
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn unusual Burkholderia gladioli double chain-initiating nonribosomal peptide synthetase assembles ‘fungal’ icosalide antibiotics†
Matthew
Jenner‡
 ab,
Xinyun
Jian‡
a,
Yousef
Dashti
a,
Joleen
Masschelein
a,
Christian
Hobson
a,
Douglas M.
Roberts
a,
Cerith
Jones
d,
Simon
Harris
e,
Julian
Parkhill
e,
Huzefa A.
Raja
f,
Nicholas H.
Oberlies
f,
Cedric J.
Pearce
g,
Eshwar
Mahenthiralingam
d and
Gregory L.
Challis
*abc
ab,
Xinyun
Jian‡
a,
Yousef
Dashti
a,
Joleen
Masschelein
a,
Christian
Hobson
a,
Douglas M.
Roberts
a,
Cerith
Jones
d,
Simon
Harris
e,
Julian
Parkhill
e,
Huzefa A.
Raja
f,
Nicholas H.
Oberlies
f,
Cedric J.
Pearce
g,
Eshwar
Mahenthiralingam
d and
Gregory L.
Challis
*abc
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: g.l.challis@warwick.ac.uk
bWarwick Integrative Synthetic Biology Centre, University of Warwick, Coventry CV4 7AL, UK
cBiomedicine Discovery Institute, Department of Biochemistry and Molecular Biology, Monash University, Victoria 3800, Australia
dOrganisms and Environment Division, Cardiff School of Biosciences, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
eWellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, UK
fDepartment of Chemistry and Biochemistry, University, of North Carolina at Greensboro, Greensboro, NC 27402, USA
gMycosynthetix, 4905 Pine Cone Drive, Durham, North Carolina 27707, USA
First published on 25th April 2019
Abstract
Burkholderia is a multi-talented genus of Gram-negative bacteria, which in recent years has become increasingly recognised as a promising source of bioactive natural products. Metabolite profiling of Burkholderia gladioli BCC0238 showed that it produces the asymmetric lipopeptidiolide antibiotic icosalide A1, originally isolated from a fungus. Comparative bioinformatics analysis of several genome-sequenced B. gladioli isolates identified a gene encoding a nonribosomal peptide synthase (NRPS) with an unusual architecture that was predicted to be responsible for icosalide biosynthesis. Inactivation of this gene in B. gladioli BCC0238 abolished icosalide production. PCR analysis and sequencing of total DNA from the original fungal icosalide A1 producer revealed it has a B. gladioli strain associated with it that harbours an NRPS with an identical architecture to that responsible for icosalide A1 assembly in B. gladioli BCC0238. Sequence analysis of the icosalide NRPS indicated that it contains two chain-initiating condensation (CI) domains. One of these is appended to the N-terminus of module 1 – a common architecture for NRPSs involved in lipopeptide assembly. The other is embedded in module 3, immediately downstream of a putative chain-elongating condensation domain. Analysis of the reactions catalysed by a tridomain construct from module 3 of the NRPS using intact protein mass spectrometry showed that the embedded CI domain initiates assembly of a second lipopeptide chain, providing key insights into the mechanism for asymmetric diolide assembly.
Introduction
Bacteria belonging to the Burkholderia genus thrive in various ecological niches, ranging from the plant rhizosphere to the human lung, and play critical roles in ecological interactions often through the secretion of specialised metabolites.1,2 The metabolic arsenal of these bacteria is employed to eliminate other bacterial competitors and suppress plant pathogenic fungi and nematodes. In recent years various Burkholderia species have been shown to produce numerous specialised metabolites, many of which are assembled by polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs) biosynthetic assembly lines.3 Notable examples include: the respiratory toxin bongkrekic acid;4 anti-proliferative agents such as thailanstatin and spliceostatin;5,6 and the antibiotics enacyloxin IIa and gladiolin.7,8Burkholderia participate in both antagonistic and mutualistic interactions with fungi. Much of the antagonistic behaviour can be attributed to an array of antifungal compounds secreted by many Burkholderia species.9–12 However, it has also been reported that Burkholderia strains have beneficial effects on fungi, suggestive of symbiotic, or at least mutualistic interactions.13–15 The exemplar study of this relationship is the symbiotic pairing of Burkholderia rhizoxinica (since reclassified as Paraburkholderia rhizoxinica) and the rice seedling blight pathogen Rhizopus microspores. B. rhizoxinica was identified as the producer of the antimitotic macrolide rhizoxin, the causative agent of rice seedling blight.10,16
In a preliminary communication, we disclosed that Burkholderia gladioli isolates from cystic fibrosis (CF) patients produce icosalide A1 (1),17 a lipopeptidiolide antibiotic originally isolated from a fungus.18 We also showed that a nonribosomal peptide synthetase (NRPS) with an unusual architecture is responsible for icosalide A1 assembly.17 Subsequently, Hertweck and co-workers reported that a B. gladioli endosymbiont of a beetle, along with several other Burkholderia species from diverse sources, also produce icosalide A1 and showed that an NRPS with the same unusual architecture as that identified in the CF isolates is responsible for its biosynthesis.19 Here we provide a full account of our work and show that a B. gladioli strain containing the icosalide A1 NRPS is associated with the originally reported fungal producer. Moreover, we demonstrate that a chain-initiating condensation (CI) domain embedded in the middle of the icosalide A1 NRPS selectively N-acylates a serinyl thioester covalently attached to the downstream peptidyl carrier protein (PCP) domain with a (3R)-3-hydroxydecanoyl panthetheine thioester, providing a mechanistic rationale for asymmetric lipopeptidiolide formation.
Results and discussion
We recently reported the discovery of gladiolin, a novel macrolide antibiotic with promising activity against Mycobacterium tuberculosis (MIC = 0.4 μg mL−1) and low mammalian cell cytotoxicity, from B. gladioli BCC0238 (LMG-P26202).7 In the course of investigations of other specialised metabolites produced by this CF clinical isolate using UHPLC-ESI-Q-TOF-MS, compounds with the molecular formulae C36H64N4O10 and C34H60N4O10 were observed (Fig S1 and S2†). These molecular formulae correspond to a set of three structurally-related lipopeptidiolides, known as icosalides A1 (1), A2 (2) and B (3), reported to be produced by fungi18 (Fig. 1A). | ||
| Fig. 1 Structures of the icosalides, identification of the NRPS-encoding gene responsible for icosalide biosynthesis in B. gladioli BCC0238 and phylogenetic comparison of the recA sequence amplified from total DNA of the Aureobasidium sp. originally reported to produce icosalide A1 with various Burkholderia species. (A) Structures of icosalides A1 (1), A2 (2) and B (3). (B) Schematic representation of the ∼500 kDa NRPS encoded by the ∼15 kb icoA gene. Domain abbreviations are as follows: C, condensation; A, adenylation; PCP, peptidyl carrier protein; TE, thioesterase. (C) Extracted ion chromatogram at m/z 713.46 ± 0.02, corresponding to [M + H]+ for icosalide A1, from LC-MS analyses of extracts of agar-grown cultures of B. gladioli BCC0238 (top) and B. gladioli BCC0238 ΩicoA, in which the NRPS-encoding gene has been disrupted (bottom). (D). Phylogenetic comparison of the recA sequence amplified from the total DNA extract of Aureobasidium sp. MSX 59166 (labelled ‘icosalide producer’) with recA sequences from 24 representative Burkholderia species, showing that the fungus-associated bacterium clades with Burkholderia gladioli. The scale bar indicates the number of substitutions per site. | ||
The icosalides possess similar 20-membered lipopeptidiolide structures consisting of two serine, leucine and 3-hydroxy acid residues. In icosalide A1 (1), one of the leucine residues is D-configured, whereas all of the amino acid residues in icosalides A2 (2) and B (3) are L-configured. Icosalides A1 (1) and A2 (2) both contain one eight-carbon and one ten-carbon 3-hydroxy acid residue, whereas icosalide B (3) contains two eight-carbon 3-hydroxy acid residues. The incorporation of a D-leucine residue into icosalide A1 (1) has a dramatic effect on its biological activity. While icosalide A1 (1) is active against Streptococcus pyogenes (MIC: 8–16 μg mL−1), icosalides A2 (2) and B (3) do not show any antibacterial activity but are cytotoxic towards replicating MDCK cells (CC50: 5 μg mL−1).18
The compound with the molecular formula C36H64N4O10 was purified using semi-preparative HPLC and its planar structure was shown to be identical to that of icosalide A1 (1) using a combination of 1H, 13C and HMBC NMR experiments (Fig. S3 and Table S1†). The absolute stereochemistry of the amino acid residues in the purified compound was determined via saponification of the esters, followed by semi-preparative HPLC purification and MS analysis of the resulting pair of 3-hydroxyacyl-dipeptides, acid hydrolysis to liberate the constituent amino acids, derivatisation using Marfey's reagent and comparison with the corresponding L and D-configured leucine and serine standards (Fig. S4 and S5†). This revealed that the Leu-2 residue is D-configured, confirming the identity of the metabolite produced by B. gladioli as icosalide A1 (1). The production level of the compound with the molecular formula C34H60N4O10 was too low to permit characterisation by NMR spectroscopy (Fig. S6†), but it seems likely that this has the same planar structure as icosalide B (3). The activity of purified icosalide A1 (1) against several Gram-positive and Gram-negative bacteria, including the ESKAPE panel of pathogens, and Candida albicans was tested. Modest activity against E. faecium (MIC: 16 μg mL−1) was observed (Table S2†).
Inspection of putative specialised metabolite biosynthetic gene clusters in the previously reported genome sequence of B. gladioli BCC0238,7 identified a single ∼15 kb gene (icoA) on chromosome 2 encoding a nonribosomal peptide synthetase (NRPS) with the requisite module and domain organisation for icosalide assembly (Fig. 1B). This NRPS contains four adenylation (A) domains, two of which are predicted to be specific for L-leucine and two for L-serine20 (Table S3†). It also contains five condensation (C) domains and a thioesterase (TE) domain, which together account for the four amide and two ester bond-forming reactions required to assemble the lipopeptidiolide structure of the icosalides21 (Fig. 2A). We identified icoA in the genomes of several other B. gladioli isolates (BCC0252, BCC1677, BCC1698 and BCC1720), all of which were shown to produce icosalide A1 (1). In contrast, no icosalide A1 (1) production was detected in B. gladioli BCC1713 (Fig. S7†), which is lacking icoA. These data are consistent with the hypothesis that the NRPS encoded by icoA is responsible for icosalide biosynthesis. To verify this, we inactivated the icoA gene in B. gladioli BCC0238 using insertional mutagenesis. UHPLC-ESI-Q-TOF-MS analysis of extracts from the icoA mutant confirmed that it is no longer able to produce icosalides (Fig. 1C).
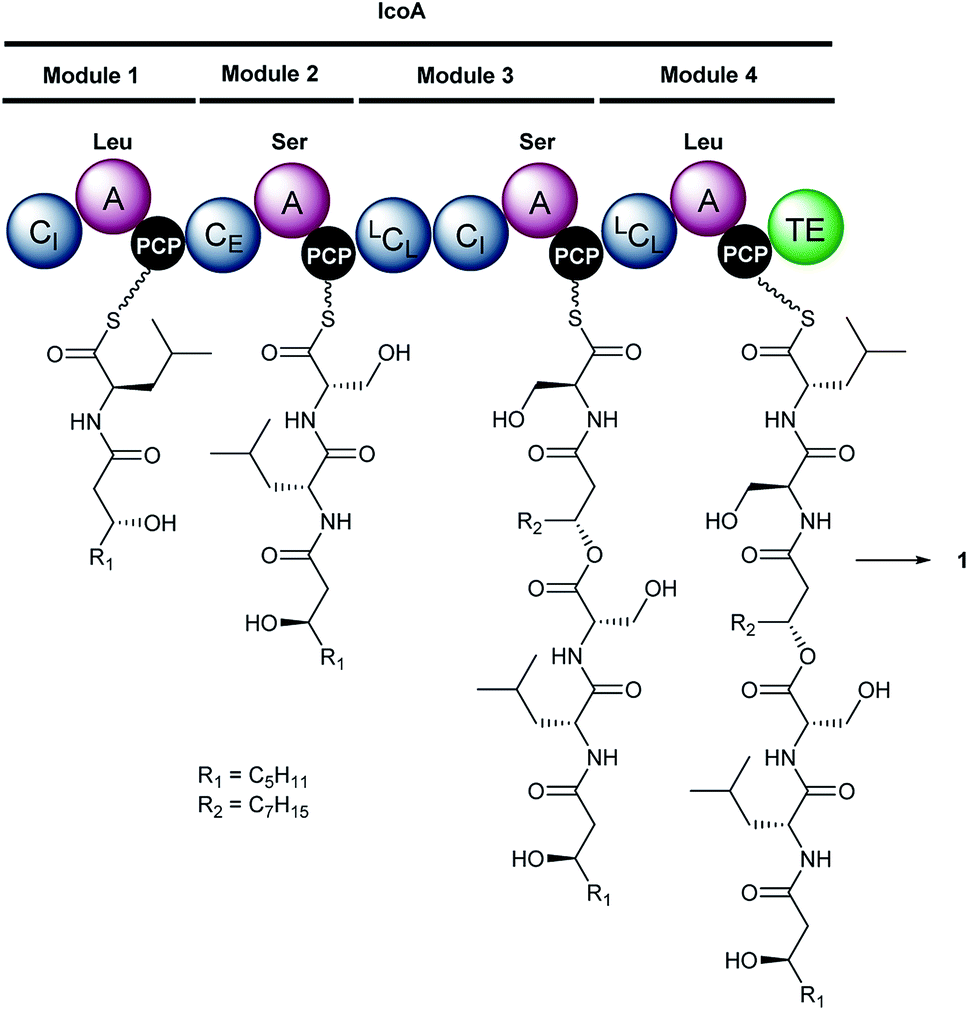 | ||
| Fig. 2 Proposed mechanism for assembly of icosalide A1 (1) by IcoA. Domain and module organisation of the NRPS, showing the PCP-bound thioester intermediates proposed to be formed by each module. The CI domains initiate lipopeptide chain assembly by N-acylating the aminoacyl thioesters attached to the downstream PCP domains with 3-hydroxyacyl thioester intermediates in fatty acid biosynthesis. Abbreviations are as follows: A, adenylation domain; CI, chain initiating condensation domain; CE, bifunctional epimerisation–condensation domain; LCL, condensation domain that catalyses condensation of L-configured aminoacyl donor and acceptor substrates; PCP, peptidyl carrier protein domain; TE, thioesterase domain. The residues used to predict the substrate specificity of A-domains are shown in Table S3†. | ||
Icosalide A1 (1) was originally isolated from extracts of an Aureobasidium species fungus (strain MSX 59166), whereas icosalides A2 (2) and B (3) were isolated from a second less well characterized fungal species (MSX 74159).18 The data presented above suggested that B. gladioli bacteria associated with these fungi might be the true producers of the icosalides isolated from them. To investigate this hypothesis, total DNA from the originally reported icosalide A1 (1) producer, Aureobasidium sp. MSX 59166, was screened by PCR for orthologues of icoA and the bacterial housekeeping gene recA. In both cases, we obtained amplimers of the expected size (Fig. S8†), confirming that a bacterium, likely harbouring an icoA orthologue, is associated with the fungus. Burkholderia species can be unambiguously identified via phylogenetic comparison of their recA sequences.22 Thus, we compared the sequence of the recA amplimer with the sequences of recA genes from 25 representative Burkholderia species. This showed that the Aureobasidium-associated bacterium is B. gladioli (Fig. 1D). Illumina sequencing of the total DNA from the Aureobasidium sp. allowed an approximately 1 Mb contiguous sequence of bacterial DNA, encompassing the icoA orthologue, to be assembled (Fig. S9†). Comparison of this contiguous sequence with the corresponding region of the B. gladioli BCC0238 genome showed a high degree of synteny (Fig. S9†), confirming that the Aureobasidium sp. harbours a B. gladioli strain containing an icoA orthologue. Although a Gram-negative bacterium could be isolated from the Aureobasidium sp., it quickly lost viability and was not identifiable.
Inspection of the structure of icosalide A1 (1) suggests it is assembled via initial formation of 3-hydroxyoctanoyl-D-leucinyl-L-serinyl and 3-hydroxydecanoyl-L-serinyl-L-leucinyl thioesters, which are joined via a pair of ester linkages to form the lipopeptidiolide. To gain insight into the role played by each of the five C domains of IcoA in the biosynthesis of icosalide A1 (1), we carried out phylogenetic analyses.23 These indicated that the first and fourth domains group with chain initiating (CI) domains, which are typically located at the N-terminus of the first module in NRPSs responsible for lipopeptide biosynthesis and initiate chain assembly via N-acylation of the amino acyl thioester bound to the downstream PCP domain with a fatty acyl thioester. The third and fifth C domains of IcoA belong to the LCL group, which catalyse formation of a peptide bond between L-configured amino acyl thioesters attached to the upstream and downstream PCP domains. In contrast, the second C domain of IcoA clusters with bifunctional epimerisation–condensation (CE) domains, which catalyse epimerisation of the α-carbon in the aminoacyl thioester attached to the upstream PCP domain, followed by condensation of the resulting D-configured intermediate with an L-aminoacyl thioester attached to the downstream PCP domain. These C domain functional assignments led us to propose that modules 1 and 2 of IcoA are responsible for assembly of the 3-hydroxyoctanoyl-D-leucinyl-L-serinyl thioester intermediate in icosalide A1 (1) biosynthesis (Fig. 2). The CI domain appended to the N-terminus of module 1 initiates chain assembly via N-acylation of the L-leucinyl thioester attached to the module 1 PCP domain with a 3-hydroxyoctanoyl thioester, likely donated by the primary metabolic fatty acid synthase (FAS). The C-domain in module 2 then catalyses epimerisation of the resulting N-acyl-leucinyl thioester, followed by condensation with the L-serinyl thioester attached to the module 2 PCP domain. Module 3 of IcoA is very unusual, because it contains an LCL domain directly followed by a CI domain. We hypothesize that the CI domain in this module catalyses a second chain initiation event, resulting in N-acylation of the L-serinyl thioester attached to the downstream PCP domain with a 3-hydroxydecanoyl group, again likely provided by the primary metabolic FAS. The LCL domain then catalyses acylation of the hydroxyl group in the resulting 3-hydroxydecanoyl-L-serinyl thioester with the 3-hydroxyoctanoyl-D-leucinyl-L-serinyl thioester attached to the module 2 PCP domain. Elongation of the resulting intermediate with the L-leucinyl thioester attached to the PCP domain in module 4, catalysed by the upstream LCL domain, followed by lactonisation catalysed by the downstream TE domain, yields icosalide A1 (Fig. 2).
To the best of our knowledge, initiation of chain assembly by an embedded CI domain like the one found in module 3 of IcoA is without precedent in NRPS enzymology. We thus sought to experimentally validate the proposed chain initiation reaction in vitro by overproducing the CI-A-PCP tri-domain from module 3 of IcoA in E. coli. The resulting N-terminal His6 fusion protein, was purified to homogeneity using immobilised metal-ion affinity chromatography and the hexahistidine tag was cleaved using thrombin (Fig. S10†). ESI-Q-TOF-MS analysis showed that the PCP domain of this protein had been partially converted to the holo form by an E. coli phosphopantetheinyl transferase (Fig. S10†). To fully convert the PCP domain into the holo form we incubated the protein with purified recombiant Sfp, a substrate tolerant phosphopantetheinyl transferase from B. subtilis,24 and coenzyme A.
The holo-CI-A-PCP tri-domain was incubated with ATP, L-Ser and 3R-configured 3-hydroxyoctanoyl or 3-hydroxydecanoyl-panthetheine thioesters, which mimic the (3R)-3-hydroxyoctanoyl and (3R)-3-hydroxydecanoyl-ACP intermediates in fatty acid biosynthesis proposed to be intercepted by the CI domains in modules 1 and 3, respectively, of the NRPS. UHPLC-ESI-Q-TOF-MS analysis of the CI-A-PCP tri-domain showed mass shifts consistent with the formation of serinyl, 3-hydroxyoctanoyl-serinyl and 3-hydroxydecanoyl-serinyl PCP-thioesters (Fig. 3). When the CI-A-PCP tri-domain was replaced with a mutant containing an Ala residue in place of the active site His residue in the CI domain, only trace amounts of products were formed (Fig. 3).
 | ||
| Fig. 3 In vitro reconstitution of aminoacyl thioester formation and N-acylation by the CI-A-PCP tridomain from module 3 of IcoA. Deconvoluted mass spectra of IcoA module 3 CI-A-PCP tri-domain. (A) Resulting from incubation with ATP, L-Ser and (3R)-3-hydroxyoctanoyl (top), (3R)-3-hydroxydecanoyl (middle), or (3R)-3-hydroxyoctanoyl and (3R)-3-hydroxydecanoyl (bottom) pantetheine thioesters. (B) Following incubation with ATP, L-Ser and (3S)-3-hydroxyoctanoyl (top), or (3S)-3-hydroxydecanoyl (bottom) pantetheine thioesters. (C) Resulting from incubation of the H164A mutant with ATP, L-Ser and (3R)-3-hydroxyoctanoyl (top), or (3R)-3-hydroxydecanoyl (bottom) pantetheine thioesters. Dashed lines indicate the peaks corresponding to each condensed species, and the holo- and L-Ser-loaded species are labelled as indicated in the bottom right corner. Loading of L-Ser onto the PCP domain results in an 87 Da mass increase. For the wild type IcoA CI-A-PCP tri-domain, N-acylation of the serinyl-PCP thioester with the (3R)-3-hydroxyoctanoyl and (3R)-3-hydroxydecanoyl thioesters resulted in additional 142 and 170 Da mass increases, respectively. Analogous mass increases were observed when the (3S)-3-hydroxyoctanoyl and (3S)-3-hydroxydecanoyl thioesters were used, but the levels of product formation were lower, indicating that the CI domain prefers R-configured 3-hydroxyacyl thioesters. A small amount of product formation was observed when the H164A mutant of the IcoA CI-A-PCP tri-domain was used in place of the wild type protein, due to uncatalysed N-acylation of the serinyl thioester with the (3R)-3-hydroxyoctanoyl and (3R)-3-hydroxydecanoyl thioesters. | ||
Icosalide A1 contains an N-((3R)-3-hydroxydecanoyl)-L-serine residue.19 The module 3 CI domain is thus expected to accept 3-hydroxydecanoyl thioesters in preference to 3-hydroxyoctanoyl thioesters. To test this hypothesis, we incubated the holo-CI-A-PCP tri-domain with ATP, L-Ser and equimolar mixture of the 3R-configured 3-hydroxyoctanoyl and 3-hydroxydecanoyl-panthetheine thioesters. UHPLC-ESI-Q-TOF-MS analysis showed that only the product resulting from condensation with the 10-carbon thioester is formed (Fig. 3). The incorporation of 3-hydroxyoctanoyl-L-serinyl-L-leucine into icosalide A2 (2), the major icosalide produced by the MSX 74159 fungus, suggests that the NRPS responsible for assembly of this metabolite contains an embedded CI domain with a preference for 8-carbon thioesters. It would therefore be interesting to investigate the mechanisms underlying icosalide A2 biosynthesis.
The module 3 CI domain might also be expected to prefer R-configured 3-hydroxyacyl thioesters to their S-configured counterparts. (Note, however, that the ketoreductase in bacterial fatty acid biosynthesis produces exclusively 3R-configured 3-hydroxyacyl-ACP thioester intermediates.25 There may therefore be no selective pressure to drive the evolution of stereoselectivity in CI domains.) To investigate whether the module 3 CI domain is stereoselective, we incubated the holo-CI-A-PCP tri-domain with ATP, L-Ser and (3S)-3-hydroxyoctanoyl or (3S)-3-hydroxydecanoyl-panthetheine thioesters. Although the reaction with the 10-carbon thioester resulted in a significant degree of product formation, it was clearly less efficient than the reaction with the corresponding 3R-configured thioester (Fig. 3). In contrast, only trace amounts of product were formed in the reaction employing the eight-carbon thioester (Fig. 3). Thus, the CI domain appears to possess a moderate to high degree of stereoselectivity, depending on the carbon chain length of the substrate. Overall, our data show that the module 3 CI domain of IcoA plays a key role in initiating the assembly of the (3R)-3-hydroxydecanoyl-L-serinyl-L-leucinyl moiety of icosalide A1.
Conclusions
We have shown that the “fungal” antibiotic icosalide A1 (1), originally isolated from an Aureobasidium sp., is produced by a B. gladioli strain closely associated with the fungus. In addition, we have shown that B. gladioli isolates from a range of sources, including CF lung infections, mushroom rot and insects also produce this metabolite. Comparative genome sequence analysis coupled with genetic manipulation allowed identification of a gene encoding the NRPS responsible for icosalide A1 biosynthesis. The third module of this NRPS has a very unusual architecture, containing a putative chain-initiating CI domain in addition to a chain elongating LCL domain. Intact protein mass spectrometry showed that this CI domain catalyses selective N-acylation of a serinyl thioester bound to the module 3 PCP domain with a (3R)-3-hydroxydecanoyl thioester. Thus, the asymmetric lipopeptidiolide core of icosalide A1 (1) is assembled by a unique mechanism involving two distinct chain initiation events on a single NRPS subunit.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants from the BBSRC (BB/L021692/1) to E. M., G. L. C., and J. P., and the Welsh Government Life Sciences Bridging Fund (LSBF R2-004) to E. M. The Bruker MaXis II instrument used in this study was funded by the BBSRC (BB/M017982/1). M. J. is the recipient of a BBSRC Future Leader Fellowship (BB/R01212/1) and X. J. was supported by fellowships the University of Warwick and the China Scholarship Council. J. M. was supported by a Marie Sklodowska-Curie Fellowship from the European Commission (656067). Y. D. and D. R. were supported by grants from the MRC (MR/N501839/1) and BBSRC (BB/K002341/1), respectively. G. L. C. is the recipient of a Wolfson Research Merit Award from the Royal Society (WM130033). We thank Thomas R. Connor and the MRC Cloud Infrastructure for Microbial Informatics (CLIMB) for providing additional computational resources for genomic analysis.Notes and references
- E. Mahenthiralingam, A. Baldwin and C. G. Dowson, J. Appl. Microbiol., 2008, 104, 1539–1551 CrossRef CAS PubMed.
- J. J. LiPuma, Clin. Microbiol. Rev., 2010, 23, 299–323 CrossRef PubMed.
- Q. Esmaeel, M. Pupin, N. P. Kieu, G. Chataigné, M. Béchet, J. Deravel, F. Krier, M. Höfte, P. Jacques and V. Leclère, MicrobiologyOpen, 2016, 5, 512–526 CrossRef CAS PubMed.
- N. Moebius, C. Ross, K. Scherlach, B. Rohm, M. Roth and C. Hertweck, Chem. Biol., 2012, 19, 1164–1174 CrossRef CAS PubMed.
- X. Liu, S. Biswas, M. G. Berg, C. M. Antapli, F. Xie, Q. Wang, M.-C. Tang, G.-L. Tang, L. Zhang, G. Dreyfuss and Y.-Q. Cheng, J. Nat. Prod., 2013, 76, 685–693 CrossRef CAS PubMed.
- H. He, A. S. Ratnayake, J. E. Janso, M. He, H. Y. Yang, F. Loganzo, B. Shor, C. J. O'Donnell and F. E. Koehn, J. Nat. Prod., 2014, 77, 1864–1870 CrossRef CAS PubMed.
- L. Song, M. Jenner, J. Masschelein, C. Jones, M. J. Bull, S. R. Harris, R. C. Hartkoorn, A. Vocat, I. Romero-Canelon, P. Coupland, G. Webster, M. Dunn, R. Weiser, C. Paisey, S. T. Cole, J. Parkhill, E. Mahenthiralingam and G. L. Challis, J. Am. Chem. Soc., 2017, 139, 7974–7981 CrossRef CAS PubMed.
- E. Mahenthiralingam, L. Song, A. Sass, J. White, C. Wilmot, A. Marchbank, O. Boaisha, J. Paine, D. Knight and G. L. Challis, Chem. Biol., 2011, 18, 665–677 CrossRef CAS PubMed.
- S. Lewenza and P. A. Sokol, J. Bacteriol., 2001, 183, 2212–2218 CrossRef CAS PubMed.
- L. P. Partida-Martinez and C. Hertweck, ChemBioChem, 2007, 8, 41–45 CrossRef CAS PubMed.
- S. Schmidt, J. F. Blom, J. Pernthaler, G. Berg, A. Baldwin, E. Mahenthiralingam and L. Eberl, Environ. Microbiol., 2009, 11, 1422–1437 CrossRef PubMed.
- C. Ross, K. Scherlach, F. Kloss and C. Hertweck, Angew. Chem., Int. Ed., 2014, 53, 7794–7798 CrossRef CAS PubMed.
- R. Nazir, J. A. Warmink, H. Boersma and J. D. van Elsas, FEMS Microbiol. Ecol., 2010, 71, 169–185 CrossRef CAS PubMed.
- J. A. Warmink, R. Nazir and J. D. van Elsas, Environ. Microbiol., 2009, 11, 300–312 CrossRef CAS PubMed.
- N. Stopnisek, D. Zühlke, A. Carlier, A. Barberán, N. Fierer, D. Becher, K. Riedel, L. Eberl and L. Weisskopf, ISME J., 2016, 10, 253–264 CrossRef CAS PubMed.
- L. P. Partida-Martinez and C. Hertweck, Nature, 2005, 437, 884–888 CrossRef CAS PubMed.
- M. Jenner, J. Masschelein, Y. Dashti, C. Jones, S. Harris, J. Parkhill, C. Pearce, E. Mahenthiralingham and G. L. Challis, Planta Med., 2016, 82, P1112 Search PubMed.
- C. Boros, C. J. Smith, Y. Vasina, Y. Che, A. B. Dix, B. Darveaux and C. Pearce, J. Antibiot., 2006, 59, 486–494 CrossRef CAS PubMed.
- B. Dose, S. P. Niehs, K. Scherlach, L. V. Flórez, M. Kaltenpoth and C. Hertweck, ACS Chem. Biol., 2018, 13, 2414–2420 CrossRef CAS PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS PubMed.
- S. A. Sieber and M. A. Marahiel, Chem. Rev., 2005, 105, 715–738 CrossRef CAS.
- G. W. Payne, P. Vandamme, S. H. Morgan, J. J. LiPuma, T. Coenye, A. J. Weightman, T. H. Jones and E. Mahenthiralingam, Appl. Environ. Microbiol., 2005, 71, 3917–3927 CrossRef CAS PubMed.
- C. Rausch, I. Hoof, T. Weber, W. Wohlleben and D. H. Huson, BMC Evol. Biol., 2007, 7, 78 CrossRef PubMed.
- L. E. Quadri, P. H. Weinreb, M. Lei M, M. M. Nakano, P. Zuber and C. T. Walsh, Biochemistry, 1998, 37, 1585 CrossRef CAS PubMed.
- P. W. Majerus, A. W. Alberts and P. R. Vagelos, J. Biol. Chem., 1965, 240, 618 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04897e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |