Fig. 1 TEM (a and c) and HAADF-STEM (b) images of Co–N–C. Normalized Co K-edge XANES spectra of Co–N–C in reference to Co foil (d). (b) k3-weighted Fourier-transform Co K-edge EXAFS spectra of Co foil (e) and Co–N–C (f), respectively. | ||
The content of Co SAs was subsequently detected by inductively coupled plasma atomic emission spectroscopy (ICP-AES). Surprisingly, an extremely high loading of 4.2 wt% of Co was determined, suggesting that this new simple in situ preparation approach is a unique method that enables the synthesis of SAs with a large metal loading. Furthermore, as determined using the N2 adsorption–desorption isotherms at 77 K, Co–N–C exhibits an attractive porous nature with a high Brunauer–Emmett–Teller (BET) surface area of 268 m2 g−1 (Fig. S7†), suggesting promising catalysis applications.
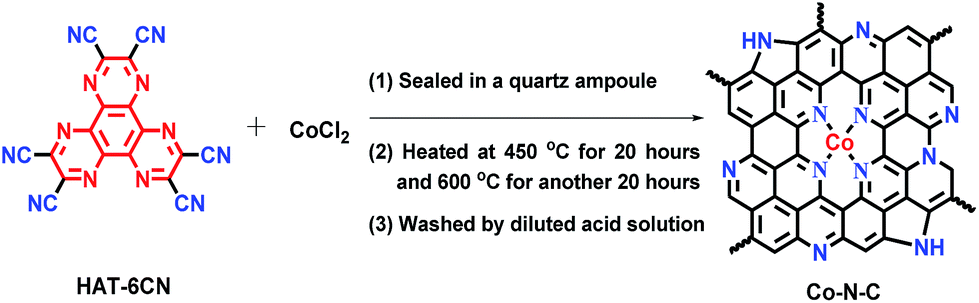
Inspired by the above success, we examined the photocatalytic H2 production, using polymeric g-C3N4 as a photosensitizer and Co–N–C as a co-catalyst. Triethanolamine (TEOA) was employed as a sacrificial agent. As shown in the UV-vis studies (Fig. S8†), pristine g-C3N4 exhibits an absorption edge at 440 nm and a corresponding band gap of ca. 2.8 eV. A slight bathochromic shift of the photoabsorption edge was observed for the Co–N–C/g-C3N4 composite, where the absorption intensity was gradually enhanced with the increase of the loading of Co–N–C. This result is similar to those of previously-developed metal phosphide and sulfide co-catalysts.44 As shown in Fig. 2a, pristine g-C3N4 exhibits weak photocatalytic activity, suggesting a fast recombination of photogenerated charges.45 In our work, g-C3N4 was synthesized through a typical thermal polymerization of urea, which exhibits photocatalytic activity with a H2 evolution of 22 μmol g−1 h−1 under LED visible light irradiation (12 W, λ = 420 ± 10 nm). This performance is lower than that of other g-C3N4 materials in the literature. We reasoned that the difference in the light source may account for this decrease since a light source higher than 300 W is widely used (ESI Table S3†). Co–N–C/g-C3N4 with 35 wt% content of Co–N–C, shows the highest H2 evolution rate, up to 920.0 μmol g−1 h−1, which is ca. 41 fold higher than that of pristine g-C3N4, despite the fact that the photocatalytic activity of Co–N–C alone is very low. A further increase in the Co–N–C loading results in a decrease of photocatalytic performance because excess Co–N–C may shield the incident light. As a result, Co–N–C/g-C3N4 with 35 wt% content of Co–N–C was used as the optimized material for the following studies.
 | ||
| Fig. 2 (a) Photocatalytic H2 evolution rate for various contents of Co–N–C/g-C3N4 composites from 10 vol% TEOA aqueous solution (b) comparison of photo-generating H2 under different conditions. | ||
The sacrificial reagent effect on the photocatalytic performance was then examined. Triethylamine (TEA) and methanol (CH3OH) were investigated. Fig. 2b summarizes the results and indicates that the sacrificial reagent plays an important role in achieving high-performance catalytic activity on Co–N–C/g-C3N4. The use of TEOA affords the best photocatalytic H2 production. In addition, negligible H2 can be detected when the sacrificial reagent and light are absent, suggesting that the generation of H2 is driven by the photocatalytic process. Theoretically, the electronic band structures of g-C3N4 are suitable for visible-light-driven overall water splitting. However, enormous studies have demonstrated that g-C3N4 suffers from rapid recombination of photogenerated carriers. To solve this problem, electron sacrificial agents have been frequently added into the reaction system to consume the photogenerated holes, thereby increasing the survival time of photogenerated electrons. The long-lived photogenerated electrons are able to reach the surface active sites to initiate the photocatalytic redox reaction. Electron sacrificial reagents are used to achieve H2 evolution in most research studies, although very few literature studies have reported that g-C3N4 can exhibit the overall water splitting reaction, as shown in ESI Table S3.† The controlled metal-free N-doped carbon sample (N–C), which was prepared in the absence of CoCl2, shows very poor catalytic activity, indicating that Co SAs are intrinsic active sites. To confirm whether the as-generated H2 is from water or TEOA, a reference evaluation using a pure TEOA solvent instead of the TEOA aqueous solution (10% in volume) was performed (Fig. S9†). As expected, there was almost no H2 evolution when TEOA alone was used, suggesting that water was the source of H2 during the photocatalytic reaction. To get a better understanding, an isotopic experiment was further conducted. When H2O was replaced with D2O, D2 was detected using He gas as the GC carrier.46 The results demonstrate that a negative GC signal for D2 gas was detected, but H2 was not generated at all (Fig. S10†). Taken together, it thus can be concluded that H2 evaluated from the photocatalytic reaction originates from the splitting of water, not TEOA. Furthermore, EIS-MS spectroscopy was employed to monitor the possible oxidation products of TEOA during photocatalytic H2 evolution (Fig. S11†). No additional peaks of the corresponding oxidation products of TEOA, such as aldehyde and carboxylic acid, were observed, suggesting that TEOA is degraded into fragmented molecules after accepting the photogenerated holes. A durability test was subsequently performed. As shown in Fig. S12,† the rate of H2 production exhibits a slight decrease after five runs. The XRD pattern of the recovered Co–N–C/g-C3N4 is consistent with that of the as-prepared sample (Fig. S13†). Moreover, ICP-AES results show that the content of the Co atom is only slightly reduced from 4.2% to 4.0%, where the change in the value is within the measurement error. HAADF-STEM of Co–N–C/g-C3N4 after the durability test was also examined. As expected, isolated Co SAs were clearly observed (Fig. S14†), further supporting the high-performance of catalytic stability on Co–N–C/g-C3N4.
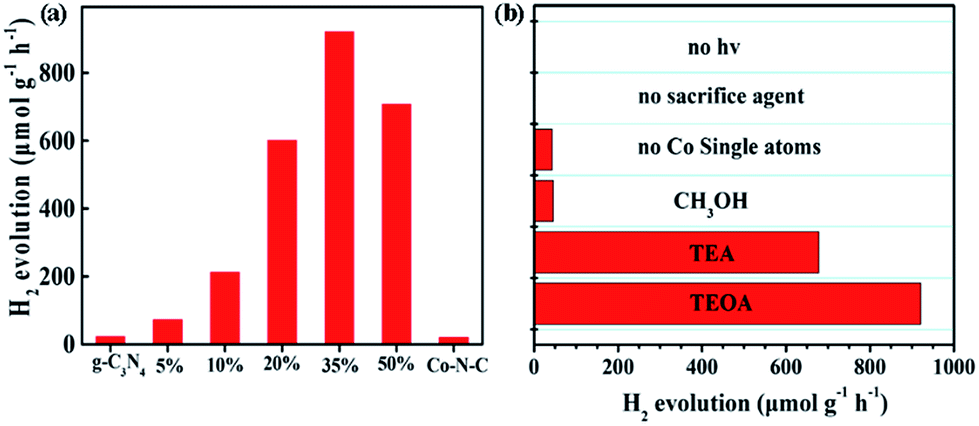
The synergistic effect between g-C3N4 and Co–N–C may account for the obtained enhancement of the photocatalytic H2 evolution. To confirm this, photoluminescence (PL) emission spectroscopy, electrochemical impedance spectroscopy (EIS) and transient IR absorption spectroscopy (TIRA) were carried out to determine the electron transfer between g-C3N4 and Co–N–C. As shown in Fig. 3a, one main emission peak, attributed to the band to band recombination, appears at about 450 nm for pure g-C3N4.47 As the intensity of the emission peak is correlated with the recombination rate of photogenerated electrons and holes, it can be found that the signal intensities of Co–N–C/g-C3N4 composites are greatly inhibited, implying that the photogenerated electron–hole pairs have a better separation at the interface between Co–N–C and g-C3N4.48,49 Moreover, an interfacial transition of charge carriers in Co–N–C/g-C3N4 is also supported by the EIS results (Fig. 3b). The semicircle diameter in the Nyquist plots of Co–N–C/g-C3N4 is smaller than that of g-C3N4. A smaller arc radius of the EIS Nyquist plot suggests an effective separation of the photogenerated electron–hole pairs and fast interfacial charge transfer, which are in good accordance with the PL results.
 | ||
| Fig. 3 (a) PL spectra excited at 420 nm for the Co–N–C, g-C3N4 and Co–N–C/g-C3N4 composite, (b) EIS spectroscopy of g-C3N4 and Co–N–C/g-C3N4, (c) TIRA spectra of Co–N–C, g-C3N4, and Co–N–C/g-C3N4 excited at 600 nm, and (d) TIRA spectra of Co–N–C, g-C3N4 and Co–N–C/g-C3N4 excited at 420 nm. (e) Schematic of photogenerated charge transfer in the Co–N–C/g-C3N4 composite under visible light irradiation. | ||
TIRA measurements are therefore performed to determine the direction of electron transfer. The time profiles of transient absorption are studied under 420 and 600 nm visible-light irradiation, respectively. For 600 nm irradiation, no photogenerated electron signals are detected for g-C3N4 (Fig. 3c). The time profiles of Co–N–C and Co–N–C/g-C3N4 could be fitted by two-exponential functions, and their lifetimes are summarized in Table S2.† Based on the calculated equation, the average lifetime of Co–N–C is only 3.9 ps. Comparing with Co–N–C, no significant change was observed for that of Co–N–C/g-C3N4 (4.4 ps), suggesting that photogenerated electrons cannot be transferred from Co–N–C to g-C3N4. When 420 nm excitation was employed, photogenerated electrons could be produced in g-C3N4, and its average lifetime was 723.9 ps (Fig. 3d). Meanwhile, the lifetime of Co–N–C is still only 3.2 ps, which is about two-hundredth of that of g-C3N4. Owing to the ultra-short lifetime of photogenerated electrons in Co–N–C, the absorption signal of photogenerated electrons in the Co–N–C/g-C3N4 composite mostly originated from the exited g-C3N4.24 Compared to pure g-C3N4, the average lifetime of Co–N–C/g-C3N4 is significantly decreased and is only 3.4 ps. Hence, the loading of Co–N–C greatly decreases the decay lifetime of g-C3N4. This effect is attributed to an additional decay channel that is opened through electron transfer from g-C3N4 to the Co atom in Co–N–C.50,51 Based on the PL, EIS and TIRA results, it can be concluded that under 420 nm irradiation, there is an electron transfer between g-C3N4 and Co–N–C, and the direction of electron transfer is from g-C3N4 to Co–N–C.
In an effort to get a deeper insight into the transfer of photogenerated electrons from g-C3N4 to Co–N–C and determine the energy levels, we then examined the flat potentials of both samples. As displayed in Fig. S15,† the positive slopes of Mott–Schottky plots indicate that both Co–N–C and g-C3N4 possess n-type semiconductor characteristics. For n-type semiconductors, it has been reported that the conduction bands (CBs) are normally 0.1–0.2 eV deeper than the flatband potential.52 As such, the difference between the CB and the flat potential value is set to be 0.1 eV. The CB potentials of Co–N–C and g-C3N4 were calculated to be −1.14 and −1.42 V (vs. SCE), respectively. Consequently, Co–N–C has a suitable redox potential for accepting the photoinduced electrons from g-C3N4. A possible photocatalytic mechanism was thus proposed and is shown in Fig. 3e. When g-C3N4 is modified by the co-catalyst Co–N–C, the photogenerated electrons in g-C3N4 will be transferred to isolated Co atoms in Co–N–C on account of the difference in the CB position. Then, the electrons will accumulate on Co atoms and participate in H2 evolution. The electron sacrificial agent will be oxidized by photogenerated holes on the valence band (VB) of g-C3N4. Therefore, an effective photogenerated charge carrier separation can be achieved, resulting in the enhanced photocatalytic H2 production.
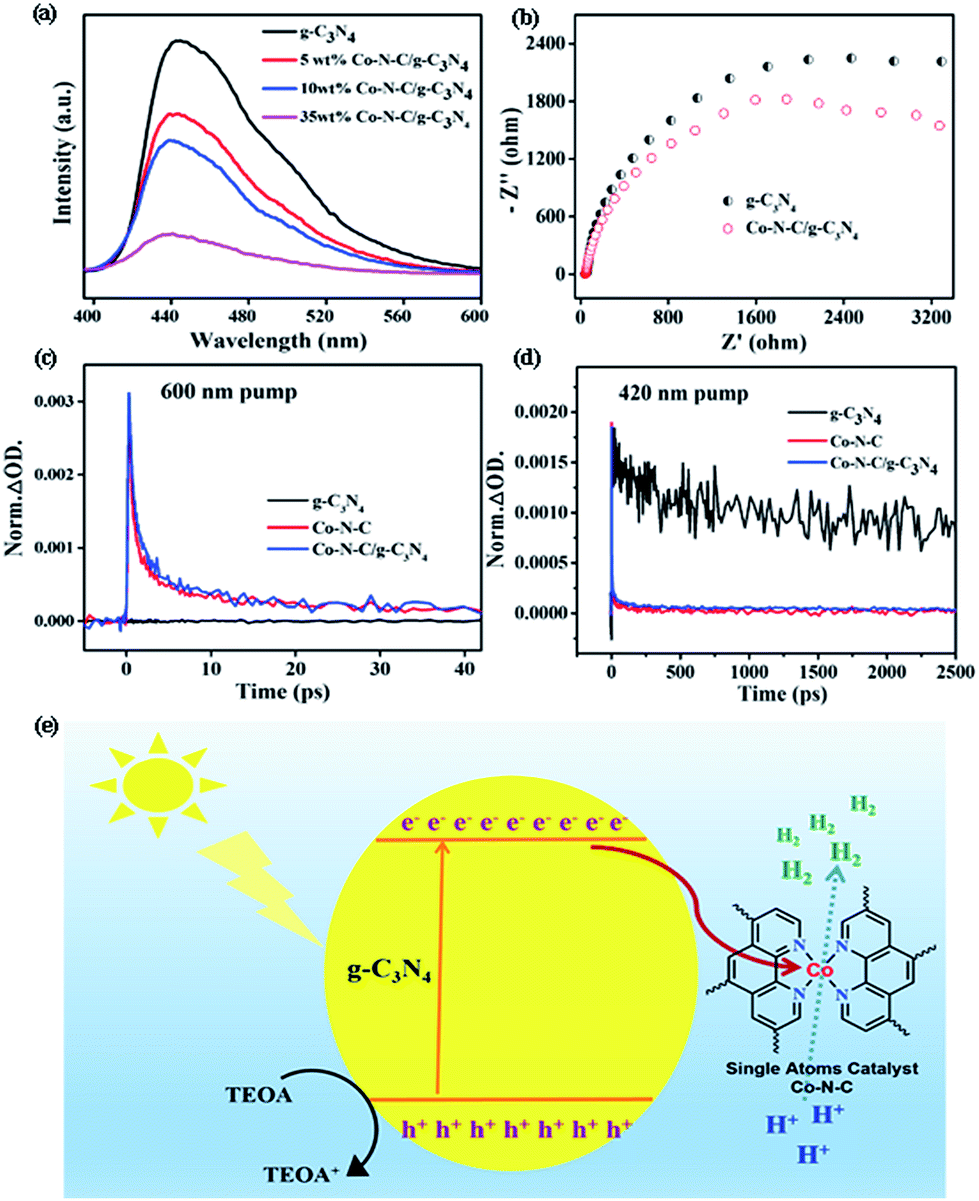
Based on the aforementioned results, we further attempted the in situ synthesis to achieve a higher loading of Co SAs by simply increasing the use of CoCl2. Significantly, the Co SA loading can be enhanced to 5.9 wt% when 1:30 molar ratio of HAT-6CN and CoCl2 was employed (Fig. 4a). The HAADF-STEM result successfully confirms the rich abundance of isolated Co SAs on the carbon matrix (Fig. 4d). As a result, the H2 evolution rate was improved from 920.0 μmol g−1 h−1 (4.2 wt% Co) to 1180 μmol g−1 h−1 (Fig. 4b). A negligible difference in the H2 evolution rate was obtained for the samples with 5.6 and 5.9 wt% Co loading. We further calculated the AQE determination at 420 nm for Co–N–C with 5.6 wt% of Co SACs. The AQE value for H2 evolution in 4 h was calculated to be 2.53%. In the meantime, the calculated turnover frequency (TOF) for single Co atom active sites reaches 3.6 h−1. A comparison of the state-of-art performance of other SACs and g-C3N4 materials is shown in the Tables S3 and S4.† The photocatalytic activity of H2 evolution of Co–N–C is better than that of most composites reported in the literature. We reasoned that there could be two factors affecting the photocatalytic performance of our Co SACs. One is the loading of Co SACs, the other is the degree of graphitization of our support. The former can improve the number of catalytic active sites, and the latter may promote the transfer rate of photogenerated electrons. As the loading of Co SACs increases, more N-doped sites within the support need to be served as anchors to stabilize Co atoms, which may affect the delocalization π bond of the support. As a result, there could be a threshold for the effect of Co loading on the photocatalytic performance. As shown in Fig. 4a and b, the difference in photocatalytic activity of Co SACs is not significant when the Co loading is over 5.6 wt%.
 | ||
| Fig. 4 (a) The synthesis of Co SAs by varying the molar ratio of HAT-6CN and CoCl2 (from 1:10 to 1:30); (b) the H2 evolution rate of Co–N–C with various Co loadings; (c) XRD patterns of Co–N–Cs prepared by varying the molar ratios and (d) HAADF-STEM image of Co–N–C with 5.9 wt% Co SACs [HAT-6CN:CoCl2 = 1:30]. | ||
Conclusions
In summary, a new in situ synthesis strategy was successfully developed for the preparation of isolated Co SAs with extremely high metal loadings. A simple one-step pyrolysis of a N-enriched molecular carbon monomer (1,4,5,8,9,12-hexaazatriphenylene hexacarbonitrile) and CoCl2 affords the generation isolated Co SAs on a porous carbon support with high loadings, up to 5.9 wt%. Furthermore, based on the successful electron transfer from the photosensitizer graphitic carbon nitride to isolated Co SAs, the resulting new material shows a high catalytic activity for visible-light-driven photocatalytic H2 production with an evolution rate of 1180 μmol g−1 h−1. We anticipate that this new study could advance the development of new SACs with high metal loadings for catalysis applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB17000000) and National Natural Science Foundation of China (21773275). SD was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, US Department of Energy. Y. C. acknowledges the financial support from the K. C. Wong Education Foundation and CAS-Croucher Funding Scheme for Joint Laboratories. X. Z. thanks the start-up financial support from the Chinese Academy of Sciences. L. H. was supported by Foundation research project of Jiangsu Province (BK20171242), and National Natural Science Foundation of China (91645118).Notes and references
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- H. Zhang, W. Zhou, T. Chen, B. Guan, Z. Li and X. Lou, Energy Environ. Sci., 2018, 11, 1980–1984 RSC.
- H. Zhang, P. An, W. Zhou, B. Guan, P. Zhang, J. Dong and X. Lou, Sci. Adv., 2018, eaao6657 CrossRef PubMed.
- H. Zhang, L. Yu, T. Chen, W. Zhou and X. Lou, Adv. Funct. Mater., 2018, 28, 1807086 CrossRef.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- A. Corma, P. Concepción, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. López-Quintela, D. Buceta, E. Mendoza, G. Guilera and A. Mayoral, Nat. Chem., 2013, 5, 775–781 CrossRef CAS PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411 CrossRef CAS PubMed.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–800 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- N. S. Lewis, Science, 2016, 351, aad5117 CrossRef PubMed.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- A. Indra, P. W. Menezes, K. Kailasam, D. Hollmann, M. Schroder, A. Thomas, A. Bruckner and M. Driess, Chem. Commun., 2016, 52, 104–107 RSC.
- A. Indra, A. Acharjya, P. W. Menezes, C. Merschjann, D. Hollmann, M. Schwarze, M. Aktas, A. Friedrich, S. Lochbrunner, A. Thomas and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 1653–1657 CrossRef CAS PubMed.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- J. Ran, B. Zhu and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS PubMed.
- F. Wen and C. Li, Acc. Chem. Res., 2013, 46, 2355–2364 CrossRef CAS PubMed.
- Y. Cao, S. Chen, Q. Luo, H. Yan, Y. Lin, W. Liu, L. Cao, J. Lu, J. Yang, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 12191–12196 CrossRef CAS PubMed.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- J. Xing, J. F. Chen, Y. H. Li, W. T. Yuan, Y. Zhou, L. R. Zheng, H. F. Wang, P. Hu, Y. Wang, H. J. Zhao, Y. Wang and H. G. Yang, Chem.–Eur. J., 2014, 20, 2088 CrossRef CAS.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H. L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893 RSC.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- A. Han, W. Chen, S. Zhang, M. Zhang, Y. Han, J. Zhang, S. Ji, L. Zheng, Y. Wang, L. Gu, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1706508 CrossRef PubMed.
- X. Zhu, C. Tian, G. M. Veith, C. W. Abney, J. Dehaudt and S. Dai, J. Am. Chem. Soc., 2016, 138, 11497–11500 CrossRef CAS PubMed.
- J. Deng, B. Wang, Y. Shi, Q. Song, A. Wang, L. Hao, B. Luo, X. Li, Z. Wang, F. Wang and L. J. Zhi, Macromol. Chem. Phys., 2012, 213, 1051–1059 CrossRef CAS.
- P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2009, 42, 319–326 CrossRef CAS.
- X. Zhu, T. Jin, C. Tian, C. Lu, X. Liu, M. Zeng, X. Zhuang, S. Yang, L. He, H. Liu and S. Dai, Adv. Mater., 2017, 29, 1704091 CrossRef PubMed.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. Dong Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS PubMed.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- S. Cao, Y. Chen, C. Hou, X. Lv and W. Fu, J. Mater. Chem. A, 2015, 3, 6096–6101 RSC.
- L. Ge and C. Han, Appl. Catal., B, 2012, 117–118, 268–274 CrossRef CAS.
- F. Dong, Z. Zhao, T. Xiong, Z. Ni, W. Zhang, Y. Sun and W.-K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CrossRef CAS PubMed.
- Q. Xu, C. Jiang, B. Cheng and J. Yu, Dalton Trans., 2017, 46, 10611–10619 RSC.
- C. Li, Y. Du, D. Wang, S. Yin, W. Tu, Z. Chen, M. Kraft, G. Chen and R. Xu, Adv. Funct. Mater., 2017, 27, 1604328 CrossRef.
- R. Shi, H. F. Ye, F. Liang, Z. Wang, K. Li, Y. Weng, Z. Lin, W. F. Fu, C. M. Che and Y. Chen, Adv. Mater., 2018, 30, 1705941 CrossRef.
- D. Liu, J. Wang, X. Bai, R. Zong and Y. Zhu, Adv. Mater., 2016, 28, 7284–7290 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization details, XPS studies, XRD patterns, Raman spectra, N2 adsorption–desorption curves, UV-vis diffuse reflectance images, durability tests, HAADF-STEM images, Mott–Schottky plots in the dark at frequencies of 2000 and 3000 Hz, Scheme S1 and lifetimes of TIRA decays under 420 nm and 600 nm irradiation, respectively. See DOI: 10.1039/c8sc05540h |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |