
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltralow-temperature photochemical synthesis of atomically dispersed Pt catalysts for the hydrogen evolution reaction†
Hehe
Wei‡
 a,
Hongbo
Wu‡
b,
Kai
Huang‡
af,
Binghui
Ge
*cd,
Jingyuan
Ma
e,
Jialiang
Lang
a,
Di
Zu
a,
Ming
Lei
f,
Yugui
Yao
b,
Wei
Guo
*b and
Hui
Wu
*a
a,
Hongbo
Wu‡
b,
Kai
Huang‡
af,
Binghui
Ge
*cd,
Jingyuan
Ma
e,
Jialiang
Lang
a,
Di
Zu
a,
Ming
Lei
f,
Yugui
Yao
b,
Wei
Guo
*b and
Hui
Wu
*a
aState Key Laboratory of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing, 100084, China. E-mail: huiwu@tsinghua.edu.cn
bSchool of Physics, Beijing Institute of Technology, 5 South Zhongguancun Street, Haidian District, Beijing, 100081, China. E-mail: weiguo7@bit.edu.cn
cBeijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China. E-mail: bhge@iphy.ac.cn
dInstitute of Physical Science and Information Technology, Anhui University, 230601, Hefei, Anhui, China
eShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201204, China
fState Key Laboratory of Information Photonics and Optical Communications & School of Science, Beijing University of Posts and Telecommunications, Beijing 100876, China
First published on 19th January 2019
Abstract
Efficient control of nucleation is a prerequisite for the solution-phase synthesis of nanocrystals. Although the thermodynamics and kinetics of the formation of metal nanoparticles have been largely investigated, fully suppressing the nucleation in solution synthesis remains a major challenge due to the high surface free energy of isolated atoms. In this article, we largely decreased the reaction temperature for ultraviolet (UV) photochemical reduction of H2PtCl6 solution to −60 °C and demonstrated such a method as a fast and convenient process for the synthesis of atomically dispersed Pt. We showed that the ultralow-temperature reaction efficiently inhibited the nucleation process by controlling its thermodynamics and kinetics. Compared with commercial platinum/carbon, the synthesized atomically dispersed Pt catalyst, as a superior HER catalyst, exhibited a lower overpotential of approximately 55 mV at a current density of 100 mA cm−2 and a lower Tafel slope of 26 mV dec−1 and had higher stability in 0.5 M H2SO4.
Introduction
Nucleation is the bridge between nanocrystals and isolated metal atoms or species1,2 and has a fundamental impact on nanocrystals, atomically dispersed metal materials and their applications in natural engineering and medicine.3,4 Very recently, the nucleation and growth of solids in solution have been directly observed by in situ studies.5,6 As confirmed by Mirsaidov et al.6 through in situ transmission electron microscopy (TEM) studies, we have noted that efficient control of the solid nucleation process is essential for the synthesis of atomically dispersed materials and homogeneous nanocrystals. The related classical nucleation theories (CNT) and several other theories, such as colloidal stability,7 precursor chemistry,8 and reaction kinetics,9 are still contradictory. Despite all this, the synthesis of atomically dispersed metal materials via suppressing the agglomeration of isolated atoms or species can be achieved by (i) directly impeding the nucleation process of individual atoms10–12 or (ii) anti-Ostwald ripening of nanocrystals.13,14 As for anti-Ostwald ripening methods, despite their improvements, these approaches have some disadvantages. For instance, in the decomposition of nanocrystals, the anti-Ostwald ripening method largely depends on a high-temperature annealing process, which is not only limited by the used clusters and nanocrystals but is also unsuitable for large-scale preparation and applications. In contrast, the atomically dispersed metal prepared by impeding the nucleation process of individual atoms can be easily controlled and synthesized, especially in solution. Recently, it has been shown that using an appropriate support material that strongly interacts with the metal species may prevent their nucleation and aggregation, to create stable and atomically dispersed metals.15–17 However, the significant high surface free energy of isolated atoms may lead to unmanageable agglomeration of atoms, which may thereby limit their applications;18,19 thus, it is significantly important that the metal atoms or species should be atomically dispersed in solution at the initial step of synthesis.The solution reaction and nuclei formation process are closely linked with the kinetics of reaction in solution and are critical factors during the preparation process. Previously, our group has developed iced-photochemical reduction, in which reactants and products were fixed in the solid phase (ice), and the nucleation process of atomic metals was successfully hindered in the special solid-phase reaction.20 Additionally, we reported that a previously frozen reactant solution could melt in another reactant solution, thus causing ultra-slow release of reactants, providing consistently low concentration and resulting in atomically dispersed metal solutions in a controlled solid–liquid reaction.21 While the diffusion and localized reactant concentration are among the most important factors for nucleation of synthetic products, we propose that temperature plays a very important role in controlling the reaction kinetics and nuclei formation process in most liquid-phase reactions. In this study, we investigate the reaction temperature, which has great thermodynamic and kinetic effects on the nucleation process of nanocrystals.22–24 Initially, inspired by the classical nucleation theory,25,26 we study an interesting and important question: how different the products will be when solution reactions run at ultralow temperatures? Meanwhile, we also find that temperature plays a significant role in the non-classical nucleation theories for nanocrystal formation.7–9 Despite this inference, several investigations on the nucleation process of metal nanocrystals at ultralow temperature have been conducted.
Herein, we developed an ultralow-temperature photochemical reaction, in which chloroplatinic acid (H2PtCl6) in an antifreeze solution containing ethyl alcohol and water was irradiated with ultraviolet light at −60 °C. Atomically dispersed platinum adsorbed in NMC (Pt1/NMC) was successfully synthesized by this ultralow-temperature reaction. Compared with the commercial platinum/carbon catalyst and Pt nanoparticle catalyst synthesized at room temperature, the Pt1/NMC exhibited superior catalytic activity and stability in electrocatalytic water-splitting reaction in acidic solutions. It also had lower overpotential (approximately 55 mV at 100 mA cm−2) and showed nearly no observable decay of HER activity after 5000 cyclic voltammetry (CV) cycles.
Results and discussion
The schematic illustration of the syntheses is displayed in Fig. 1. Here, a typical solution synthesis in which Pt4+ is reduced to Pt (eqn (1)) is used as an example.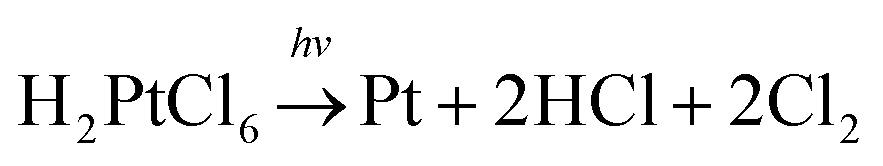 | (1) |
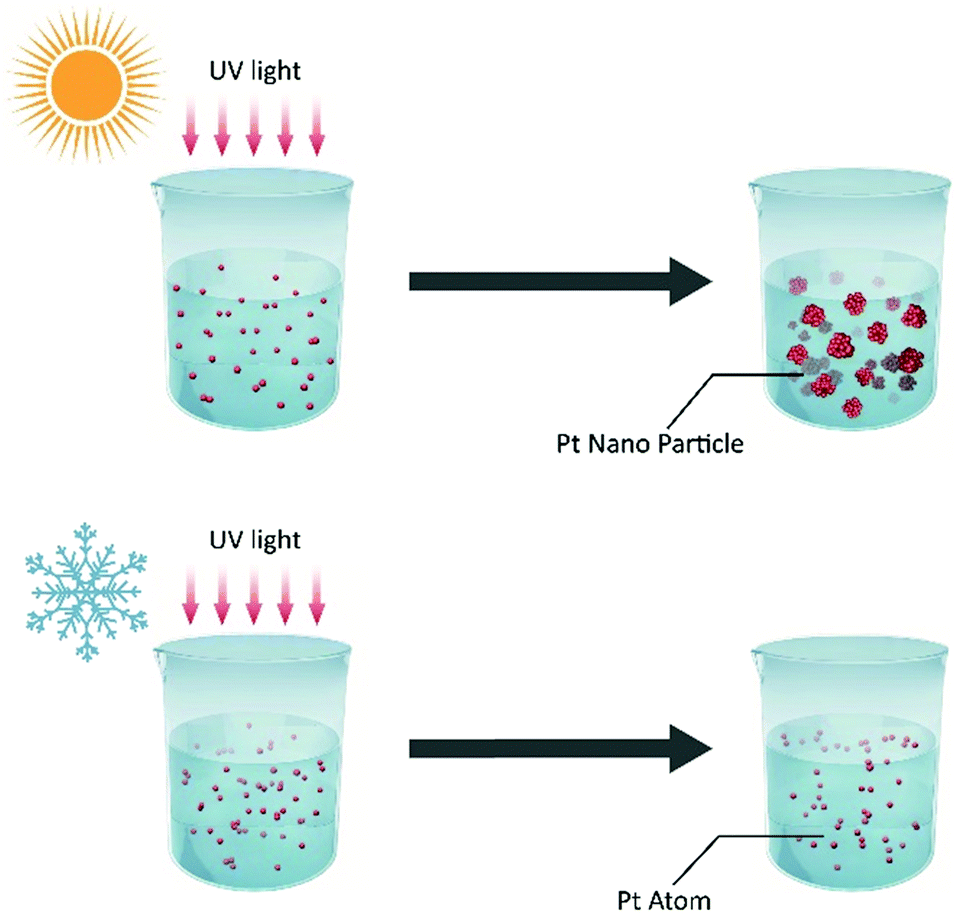 | ||
| Fig. 1 Schematic illustration of the synthesis of atomically dispersed Pt. Comparison of the conventional photochemical reaction at room temperature (top) and the designed ultralow-temperature (−60 °C) photochemical reaction (bottom); the nucleation process of isolated atoms was prevented and atomically dispersed Pt was synthesized at −60 °C. | ||
H2PtCl6 can be reduced by ultraviolet light. In the conventional solution-photochemical reaction, platinum nanoparticles are synthesized at room temperature following photochemical reduction. During the reaction, the reduced Pt atoms can nucleate, grow, and form Pt nanocrystals; this is usually regarded as the general synthesis method for metal nanocrystals. In contrast to the conventional photochemical reduction, our photochemical reaction was designed to be carried out at ultralow temperature. After the H2PtCl6 antifreeze aqueous solution containing ethyl alcohol was incubated at −60 °C, it was irradiated with UV light at −60 °C (Fig. 1). Because the reaction temperature can play a significant role in the nucleation process, its kinetics, i.e. the nucleation barrier, increase with decreasing temperature. In such an ultralow-temperature (−60 °C) reaction, nucleation was suppressed by the ultralow temperature, and atomically dispersed Pt was thus obtained at ultralow reaction temperature.
The aberration-corrected high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) images of Pt1/NMC are shown in Fig. 2b. The Pt1/NMC was synthesized after the prepared atomically dispersed Pt solution was mixed with nitrogen-doped mesoporous carbon and then stirred for one hour (to allow complete adsorption and binding), compared with Pt nanoparticles prepared at room temperature (Fig. 2a). A large number of atomically dispersed Pt atoms adsorbed on the NMC surface (dots exhibiting bright contrast) could be seen, and the examination of different regions revealed that almost no clusters or small particles were present in the vicinity of the isolated atoms (Fig. S1†). The isolated Pt atoms were approximately 0.1 nm in size (Fig. S2†). The X-ray diffraction (XRD) pattern of Pt1/NMC is also in good agreement with that of the NMC, in which the Pt phase was not detected (Fig. S3†). Moreover, Pt nanocrystals were not observed in the high-resolution (HR) TEM images, indicating that the Pt species are homogeneously and uniformly dispersed in NMC (Fig. S4†). This is consistent with the above STEM results. To determine the chemical state and coordination environment of the prepared samples, X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) were performed. Fitted peaks of the dominant nitrogen species of Pt1/NMC in N 1s XPS spectra centered at the binding energies of 400.8 and 398.5 eV were assigned to pyridinic-N and graphitic-N, respectively (Fig. S5†), which may serve as anchor points for isolated Pt atoms. In agreement with the N 1s spectra, the Pt 4f binding energy of Pt1/NMC in Pt 4f XPS spectra (Fig. S6†) was red-shifted (by 2.5 eV) to 72.7 eV, which was higher than the energy of Pt foil (71.2 eV). Such a positive shift is similar to the shift observed in Pt 4f from the bulk state to isolated Pt atoms, which is due to the well-known strong interaction between isolated Pt atoms and NMC.13,27 X-ray absorption fine structure (XAFS) can probe the local atomic and electronic structure of absorbing atoms. The white-line intensities in the normalized X-ray absorption near edge structure (XANES) spectra reflect the oxidation state of Pt. As shown in Fig. 2c, the atomically dispersed Pt adsorbed in NMC had increased white-line intensity compared with Pt nanoparticles and Pt foil, indicating the oxidation of Pt atoms in Pt1/NMC. In the extended XAFS (EXAFS) spectra, there is only one prominent peak centered at approximately 2.1 Å which originated from the Pt–C/N/O contribution in Pt1/NMC (Fig. 2d and Table S1†). In contrast to Pt1/NMC, an obvious peak at approximately 2.76 Å from the Pt–Pt contribution in Pt/NMC and Pt foil was observed. It is an indication that Pt1/NMC contains only single Pt atoms, consistent with the STEM results.
 | ||
| Fig. 2 Characterization and AIMD simulation of atomically dispersed Pt. (a) STEM images of Pt nanoparticles prepared at room temperature. (b) STEM images of atomically dispersed Pt synthesized via the ultralow-temperature photochemical reaction. (c) Normalized XANES spectra at the Pt L3-edge. (d) Extended X-ray absorption fine structure spectra of bulk Pt foil, Pt nanoparticles and atomically dispersed Pt adsorbed in NMC. (e) Pt-three-coordinate configuration in liquid ethanol. (f) Pt-four-coordinate configuration in liquid ethanol. (g) Energy diagram for Pt–Pt dimer formation at 213 K. (h) Energy diagram for Pt–Pt dimer formation at 298 K. The initial state (IS), transition state (TS) and final state (FS) are shown from left to right. C, H, O and Pt atoms are depicted as gray, white, red and light blue spheres, respectively. | ||
The experimental results demonstrated that the atomically dispersed Pt was successfully synthesized in 90% liquid ethanol or pure ethanol solution (Fig. S7†) at ultralow temperature (213 K or −60 °C). However, once the temperature was raised to 298 K (or 25 °C), Pt atoms were aggregated, in turn forming Pt nanoparticles (Fig. 2a). To understand the mechanism underlying such temperature-controlled syntheses, the atomically dispersed Pt and its initial nucleation state (Pt–Pt dimer) were analyzed in liquid ethanol at both ultralow temperature (213 K) and room temperature (298 K) using AIMD simulations. First, one Pt atom was placed in liquid ethanol in AIMD simulations at both 213 K and 298 K and the interactions between the isolated Pt atoms and the ethanol molecules were observed. As shown in Fig. 2e and f, the simulations indicated that the single Pt atom automatically forms either a three-coordinate configuration (Pt3c) (Fig. S8†) or a four-coordinate configuration (Pt4c) (Fig. S9†) with the surrounding ethanol molecules at both temperatures. The Pt atom in the Pt4c configuration is more stable than that in the Pt3c configuration and has bond energies of 0.249 eV and 0.437 eV at 213 K and 298 K, respectively. In the Pt3c configuration, one ethanol molecule releases a proton from the methyl group, while the other provides an oxygen atom. By contrast, in the Pt4c configuration, whereas one ethanol molecule releases a proton from the methyl group, both nearby ethanol molecules provide an oxygen atom. The specific configuration is highly dependent on the orientation of hydroxyl groups around Pt atoms. During the AIMD simulations, the Pt4c configuration was observed when a Pt atom was surrounded by two hydroxyl groups or the Pt3c configuration was observed otherwise.
We calculated the partial density of states (PDOS) for atoms in both Pt3c (Fig. S8,† right panel) and Pt4c (Fig. S9,† right panel) configurations. The data clearly showed that the Pt–ethanol interaction in both cases was mainly through the hybridization between the d orbitals of Pt and the p orbitals of C and O atoms below the Fermi level. However, the d-band center of Pt in Pt4c is higher (closer to the Fermi-level) than that in Pt3c, which is in good agreement with the fact that the Pt binding energy in the Pt4c configuration is stronger than that in the Pt3c configuration (Table S2†). Due to the large binding strength and high stability, Pt in the Pt4c configuration may possess a greater percentage than Pt in the Pt3c configuration in ethanol liquid. It should be noted that the absolute oxidation state is difficult to determine by computational methods.28 Here, the relative oxidation state can be determined based on Bader charge analysis (Table S3†). As for Pt in the Pt3c configuration, Pt possesses weak binding strength, and only relatively few electrons (+0.063 eV/+0.024 eV at 213 K/298 K) are transferred from the Pt atom to the surrounding ethanol molecules. On the other hand, for Pt in the Pt4c configuration, the Bader charge of Pt is +0.167 eV/0.134 eV at 213 K/298 K, respectively, indicating stronger Pt binding strength in Pt4c. Both Pt in Pt3c and Pt4c configurations possess a positive valence state of Pt atoms in Pt1/NMC, which is in agreement with the XPS results and previous reports.13,27
Based on Bader charge analysis (Table S3†), for Pt in Pt3c and Pt4c configurations, partial electrons are transformed from Pt to the surrounding ethanol molecules, which indicated the electrostatic stabilization of the Pt atom. The Pt3c and Pt4c configurations were 3.830 eV/3.985 eV and 4.079 eV/4.372 eV energetically more stable than the dispersive Pt atom in ethanol solution at 213 K/298 K (Table S2†), thus verifying a good thermodynamic stability of the Pt3c and Pt4c configurations. At the electronic level, the formation mechanisms of Pt3c and Pt4c structures were mainly through the orbital hybridization between the d orbitals of Pt and the p orbitals of C and O atoms (Fig. S8,† right panel and Fig. S9,† right panel). Then, the nucleation barriers for all possible cases of two Pt atoms, including configurations of Pt4c + Pt3c, Pt4c + Pt4c and Pt3c + Pt3c, were systematically investigated (Fig. 2 and Fig. S10 and S11†) by using the constrained minimization technique to obtain the dimerization barriers.29,30 The larger nucleation barriers at 213 K can effectively inhibit the agglomeration of Pt atoms, thus guaranteeing the stabilization of single Pt atoms at low temperature and further confirming that the low temperature can effectively suppress the nucleation processes of metal atoms. Therefore, the isolated Pt atoms were stabilized as a Pt3c or Pt4c structure with larger barrier to agglomeration at 213 K.
To further reveal the mechanism of temperature-controlled synthesis of atomically dispersed Pt, two separated Pt atoms were placed at different configurations in liquid ethanol, and the initial Pt–Pt nucleation mechanism was observed at both 213 K and 298 K using AIMD simulations and transition state search. A few interesting results were obtained as follows: (i) when the two Pt atoms in liquid ethanol were within the proximity of the Pt–Pt bonding state, they spontaneously formed clusters, due to the fact that clusters can easily be formed in high concentration of Pt atoms in ethanol at both high and low temperatures; and (ii) all possible initial configurations of two Pt atoms, including Pt4c + Pt4c, Pt4c + Pt3c and Pt3c + Pt3c, were observed prior to the formation of Pt–Pt dimers (Table S2†). In each case, the calculation results revealed that the barriers for agglomeration for Pt4c + Pt3c, Pt4c + Pt4c and Pt3c + Pt3c at 213 K were 0.422 eV, 0.305 eV and 0.219 eV larger than those at 298 K (Fig. 2g and h and S10 and S11†). The reason is mainly due to the structural effect; the volume of ethanol liquid is negatively correlated with the temperature, such that Pt atoms are more tightly surrounded by ethanol molecules when the temperature is decreased. Thus, the nucleation process at 298 K proceeded with an extremely high rate constant compared to that at 213 K in liquid ethanol. These simulation data not only verify the experimental observations but also confirm that the atomically dispersed Pt can be synthesized at ultralow temperature, whereas Pt clusters and nanoparticles with various large sizes are fabricated at room temperature.
The implementation of high-performance and durable platinum-based metal HER catalysts with lower noble metal loading may greatly enable large-scale commercialization of hydrogen evolution.31–34 To achieve this goal, atomically dispersed Pt in NMC can be considered as an ideal catalyst for the HER, because Pt nanocrystals can be downsized to the atomic level and Pt exists as an isolated atom, which is important for water splitting and hydrogen evolution. The HER was studied by linear sweep voltammetry (LSV) versus the reversible hydrogen electrode (RHE) on a rotating disk electrode (RDE) in 0.5 M H2SO4. The catalytic performance of Pt1/NMC in the HER was compared with that of commercial Pt/C, Pt/NMC and NMC under the same testing conditions, respectively. The Pt1/NMC showed a highly low overpotential of approximately 55 mV at a current density of 100 mA cm2 and a rapid increase of the cathodic current with applied potential (Fig. 3a and S12†). In contrast, the atomically dispersed Pt absorbed in NMC exhibited HER activity an order of magnitude higher than that of commercial Pt/C and Pt/NMC. NMC exhibited negligible HER activity, confirming that the Cx–Ny structure in NMC is not the active site for HER activity. The overpotential of Pt1/NMC is much lower than the best values achieved by other atomically dispersed metal catalysts, such as atomically dispersed Co, Ni and Pt.35–38 Moreover, it is comparable to those of the best Pt-based catalysts at the same Pt-loading39–42 (Fig. 3c and Table S4†). To further test the HER stability of Pt1/NMC, 1000 and 5000 cyclic voltammetry cycles from +0.3 to −0.1 V versus RHE were conducted. The Pt1/NMC after 5000 CV cycles was stable with nearly 100% retention (Fig. 3d). This indicates that the atomically dispersed Pt adsorbed in NMC is highly stable. On the other hand, the overpotential after 1000 CV cycles of Pt/NMC was increased by 20 mV, whereas that of Pt/C showed obvious decay (increased by approximately 30 mV) (Fig. S13 and S14†). The data further showed that the atomically dispersed Pt in nitrogen-doped mesoporous carbon had a similar density and almost no formation of clusters or nanoparticles after the stability test (Fig. S15†). This suggests that the deceased activities of Pt/NMC and Pt/C are caused by the irreversible detachment and/or agglomeration of Pt nanoparticles, while the high stability of Pt1/NMC is due to the strong interaction between the atomically dispersed Pt and NMC. Moreover, the mass activity (from 40 mV to 60 mV overpotential versus RHE) of Pt1/NMC was at least three times greater than that of Pt/C (Fig. S16 and S17†). This indicates that Pt1/NMC exhibited significantly higher mass activity than Pt/NMC and commercial Pt/C. The HER kinetics of Pt1/NMC were analyzed using Tafel plots, the linear portions of which were fitted with the Tafel equation η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(j) + a, where η is the overpotential, j is the current density, and b is the Tafel slope. The Tafel plot for Pt1/NMC (Fig. 3b) showed a highly low value of approximately 26 mV dec−1, which is lower than the values for Pt/NMC (32 mV dec−1) and commercial Pt/C (31 mV dec−1), suggesting that the HER of Pt1/NMC may proceed by the Volmer–Tafel mechanism. Additionally, the relatively low Tafel slope of Pt1/NMC, derived from the initial HER polarization curve, indicated that Pt1/NMC has superior HER kinetics and performances. These findings indicated that the atomically dispersed Pt adsorbed in nitrogen-doped mesoporous carbon (with low consumption of Pt) has significantly enhanced HER activity, mass activity and stability compared with commercial Pt/C.
log(j) + a, where η is the overpotential, j is the current density, and b is the Tafel slope. The Tafel plot for Pt1/NMC (Fig. 3b) showed a highly low value of approximately 26 mV dec−1, which is lower than the values for Pt/NMC (32 mV dec−1) and commercial Pt/C (31 mV dec−1), suggesting that the HER of Pt1/NMC may proceed by the Volmer–Tafel mechanism. Additionally, the relatively low Tafel slope of Pt1/NMC, derived from the initial HER polarization curve, indicated that Pt1/NMC has superior HER kinetics and performances. These findings indicated that the atomically dispersed Pt adsorbed in nitrogen-doped mesoporous carbon (with low consumption of Pt) has significantly enhanced HER activity, mass activity and stability compared with commercial Pt/C.
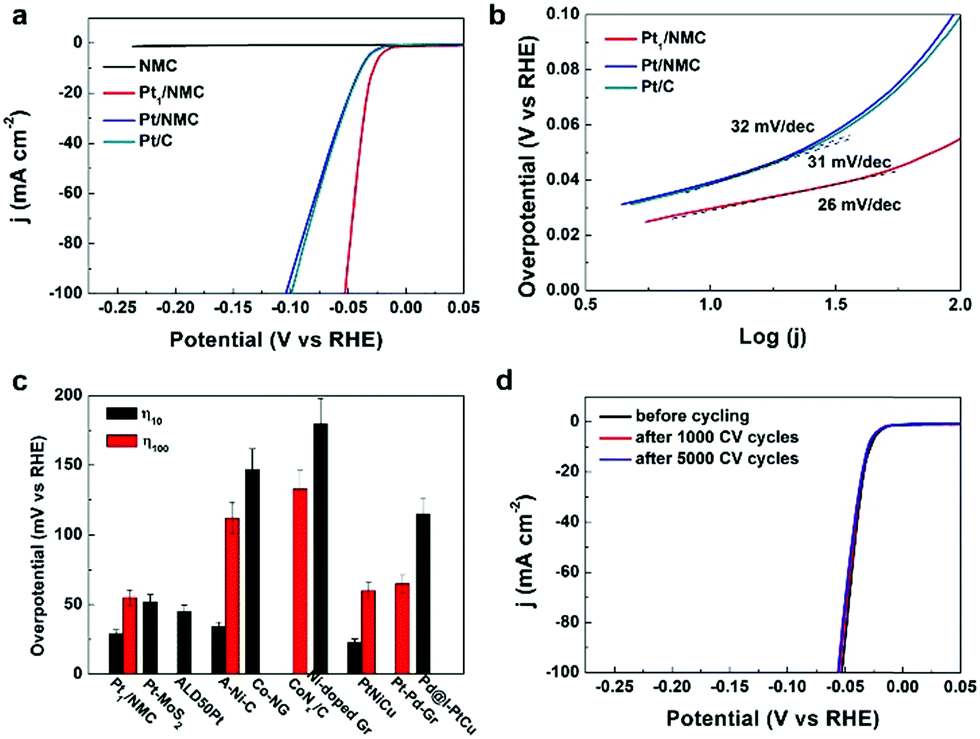 | ||
| Fig. 3 HER activities of Pt1/NMC. (a) Polarization curves for Pt1/NMC, Pt/NMC, commercial Pt/C and NMC. (b) Tafel plots for Pt1/NMC, Pt/NMC and Pt/C. (c) Overpotential of different atomically dispersed metal catalysts and Pt-based catalysts in acid solution. (d) Stability of Pt1/NMC after 1000 and 5000 CV cycles. The overpotential of catalysts at current densities of 10 mA cm−2 and 100 mA cm−2 is presented as η10 and η100, respectively. | ||
The NMC substrate normally contains various types of defects such as single vacancy (SV), double vacancy (DV) and edge vacancy (EV) defects (Fig. S18†). By using DFT calculations, the stabilities of isolated Pt atoms located in various sites (including SV, DV and EV sites) of the NMC substrate were carefully examined, in which a Pt atom in liquid ethanol was used as the reference state. The DFT calculations indicated that Pt atoms favor the NMC substrate over ethanol; the energy differences ΔEPD (Pt detaching from ethanol) for single Pt atoms in NMC-SV, NMC-DV, and NMC-EV sites for Pt3c/Pt4c configurations were −1.959/−1.710 eV, −4.681/−4.432 eV and −5.326/−5.077 eV, respectively (Table. S5†). These negative values clearly support the experimental results showing that the atomically dispersed Pt in liquid ethanol can easily be transferred to the NMC substrate.
According to the Pt binding energy calculations and Bader charge analysis (Table S6†), NMC substrates with defects (NMC-EV, NMC-SV and NMC-DV) can bind Pt atoms more strongly than the surface with no defects (NMC-ND). This suggests that the larger the binding strength of Pt, the greater the charge transferred from Pt to C and N atoms of the NMC substrate. Furthermore, as shown in Fig. 4a, DFT calculations revealed that the atomically dispersed Pt is more stable than Pt dimers (with ΔE in NMC-EV, NMC-SV and NMC-DV of −1.319, −1.765 and −5.026 eV, respectively) at the corresponding defective point/edge sites, whereas the atomically dispersed Pt in NMC-ND can easily form clusters. These data are in good agreement with the experimental observation that Pt atoms on defective NMC preferentially remained isolated rather than forming clusters.
 | ||
| Fig. 4 DFT analysis of HER activity. (a) Schematic diagrams of sites for atomically dispersed Pt and Pt–Pt dimers in NMC substrates. (b) The free energy diagrams for different H concentrations on a Pt atom in NMC-DV and NMC-EV sites. (c) Calculated activation barriers of H–H association reactions near Pt atoms at NMC-SV, NMC-DV and NMC-EV sites. IS, TS and FS are illustrated, and the energy difference (ΔE) between the Pt dimer and two dispersive Pt atoms in the NMC substrate is shown. C, N and Pt atoms are depicted as brown, blue and light blue spheres, respectively. The DFT-optimized structures are illustrated, and ΔGH of hydrogen evolution on Pt (111) is shown for comparison. The calculated activation barriers of H–H association reaction in Pt1/NMC-SV are in good agreement with the previously published data.26 C, N, Pt and H atoms are represented as brown, blue, light blue and red spheres, respectively. | ||
The Gibbs free energy (ΔGH) of the adsorbed hydrogen atom is widely used to assess the HER catalytic activity of a catalyst, and a catalyst with high HER performance normally binds a H atom neither too weakly nor too strongly. With a catalyst, whose size is reduced to the atomic scale, each Pt single atom may offer an active site. To better understand the higher HER performance of the Pt1/NMC system, compared to the commercial Pt/C catalyst, we systematically calculated the ΔGH for H adsorption on Pt1/NMC-DV and Pt1/NMC-EV sites as well as on the Pt (111) surface for comparison. As shown in Fig. 4b, the ΔGH for a H atom adsorbed on the Pt1/NMC-DV/EV system was nearly zero. Specifically, the optimal ΔGH for Pt at NMC-DV sites was PtDV(1H) ∼ −0.011 eV, while those at NMC-EV sites were PtEV(2H) ∼ 0.006 eV and PtEV(3H) ∼ −0.043 eV (Fig. S19–S20†). The atomic H adsorbed onto PtDV/PtEV clearly exhibited better ΔGH than that adsorbed onto the Pt(111) surface, thus confirming that the atomically dispersed Pt has enhanced HER catalytic activity, which is in accordance with the electrochemical experiments. Interestingly, the active sites on NMC-DV, NMC-EV and NMC-SV surfaces near the single Pt atom may bind several H atoms, which indicates that the hydrogen production on Pt1/NMC-DV and Pt1/NMC-EV systems may also follow the Tafel mechanism. This is especially feasible for the Pt1/NMC-SV system, whose site had a relatively larger binding strength (Fig. S21†), which however, became weaker from −1.187 eV to −0.486 eV when H coverage was increased.
The reaction kinetics shown in Fig. 4c indicate that the HER activation barrier on the defective NMC substrate was lower than that on Pt (111). Compared to the barrier of 0.85 eV on Pt (111),43,44 the barrier on Pt1/NMC-SV was decreased to 0.641 eV, whereas that on Pt1/NMC-DV was decreased to 0.414 eV. Notably, the H–H association reaction on the Pt1/NMC-EV surface had no barrier at low H coverage. Based on these experimental observations and DFT calculations, the HER performance of atomically dispersed Pt in the NMC substrate is superior to that of commercial Pt/C, and nearly every single Pt atom on the defective NMC surface contributes to the HER activity. To further understand the high HER performance of the atomically dispersed Pt adsorbed onto the defective NMC substrate at the electronic level, the PDOS of Pt1/NMC-DV (Fig. S22†) and Pt1/NMC-EV (Fig. S23†) before and after H adsorption was calculated. The data showed that before H atom adsorption, the 5d-orbitals of Pt are strongly hybridized with C 2p or N 2p orbitals, and charges are transferred from Pt to the corresponding C and N atoms, while unoccupied 5d-orbitals of Pt provide a good platform to promote HER performances. Upon H adsorption, electrons are transferred from H to Pt1/NMC-DV and Pt1/NMC-EV, and the 5d-orbitals of Pt shift towards low energy and in turn interact strongly with the 1s orbital of the H atom below the Fermi level, thus giving rise to electron pairing and the formation of reactive hydrogen intermediates (Table S7†). However, based on Bader charge analysis, the number of electrons transferred to the substrate in Pt1/NMC-EV is increased (Table S8†), thereby enhancing the stability of the Pt1/NMC-EV system for durable H2 production. Overall, the significant changes in the electronic characteristics of Pt1/NMC-DV and Pt1/NMC-EV systems are responsible for the enhanced HER activity.
Conclusions
In conclusion, we designed and carried out ultralow-temperature photochemical reactions. We discovered that ultralow reaction temperature could efficiently suppress the nucleation and aggregation of isolated metal atoms in solution and in turn generate atomically dispersed metal materials. HADDF-STEM and XAFS confirmed that the isolated Pt atoms were dominant in NMC. The evaluation of the electrocatalytic hydrogen evolution reaction revealed that the atomically dispersed Pt adsorbed on the NMC catalyst had superior catalytic performances with excellent durability, compared with commercial Pt/C. Low overpotential (approximately 55 mV) was achieved at a current density of 100 mA cm−2, and almost no decay of Pt1/NMC was observed after 5000 CV cycles in 0.5 M H2SO4. This ultralow reaction temperature design can further be applied to other reactions of nanocrystals and products, from which atomically dispersed metal materials and special-structured products with enhanced performances can be obtained.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Basic Research of China (Grants 2015CB932500, 2016YFE0102200 and 2018YFB0104404), the National Natural Science Foundation of China (Grant 51661135025, 51522207, 21503014, 11574029, 51788104, 51706117 and U1564205), and Graduate Technological Innovation Project Beijing Institute of Technology (2018CX10028). We express our gratitude to Beijing JinBeiNuo Technology Co. Ltd. for designing and assembling the multi-channel flow-through test system for the field test. We thank the XAFS station (BL14W1) of the Shanghai Synchrotron Radiation Facility and acknowledge computation support from Tianhe-JK at the Beijing Computational Science Research Center (CSRC).Notes and references
- X. Wang, J. Zhuang, Q. Peng and Y. Li, Nature, 2005, 437, 121–124 CrossRef CAS PubMed.
- A. S. Myerson and B. L. Trout, Science, 2013, 341, 855–856 CrossRef CAS PubMed.
- P. M. Winkler, G. Steiner, A. Vrtala, H. Vehkamki, M. Noppel, K. E. J. Lehtinen, G. P. Reischl, P. E. Wagner and M. Kulmala, Science, 2008, 319, 1374–1377 CrossRef CAS PubMed.
- Y. Xia, K. D. Gilroy, H. C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS PubMed.
- M. H. Nielsen, S. Aloni and J. J. D. Yoreo, Science, 2014, 345, 1158–1162 CrossRef CAS PubMed.
- N. D. Loh, S. Sen, M. Bosman, S. F. Tan, J. Zhong, C. A. Nijhuis, P. Kral, P. Matsudaira and U. Mirsaidov, Nat. Chem., 2017, 9, 77–82 CAS.
- J. Polte, CrystEngComm, 2015, 17, 6809–6830 RSC.
- E. D. Bøjesen and B. B. Iversen, CrystEngComm, 2016, 18, 8332–8353 RSC.
- R. Xie, Z. Li and X. Peng, J. Am. Chem. Soc., 2009, 131, 15457–15466 CrossRef CAS PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- L. Liu, U. Diaz, R. Arenal, G. Agostini, P. Concepcion and A. Corma, Nat. Mater., 2016, 16, 132–138 CrossRef PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernandez, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- Z. Huang, X. Gu, Q. Cao, P. Hu, J. Hao, J. Li and X. Tang, Angew. Chem., Int. Ed., 2012, 51, 4198–4203 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- B. Bayatsarmadi, Y. Zheng, A. Vasileff and S.-Z. Qiao, Small, 2017, 1700191, 11–19 Search PubMed.
- H. Wei, K. Huang, D. Wang, R. Zhang, B. Ge, J. Ma, B. Wen, S. Zhang, Q. Li, M. Lei, C. Zhang, J. Irawan, L.-M. Liu and H. Wu, Nat. Commun., 2017, 8, 1490 CrossRef PubMed.
- H. Wei, K. Huang, L. Zhang, B. Ge, D. Wang, J. Lang, J. Ma, D. Wang, S. Zhang, Q. Li, R. Zhang, N. Hussain, M. Lei, L.-M. Liu and H. Wu, Angew. Chem., Int. Ed., 2018, 57, 3354 CrossRef CAS PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- M. Liu, K. Wang, L. Wang, S. Han, H. Fan, N. Rowell, J. A. Ripmeester, R. Renoud, F. Bian, J. Zeng and K. Yu, Nat. Commun., 2017, 8, 15467 CrossRef CAS PubMed.
- H. Zheng, R. K. Smith, Y. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309–1312 CrossRef CAS PubMed.
- T. J. Woehl, J. E. Evans, I. Arslan, W. D. Ristenpart and N. D. Browning, ACS Nano, 2012, 6, 8599–8610 CrossRef CAS PubMed.
- Y. T. Kim, K. Ohshima, K. Higashimine, T. Uruga, M. Takata, H. Suematsu and T. Mitani, Angew. Chem., Int. Ed., 2006, 45, 407–411 CrossRef CAS PubMed.
- H. Raebiger, S. Lany and A. Zunger A, Nature, 2008, 453, 763–766 CrossRef CAS PubMed.
- S. R. Bahn and K. W. Jacobsen, Comput. Sci. Eng., 2002, 4, 56–66 CrossRef CAS.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–800 CrossRef CAS PubMed.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xioa, R. Li, T.-K. Sham, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- J. C. Meier, I. Katsounaros, C. Galeano, H. J. Bongard, A. A. Topalov, A. Kostka, A. Karschin, F. Schuth and K. J. J. Mayrhofer, Energy Environ. Sci., 2012, 5, 9319–9330 RSC.
- J. Deng, H. Li, J. Xioa, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- L. Fan, P. F. Liu, X. Yan, L. Gu, Z. Z. Yang, H. G. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 10667 CrossRef CAS PubMed.
- H. Fei, J. Dong, A. J. Arellano-Jimenez, G. Ye, N. D. Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- H. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS PubMed.
- H.-W. Liang, S. Bruller, R. Dong, J. Zhang, X. Feng and K. Mullen, Nat. Commun., 2015, 6, 7992 CrossRef CAS PubMed.
- X. Cao, Nano Energy, 2014, 9, 301–308 CrossRef CAS.
- S. Bai, C. Wang, M. Deng, M. Gong, Y. Bai, J. Jiang and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 12120–12124 CrossRef CAS PubMed.
- F. Ren, H. Lu, Z. Wang, Y. Wu and Y. Li, J. Mater. Chem. A, 2015, 3, 23660–23663 RSC.
- G. R. Xu, J. J. Hui, T. Huang, Y. Chen and J. M. Lee, J. Power Sources, 2015, 285, 393–399 CrossRef CAS.
- G. F. Wei and Z. P. Liu, Chem. Sci., 2015, 6, 1485–1490 RSC.
- E. Skúlason, F. Trpkovic, M. E. Björketun, S. Gudmundsdóttir, G. Karlberg and J. Rossmeis, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04986f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |