
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective C–C bond formation from rhodium-catalyzed C–H activation reaction of 2-arylpyridines with 3-aryl-2H-azirines†
Yonghyeon
Baek‡
a,
Jinwoo
Kim‡
cd,
Hyunseok Kim
a,
Seung Jin
Jung
a,
Ho
Ryu
 cd,
Suyeon
Kim
cd,
Jeong-Yu
Son
a,
Kyusik
Um
a,
Sang Hoon
Han
a,
Hyung Jin
Seo
a,
Juyoung
Heo
a,
Kooyeon
Lee
b,
Mu-Hyun
Baik
*cd and
Phil Ho
Lee
*a
cd,
Suyeon
Kim
cd,
Jeong-Yu
Son
a,
Kyusik
Um
a,
Sang Hoon
Han
a,
Hyung Jin
Seo
a,
Juyoung
Heo
a,
Kooyeon
Lee
b,
Mu-Hyun
Baik
*cd and
Phil Ho
Lee
*a
aNational Creative Research Initiative Center for Catalytic Organic Reactions, Department of Chemistry, Kangwon National University, Chuncheon 24341, Republic of Korea. E-mail: phlee@kangwon.ac.kr
bDepartment of Bio-Health Technology, Kangwon National University, Chuncheon 24341, Republic of Korea
cDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: mbaik2805@kaist.ac.kr
dCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
First published on 8th January 2019
Abstract
A novel method for the synthesis of acylmethyl-substituted 2-arylpyridine derivatives using 3-aryl-2H-azirines was developed by exploring a prototype reaction using DFT-calculations and carrying out targeted experiments guided by the calculated mechanism. 2H-Azirine was initially hypothesized to ring-open at the metal center to furnish familiar metal nitrene complexes that may undergo C–N coupling. Computational studies quickly revealed and prototype experimental work confirmed that neither the formation of the expected metal nitrene complexes nor the C–N coupling were viable. Instead, azirine ring-opening followed by C–C coupling was found to be much more favorable to give imines that readily underwent hydrolysis in aqueous conditions to form acylmethyl-substituted products. This new method was highly versatile and selective toward a wide range of substrates with high functional group tolerance. The utility of the new method is demonstrated by a convenient one-pot synthesis of biologically relevant heterocycles such as pyridoisoindole and pyridoisoqunolinone.
Introduction
Controlling the chemo-, regio- and stereoselectivities of reactions is of central importance in general, but particularly so in organic chemistry.1 And C–H bond activation reactions2 are among the most desirable to carry out with high degrees of selectivity and significant research efforts were made in the past to couple C–H activation to C–C and C–N bond forming transformations. Alkyl and aryl azides featured prominently as valuable reagents in this endeavor and many C–N bond forming reagents based on C–H activation reactions have been developed (Scheme 1, eqn (1)).3 Acyl azides are attractive components,4 as they are capable of forming C–C bonds through isocyanates generated in situ within the framework of well-studied Curtius rearrangement pathways.5 Most notably, Chang recently developed a method for forming C–N bonds using acyl azides via C–H activation employing a ruthenium catalyst.6 Stable sulfonyl azides have also been used to form C–N bonds in C–H activation reactions7 and, most recently, dioxazolones were identified as highly versatile surrogates of azides for installing C–N bonds under mild reaction conditions.8 These studies highlight that the choice of an appropriate reagent that will mediate the formation of C–N and/or C–C bonds is critical. Thus, we sought to expand the scope of C–H activation reactions by identifying new reagents that may act as surrogates of azides in a C–H activation reaction. Given our previous experience with azirines,9 we wondered if they can be employed to furnish vinyl nitrenoids that may be exploited to form C–C and C–N bond selectively (Scheme 1, eqn (2)). | ||
| Scheme 1 Selective C–C and C–N bond formation using azides and its surrogates in C–H activation. | ||
To increase the efficiency of the new reaction discovery process, we used density functional theory (DFT) based calculations10 to first examine the feasibility of putative reaction mechanisms and identify the most promising strategy for reaction design. As shown in eqn (3) (Scheme 1), we initially speculated that the 3-aryl-2H-azirine substrate may coordinate to a metallacycle intermediate that should be readily accessible via C–H activation using a well-known Ir, Rh or Co catalyst. Subsequent ring-opening of the azirine11 was envisioned to give a vinyl metal-nitrene,12 from which a migratory insertion may result in a C–N coupled product. Surprisingly, DFT-calculations showed that the familiar C–N bond formation is not likely in this case, but suggested that C–C bond formation can be furnished efficiently. Specifically, the barrier for the formation of the eight-membered metallacycle was computed to be low. In good agreement with this DFT-based proposal, treatment of 2-phenylpyridine with 3-(4-methoxyphenyl)-2H-azirine in the presence of a Rh catalyst gave the acylmethylated product in 17% yield, whereas no evidence for C–N coupling was detectable.13 Based on these initial results, we developed an efficient and generally applicable new method that allows for the acylmethylation of 2-arylpyridine derivatives through selective C–C bond formation employing 2-arylpyridine and 3-aryl-2H-azirine using a Rh catalyst for C–H activation.
Results and discussion
Although our initial reaction design aimed to use azirines to form a nitrenoid intermediate that can be employed to facilitate C–N coupling, our DFT-calculations suggested that azirines cannot be used in this capacity. This result is surprising, as the steric energy stored in the three-membered ring is expected to lead to a rapid ring-opening during a reaction with metal catalysts capable of performing similar reactions with azides that are driven by the similarly favorable release of N2. As a likely successful catalyst platform, we chose the ubiquitous Rh(III)-pentamethylcyclopentadienyl ([Cp*Rh]2+) platform carrying labile acetate ligands that is well-known to be an effective catalyst for C–H activation, nitrenoid formation and C–N coupling reactions.Fig. 1 shows the computed reaction energy profile of a plausible catalytic cycle employing the prototype substrates phenylpyridine and 2H-azirine. First, C–H activation via a concerted metalation deprotonation (CMD) step moderated by a Brønsted base such as acetate leads to the cyclometalation. Our calculations show in good agreement with previous work14 that this process is relatively easy with a barrier of only 17.6 kcal mol−1. Ligand exchange transforms the cyclometalated intermediate B to the N-bound azirine complex C, which is predicted to ring-open the azirine readily traversing the transition state C-TS with a barrier of 22.8 kcal mol−1 to give the singlet intermediate 1D. It is at this juncture that the azirine substrate displays a decisive difference compared to the azides. In azides, there is a driving force to completely cleave the RN–N2 bond and release a neutral nitrogen molecule and form a formally dianionic imido (R–N2−) functionality upon oxidative coupling to the metal-center. In the case of azirine, ring-opening followed by rearrangement of the double bond can produce the vinyl-nitrene moiety that could undergo a similar oxidative coupling to the metal to produce a vinyl–imido complex, as envisioned (Scheme 1, eqn (2)). Our calculations show, however, that azirine ring-opening and oxidative addition gives the four-membered rhodaheterocycle 1D that is relatively high in energy and readily attacks the phenyl-carbon of the phenylpyridine ligand and rapidly inserts into the metal–carbon bond to afford the C–C coupled product E. The azirine ring-opening can be imagined to involve a homolytic C–N bond cleavage, which may lead to a triplet pathway after rapid spin-inversion promoted by the strong spin–orbit coupling provided by the Rh-center. We examined this possibility, but rejected it based on the much higher energy of the 3D intermediate shown in red in Fig. 1.
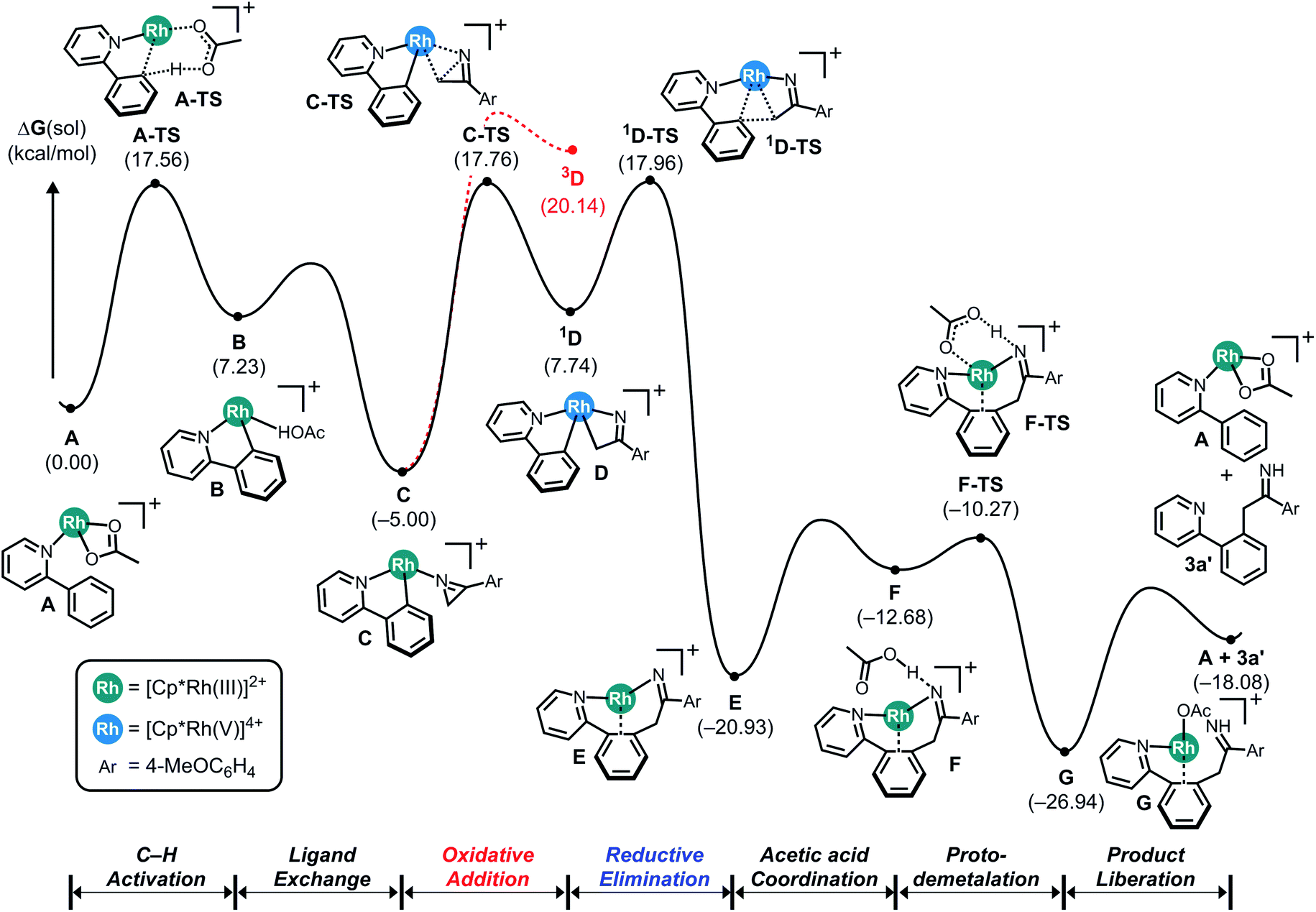 | ||
| Fig. 1 Computed energy profile for the Cp*Rh(III)-catalyzed C–C coupling reaction using 2-phenylpyridine and 2H-azirine. | ||
As anticipated, the release of the strain energy renders this last step exergonic with the free energy of −20.9 kcal mol−1 being assigned to E. Formerly an imido-complex, intermediate E can readily undergo proto-demetalation assisted by acetic acid, where the protonation of the imido functionality gives the corresponding imine product complex G, as highlighted in Fig. 1.
Fig. 2 illustrates the reaction energy profile of a putative C–N coupling mechanism. The initial phase of C–H activation of phenylpyridine is of course identical with what was shown in Fig. 1. To ring-open the azirine and facilitate the C–N bond formation with the phenylpyridine substrate, the insertion described above must be carried out by the N-atom of the metallaheterocycle, leading to the alternative insertion product H, which can be accomplished either on a singlet or a triplet pathway. Our calculations indicate that neither reaction trajectories afford a reasonable transition state, the migratory insertion to form the C–N bond requires passage through the transition states 1D-TS′ and 3D-TS, located at 31.3 and 32.5 kcal mol−1, giving rise to activation barriers of 36.3 and 37.5 kcal mol−1, respectively. These energies are too high for the reaction to be viable under realistic conditions and considering the C–C bond forming reaction discussed above, the conclusion can be drawn that the azirine substrate will give exclusively C–C coupled products and C–N coupling is not possible. Thermodynamically, the C–N coupled product H at −21.4 kcal mol−1 is slightly lower in energy than the C–C coupled intermediate E, which was found at −20.9 kcal mol−1. And our calculations show that the final portion of the putative catalysis consisting of acetic acid promoted demetalation to form the final product is reasonable. Thus, it is the kinetic inhibition of inserting the imido functionality that makes the C–N coupling pathway impossible.
 | ||
| Fig. 2 Computed energy profile for the Cp*Rh(III)-catalyzed C–N coupling reaction using 2-phenylpyridine and 2H-azirine. | ||
On the basis of these DFT-calculations, experimental studies were carried out using 2-phenylpyridine (1a) and 3-(4-methoxyphenyl)-2H-azirine (2a) in the presence of [Cp*RhCl2]2 with a number of additives and solvents, as summarized in Table 1. In good agreement with the mechanism discussed above, substrate 1a (0.2 mmol, 1.0 equiv.) reacted with 2a (1.2 equiv.) in the presence of [Cp*RhCl2]2 (4.0 mol%), AgSbF6 (16.0 mol%), H2O (1.0 equiv.), and acetic acid (1.0 equiv.) in dichloroethane (DCE). In 3 hours at 80 °C and after workup, we obtained the acylmethylated product 1-(4-methoxyphenyl)-2-(2-(pyridin-2-yl)phenyl)ethan-1-one (3a) in 35% yield (entry 2), which we propose is the hydrolyzed product of the computationally identified imine product. To test the role of acetate/acetic acid, which plays a prominent role in the computed mechanism, the experiments were repeated under identical conditions without adding acetic acid. As anticipated, no acylmethylated product was observed (entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | Yieldb (%) |
| a Reactions were carried out with 1a (0.2 mmol, 1.0 equiv.) and 2a (1.2 equiv.) in the presence of catalyst (4.0 mol%), additive (16.0 mol%), acetic acid (1.0 equiv.), and H2O (1.0 equiv.) in solvent (2.0 mL) at 80 °C for 3 h under a nitrogen atmosphere. b NMR yield using dibromomethane as an internal standard. c Acetic acid was not used. d Isolated yield. e 1a (0.4 mmol, 2.0 equiv.) was used. f Water was not used. | ||||
| 1 | [Cp*RhCl2]2 | AgSbF6 | DCE | 0c |
| 2 | [Cp*RhCl2]2 | AgSbF6 | DCE | 35 |
| 3 | [Cp*RhCl2]2 | AgSbF6 | MeOH | 20 |
| 4 | [Cp*RhCl2]2 | AgSbF6 | TFE | 73 |
| 5 | [Cp*RhCl2]2 | AgSbF6 | HFIP | 46 |
| 6 | [Cp*RhCl2]2 | AgSbF6 | THF | 28 |
| 7 | [Cp*RhCl2]2 | AgNTf2 | TFE | 37 |
| 8 | [Cp*RhCl2]2 | AgPF6 | TFE | 70 |
| 9 | [Cp*RhCl2]2 | AgSbF6 | TFE | 89(86)d,e |
| 10 | [Cp*IrCl2]2 | AgSbF6 | TFE | 0 |
| 11 | [Cp*CoCl2]2 | AgSbF6 | TFE | 0 |
| 12 | [Ru(p-cymene)Cl2]2 | AgSbF6 | TFE | 65 |
| 13 | [Cp*RhCl2]2 | AgSbF6 | TFE | 17f |
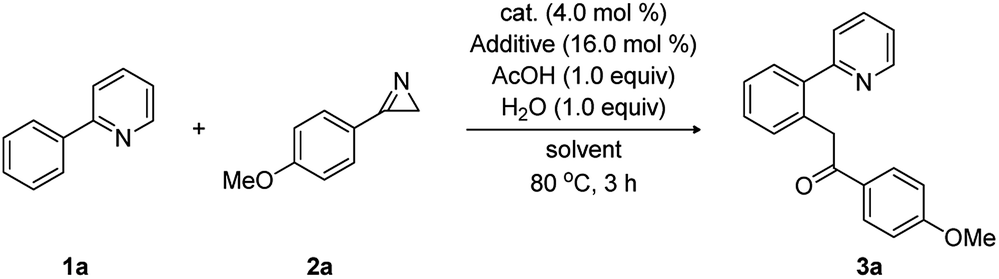
Whereas computer models are powerful in suggesting, identifying and comparing possible reaction mechanisms, as we demonstrated above, predicting the impact of subtle environmental changes, such as the nature of the solvent or counter ions to the overall yield, is exceedingly difficult.15 Unsatisfied with the relatively low yield of only 35% in DCE, we complemented our initial findings by more classical screening efforts and identified trifluoroethanol (TFE) as the optimal solvent. Other solvents, such as DCE, MeOH, hexafluoroisopropanol (HFIP), and tetrahydrofuran (THF), gave inferior results (entries 2–6). Several counter ion additives, including AgSbF6, AgNTf2, and AgPF6, were also examined and AgSbF6 gave the desired product in 73% yield (entries 4, 7, and 8). Interestingly, when [Cp*IrCl2]2 (4.0 mol%) and [Cp*CoCl2]2 (4.0 mol%) were used as catalyst, the reaction was totally ineffective (entries 10 and 11). However, [Ru(p-cymene)Cl2]2 (4.0 mol%) provided 3a in 65% yield (entry 12). The best result was obtained from the reaction of 1a (2.0 equiv.) with 2a (0.2 mmol, 1.0 equiv.) in the presence of [Cp*RhCl2]2 (4.0 mol%) with AgSbF6 (16.0 mol%), water (1.0 equiv.), and AcOH (1.0 equiv.) in TFE at 80 °C for 3 h, affording 3a in 86% yield (entry 9). Interestingly, attempts towards isolating the imine product under the anhydrous conditions were not successful (entry 13).
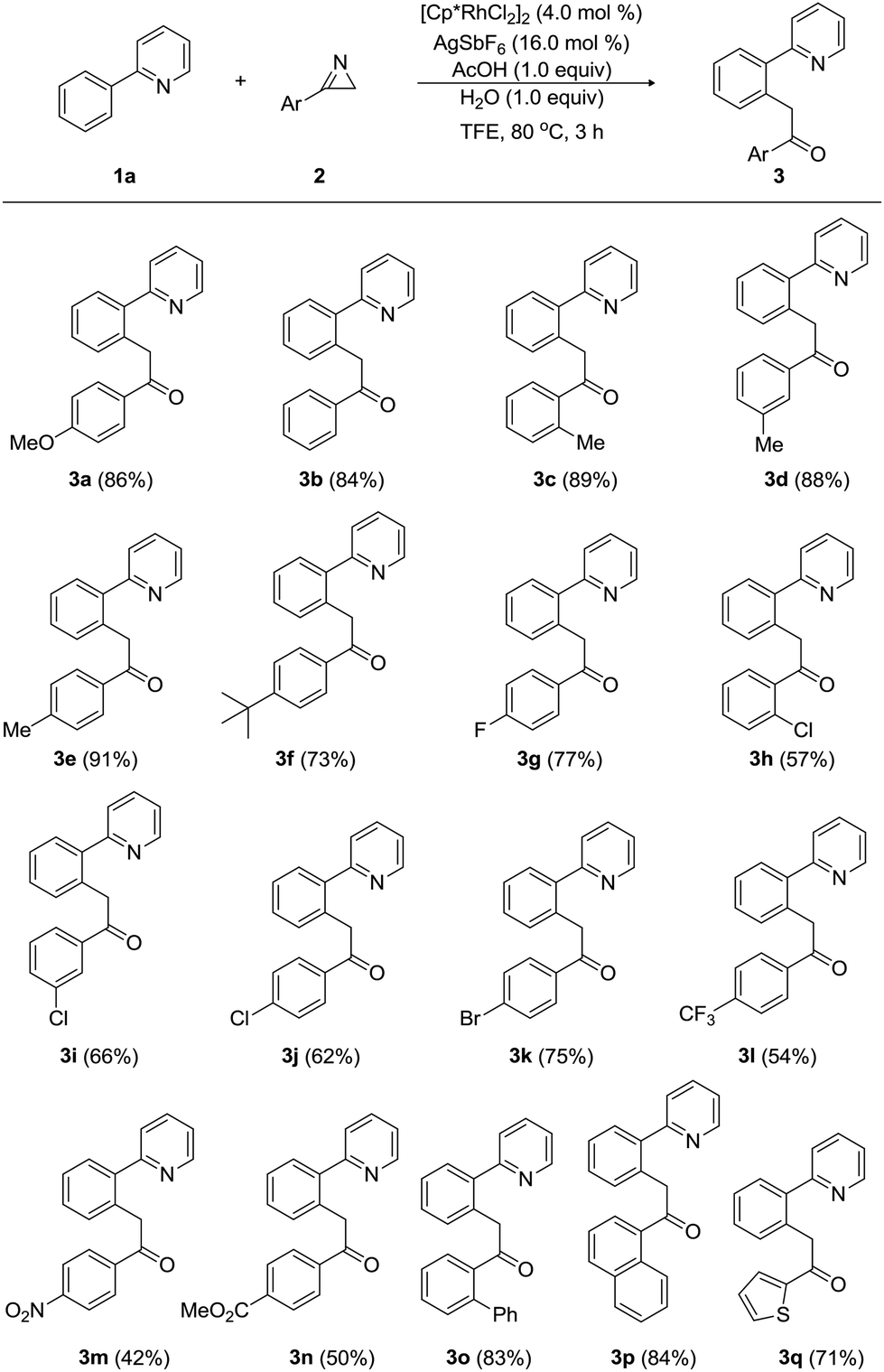
Next, we investigated the scope and limitation of Rh-catalyzed acylmethylation of 2-phenylpyridine (1a) with a wide range of 3-aryl-2H-azirines (2), as summarized in Table 2. When 3-phenyl-2H-azirine was treated with 1a under the optimum reaction conditions, product 3b was obtained in 84% yield. Electronic variations in the substituents on the aryl group of 3-aryl-2H-azirines (2) did not influence the reaction efficiency notably. Electron-donating groups, including 2-methyl, 3-methyl, and 4-methyl substituent, afforded the corresponding acylmethylated products (3c, 3d, and 3e) in high yields ranging from 88% to 91%. These results indicate that both steric and electronic effects were negligible in the case of 3-aryl-2H-azirines bearing electron-donating groups. Likewise, an electron-donating 4-tert-butyl group provided the desired pyridine (3f) in 73% yield. However, acylmethylation was slightly affected by electron-withdrawing groups on the aryl substituents of 3-aryl-2H-azirines (2). For example, a variety of electron-withdrawing groups, including 4-fluoro, 2-chloro, 3-chloro, 4-chloro, and 4-bromo groups, gave the corresponding acylmethylated 2-arylpyridines (3g, 3h, 3i, 3j, and 3k) in yields ranging from 57% to 77%. Strongly electron-withdrawing 4-trifluoromethyl, 4-nitro, and 4-ethoxycarbonyl-substituted 3-aryl-2H-azirines were less reactive and the products 3l, 3m, and 3n were produced in moderated yields ranging from 42% to 54%. Biphenyl- and 1-naphthyl-substituted 2H-azirines were found to be compatible with the reaction conditions, leading to the formation of 3o and 3p in 83% and 84% yields, respectively. The present method worked equally well with 2H-azirines possessing a thiophen-1-yl group to furnish 3q in 71% yield.
| a Reactions were carried out with 1a (2.0 equiv.) and 2 (0.2 mmol, 1.0 equiv.) in the presence of catalyst (4.0 mol%), additive (16.0 mol%), acetic acid (1.0 equiv.), and H2O (1.0 equiv.) in TFE (2.0 mL) at 80 °C for 3 h under a nitrogen atmosphere. |
|---|
|
Next, the substrate scope and the functional group tolerance with a variety of 2-arylpyridines (1) were investigated, as shown in Table 3. Modification of the substituents on the aryl ring at 2-position of 2-arylpyridines 1 did not influence the efficiency of the acylmethylation. Both electron-donating and electron-withdrawing groups on the 2-arylpyridines were compatible, thus equally affording the corresponding 2-acylmethyl-substituted arylpyridines in good yields. When 2-(m-tolyl)pyridine and 2-(p-tolyl)pyridine were treated with 3-(p-tolyl)-2H-azirine (2e), the desired acylmethylated products 5a and 5b were produced in 79% and 88% yields, respectively. The strongly electron-donating 4-methoxy group did not affect the efficiency of the reaction, and the desired product 5c was obtained in 89% yield. The transformation of 2-arylpyridine having 3,4-methylenedioxy group was also highly facile, providing 5d in 89% yield. Substrates with a halogen atom provided the desired compounds in good yields. Indeed, the 4-fluoro-substituted 2-phenylpyridine was reacted with 3-(p-tolyl)-2H-azirine (2e), furnishing the arylmethylated product 5e in 83% yield. 2-Arylpyridines possessing 3-chloro, 4-chloro, 4-trifluoromethyl, and 4-acetyl groups were smoothly acylmethylated to produce 5f–5i in good yields ranging from 61% to 74%. The tolerance of fluoro, chloro, and acetyl groups is very important, because these functional groups will allow further decoration and processing of the products. Pyridine substrates bearing 2-biphenyl and 2-naphthyl groups underwent the acylmethylation reaction, affording the corresponding products (5j and 5k) in 74% and 83% yields, respectively. Moreover, the arene is not limited to a benzene skeleton. 2-(Thiophene-1-yl)pyridine was applied to the present Rh-catalyzed acylmethylation, resulting in the production of 5l in 60% yield. When 2-arylpyridine possessing an estrone moiety was employed as the substrate, the acylmethylated product 5m was produced in 80% yield.
| a Reactions were carried out with 1 (2.0 equiv.) and 2e (0.2 mmol, 1.0 equiv.) in the presence of catalyst (4.0 mol%), additive (16.0 mol%), acetic acid (1.0 equiv.), and H2O (1.0 equiv.) in TFE (2.0 mL) at 80 °C for 3 h under a nitrogen atmosphere. |
|---|

|
Furthermore, tolerance of a wide range of substituents, including methyl, fluoro, ethoxycarbonyl, and acetyl, on the pyridine moiety was examined (Table 4). 5-Methyl-2-phenylpyridine underwent the acylmethylation with 3-(p-tolyl)-2H-azirine (2e), providing the desired product 5n in 83% yield without notably affecting the catalytic effectiveness. Substrate bearing 4-fluoro group on the pyridine ring of 2-phenylpyridine gave rise to 5o in acceptable yield. To our delight, the present acylmethylation proceeded despite the presence of an ethoxycarbonyl and acetyl group on the phenyl ring and afforded 5p and 5q in 67% and 68% yields, respectively.
| a Reactions were carried out with 1 (2.0 equiv.) and 2e (0.2 mmol, 1.0 equiv.) in the presence of catalyst (4.0 mol%), additive (16.0 mol%), acetic acid (1.0 equiv.), and H2O (1.0 equiv.) in TFE (2.0 mL) at 80 °C for 3 h under a nitrogen atmosphere. |
|---|

|
Although the DFT-calculated mechanism discussed above serves as a useful guide for understanding our experimental results and offers a plausible concept for the reaction mechanism, there are a number of unresolved issues: (i) we assumed that hydrolysis affords the final acylmethylation product, which should be confirmed. (ii) Our DFT-calculations employ a model Rh-catalyst carrying acetate ligands for convenience to start the catalytic cycle. This putative resting state of the catalyst also serves as the reference state with a relative free energy of zero kcal mol−1, which is used to evaluate the barriers of the initial C–H activation.16 This model reference state leads to a C–H activation barrier of only 17.6 kcal mol−1, whereas the free energy of the azirine-bound intermediate C is −5.0 kcal mol−1 and that affords a barrier for the concerted C–C coupling and azirine ring-opening to be 22.8 kcal mol−1. It is not clear how representative and reliable these barrier estimates are. Given that the reaction typically requires 80 °C and 3 hours, these barriers seem underestimated.
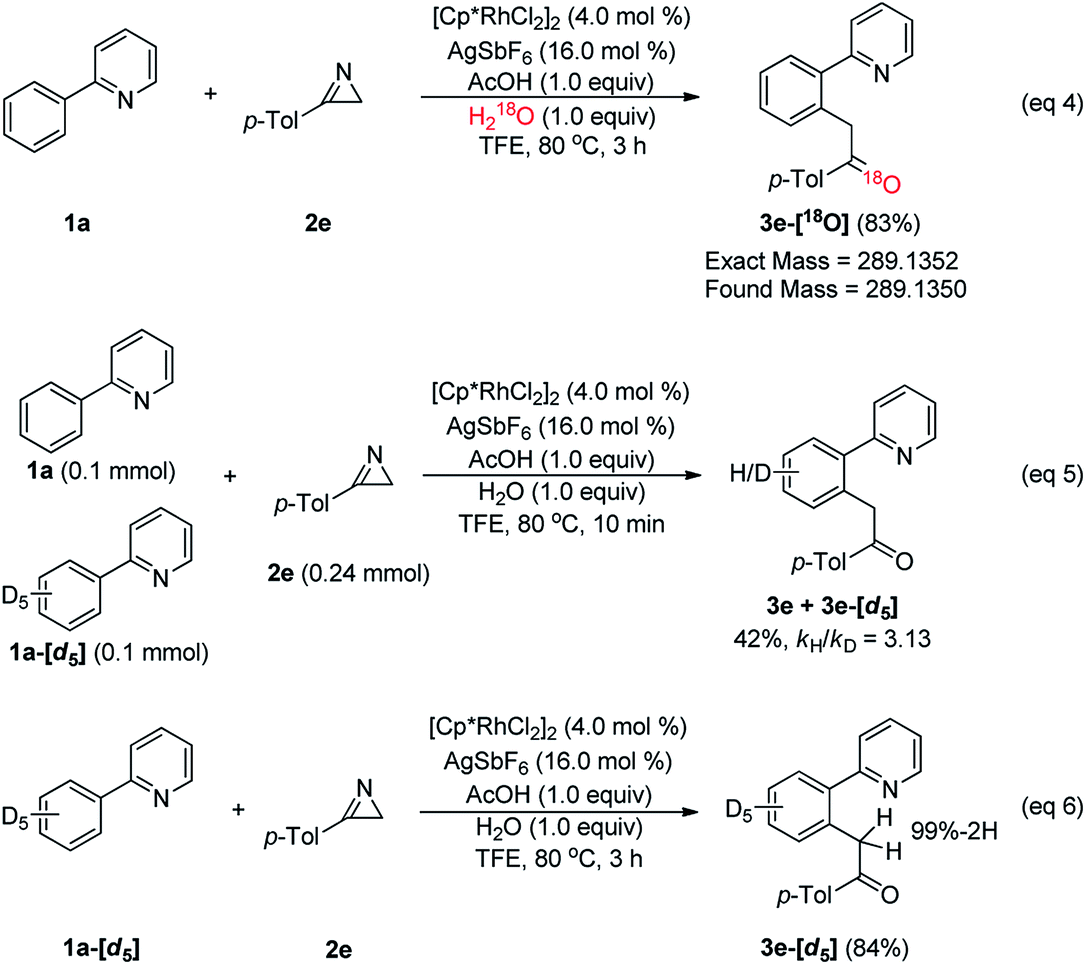
To answer these mechanistic questions that are challenging to answer using computations, a series of experiments were designed, as summarized in Scheme 2. First, isotopic labeling studies were performed to examine the source of oxygen in 3e. When 1a (2.0 equiv.) was treated with 2a (0.2 mmol, 1.0 equiv.) in the presence of [Cp*RhCl2]2 (4.0 mol%) with AgSbF6 (16.0 mol%), H218O (1.0 equiv.), and AcOH (1.0 equiv.) in TFE at 80 °C for 3 h, the corresponding 18O-inserted-ketone 3e-[18O] was produced in 83% yield (eqn (4)). These results confirm that the oxygen in the acylmethylation reaction originates from water, as we had assumed. Next, the kinetic isotope effect (KIE) was determined by utilizing the deuterated phenylpyridine substrate (eqn (5)). The KIE was found to be kH/kD = 3.13 from the intermolecular competition reaction17 between 1a and 1a-[d5]. These results indicate that the C–H cleavage at the ortho-position of 2-phenylpyridine is most likely involved in the rate-determining step. Although 1a-[d5] was treated with 2e under the optimum reaction conditions, the corresponding deuterium-inserted product at benzylic position was not detected (eqn (6)). This result implies that deuterium transfer through C–H activation did not occur.
 | ||
| Scheme 2 Experimental mechanistic studies. | ||
The KIE of 3.13 provides significant insight that our DFT-calculations are unable to capture. As discussed above, we employ the Rh-acetate model for convenience and stoichiometric consistency during the computer simulation. For the KIE study, the chloride derivative of the Rh-catalyst is used in combination with a silver salt, which removes the chloride by forming poorly soluble silver chloride deposits. The counter anion of the silver salt has a notable impact on the yield, as we showed above. DFT-calculations are not capable of incorporating these effects accurately into the mechanistic model. One meaningful result that can be utilized is the DFT-predicted energy difference of 8.9 kcal mol−1 between the product complex G and the recovered catalyst A upon release of the imine product 3a′. Ignoring the aforementioned complications with the silver salt and the chloride ligands, the calculated energy difference between G and A suggests that after the first catalytic cycle, the product complex G becomes the resting state of the catalytic cycle and, thus, should be taken as the reference for evaluating the C–H activation barrier. And because the energy of G is lower than that of the azirine-adduct C, the barrier of the C–C coupling reaction must also be referenced against G. The adjusted barriers for the C–H and C–C activations become 26.5 and 26.7 kcal mol−1, respectively. We note that these numbers are much more consistent with the observed reaction conditions of 80 °C. And because the computed barriers are nearly identical and our DFT-calculations are fundamentally unable to offer information about the collision factor of the Arrhenius equation that would allow for computing the rate of reactions, our DFT-model is unable to clearly identify the rate determining step. The KIE of 3.13 clarifies this point and strongly suggests that the C–H bond activation step is rate determining.
Scheme 3 summarizes the proposed mechanism incorporating the insights derived both from computations and experiments. We propose that the rate determining step is the C–H activation, giving rise to an experimental KIE value of 3.13 and turns the reactant complex A to the cyclometalated intermediate B. Coordination on 2H-azirine to Rh affords key intermediate C, which can ring-open the azirine via oxidative addition to form the rhodaheterocycle intermediate D. Subsequent insertion into the Rh–phenyl bond leads to the C–C coupling to give the product complex E. The alternative C–N coupling is found to be much less likely, as the migratory insertion for that process has a barrier that is ∼15 kcal mol−1 higher than the C–C coupling. The catalytic cycle closes via proto-demetalation with acetic acid to give the imine product 3a′, which is readily hydrolyzed to give the acylmethylated product 3a.
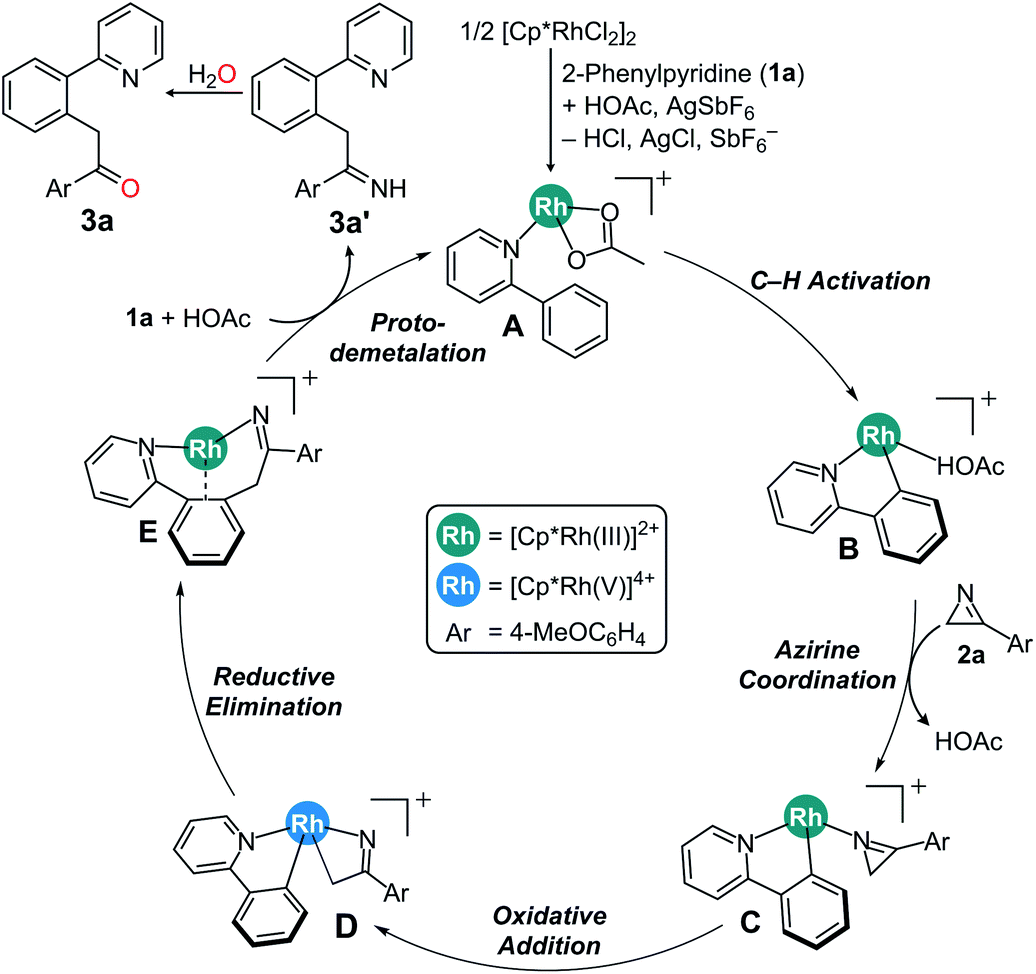 | ||
| Scheme 3 Proposed mechanism. | ||
This new methodology is attractive for the synthesis of biologically relevant heterocycles such as pyridoisoindole and pyridoisoqunolinone through cyclization of the acylmethylated compounds (Scheme 4). Reactant 3j was smoothly cyclized in the presence of Cu(OAc)2 (50.0 mol%) to provide pyridoisoindole 6a in 91% yield (eqn (7)).
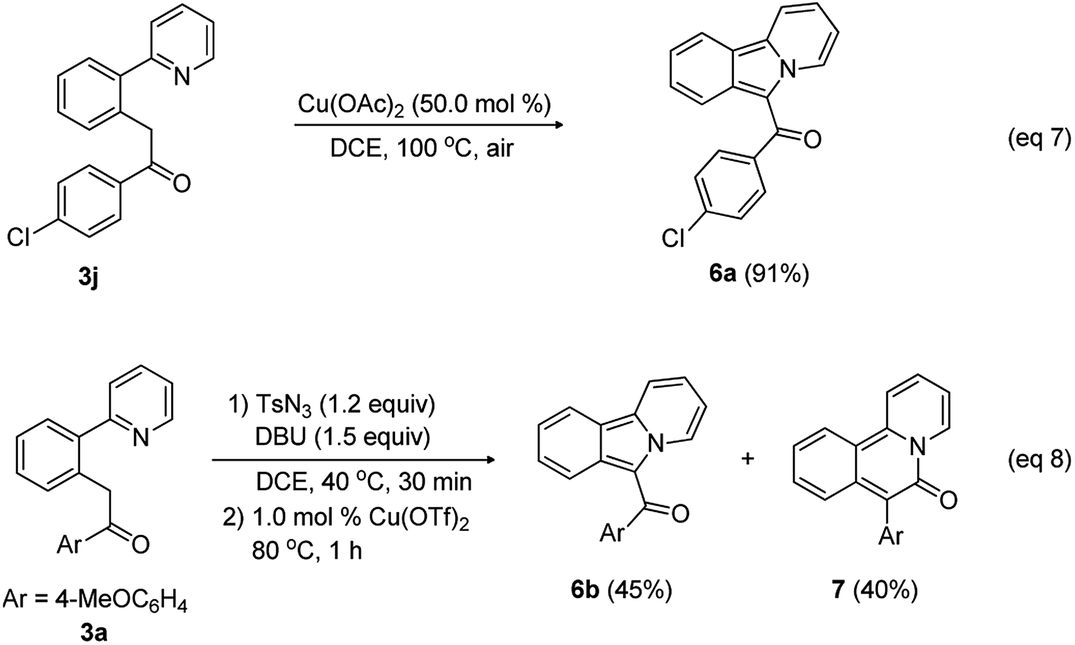 | ||
| Scheme 4 Synthetic applications. | ||
Because 2-arylpyridine bearing an acyldiazo group can be easily obtained from the diazotization of the corresponding acylmethyl-substituted 2-arylpyridine, we envisioned that the intramolecular cyclization using in situ generated diazo intermediate should be possible in a convenient one-pot procedure. To test this idea, 1-(4-methoxyphenyl)-2-(2-(pyridin-2-yl)phenyl)ethan-1-one (3a), tosyl azide, DBU, and DCE were placed in a reaction vessel, and the reaction mixture was stirred at 40 °C. After 30 min, the reaction mixture was treated with 1.0 mol% Cu(OTf)2 and stirred at 80 °C for 1 h, leading to the formation of pyridoisoindole 6b (45%) and pyridoisoquinolinone 7 (40%) through Curtius rearrangement (eqn (8)).
Conclusions
In conclusion, we developed a novel Rh-catalyzed synthetic method for a wide range of acylmethyl-substituted 2-arylpyridine derivatives using 3-aryl-2H-azirines as the reaction partner. The oxygen in the acylmethylation reaction was confirmed to originate from water, which hydrolyzes the initial imine product. C–H cleavage at the ortho-position of 2-phenylpyridine is most likely the rate-limiting step. Initially, we had aimed at developing a C–N coupling reaction, but DFT-calculations and screening results clearly suggest that C–C coupling is much faster and preferable after the azirine substrate is ring-opened at the metal center of the catalyst. The present method is highly efficient and selective, displays a broad substrate scope and a high tolerance of various functional groups, including fluoro, chloro, bromo, ketone, ester, nitro, alkyl, alkoxy, trifluoromethyl, 2-naphthyl-, and thiophen-1-yl. The mechanism was elucidated by combining computational and experimental studies, where the weaknesses and strengths of the two complementary methods of mechanistic inquiries were combined to obtain a precise and convincing mechanism for this novel reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is dedicated to Professor Chul-Ho Jun (Yonsei University) for his honorable retirement. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (2011-0018355 and 2017R1A4A1015405). We thank the Institute for Basic Science in Korea for financial support (IBS-R10-A1).Notes and references
- (a) J. Royer, Asymmetric Synthesis of Nitrogen Heterocycles, Wiley-VCH Verlag, Weinheim, 2009 CrossRef; (b) X.-F. Wu, Transition Metal-Catalyzed Heterocycle Synthesis via C–H Activation, John Wiley & Sons, Weinheim, 2015 Search PubMed.
- (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed.
- For selected literatures on the usage of acyl azide, see: (a) J. Ryu, J. Kwak, K. Shin, D. Lee and S. Chang, J. Am. Chem. Soc., 2013, 135, 12861 CrossRef CAS PubMed; (b) J. Peng, Z. Xie, M. Chen, J. Wang and Q. Zhu, Org. Lett., 2014, 16, 4702 CrossRef CAS.
- (a) T. Curtius, Ber. Dtsch. Chem. Ges., 1890, 23, 3023 CrossRef; (b) E. F. V. Scrivenk and K. Turnbull, Chem. Rev., 1988, 88, 297 CrossRef; (c) D. Li, T. Wu, K. Liang and C. Xia, Org. Lett., 2016, 18, 2228 CrossRef CAS; (d) H. Lebel and O. Leogane, Org. Lett., 2005, 7, 4107 CrossRef CAS PubMed.
- K. Shin, J. Ryu and S. Chang, Org. Lett., 2014, 16, 2022 CrossRef CAS PubMed.
- For selected examples on the usage of sulfonyl azide, see: (a) E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797 CrossRef CAS PubMed; (b) J. Y. Kim, S. H. Park, J. Ryu, S. H. Cho, S. H. Kim and S. Chang, J. Am. Chem. Soc., 2012, 134, 9110 CrossRef CAS; (c) B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1491 CrossRef CAS; (d) D. Lee, Y. Kim and S. Chang, J. Org. Chem., 2013, 78, 11102 CrossRef CAS PubMed; (e) B. Zhu, X. Cui, C. Pi, D. Chen and Y. Wu, Adv. Synth. Catal., 2016, 358, 326 CrossRef CAS.
- For selected literature references on the usage of dioxazolone, see: (a) Y. Park, K. T. Park, J. G. Kim and S. Chang, J. Am. Chem. Soc., 2015, 137, 4534 CrossRef CAS PubMed; (b) Y. Park, J. Heo, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2016, 138, 14020 CrossRef CAS PubMed; (c) H. Wang, G. Tang and X. Li, Angew. Chem., Int. Ed., 2015, 54, 13049 CrossRef CAS PubMed; (d) F. Wang, H. Wang, Q. Wang, S. Yu and X. Li, Org. Lett., 2016, 18, 1306 CrossRef CAS PubMed; (e) Y. Liang, Y.-F. Liang, C. Tang, Y. Yuan and N. Jiao, Chem.–Eur. J., 2015, 21, 16395 CrossRef CAS PubMed; (f) R. Mei, J. Loup and L. Ackermann, ACS Catal., 2016, 6, 793 CrossRef CAS; (g) H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 10386 CrossRef CAS PubMed; (h) X. Wang, A. Lerchen and F. Glorius, Org. Lett., 2016, 18, 2090 CrossRef CAS PubMed; (i) N. Barsu, M. A. Rahman, M. Sen and B. Sundararaju, Chem.–Eur. J., 2016, 22, 9135 CrossRef CAS; (j) J. Wang, S. Zha, K. Chen, F. Zhang, C. Song and J. Zhu, Org. Lett., 2016, 18, 2062 CrossRef CAS; (k) B. Jeon, U. Yeon, J.-Y. Son and P. H. Lee, Org. Lett., 2016, 18, 4610 CrossRef CAS.
- (a) T. Ryu, Y. Baekk and P. H. Lee, J. Org. Chem., 2015, 80, 2376 CrossRef CAS; (b) Y. Baek, C. Maeng, H. Kim and P. H. Lee, J. Org. Chem., 2018, 83, 2349 CrossRef CAS PubMed.
- See ESI for details.†.
- (a) A. Padwa and T. Stengel, Tetrahedron Lett., 2004, 45, 5991 CrossRef CAS; (b) F. Palacios, A. M. O. de Retana, E. M. de Marigorta and J. M. de los Santos, Eur. J. Org. Chem., 2001, 2001, 2401 CrossRef; (c) S. Chiba, G. Hattori and K. Narasaka, Chem. Lett., 2007, 36, 52 CrossRef CAS; (d) D. A. Candito and M. Lautens, Org. Lett., 2010, 12, 3312 CrossRef CAS PubMed; (e) T. Izumi and H. Alper, Organometallics, 1982, 1, 322 CrossRef CAS.
- (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (b) J. L. Roizen, M. E. Harvey and J. D. Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed; (c) S. Jana, M. D. Clements, B. K. Sharp and N. Zheng, Org. Lett., 2010, 12, 3736 CrossRef CAS PubMed.
- (a) M. Barday, C. Janot, N. H. Halcovitch, J. Muir and C. Aïssa, Angew. Chem., Int. Ed., 2017, 56, 13117 CrossRef CAS PubMed; (b) Y. Xu, X. Zhou, G. Zheng and X. Li, Org. Lett., 2017, 19, 5256 CrossRef CAS PubMed; (c) H. Oh, S. Han, A. K. Pandey, S. H. Han, N. K. Mishra, S. Kim, R. Chun, H. S. Kim, J. Park and I. S. Kim, J. Org. Chem., 2018, 83, 4641 CrossRef PubMed; (d) G. Zheng, M. Tian, Y. Xu, X. Chen and X. Li, Org. Chem. Front., 2018, 5, 998 RSC; (e) S. Ji, K. Yan, B. Li and B. Wang, Org. Lett., 2018, 20, 5981 CrossRef CAS PubMed.
- (a) S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492 CrossRef CAS PubMed; (b) D. L. Davies, S. M. A. Donald, O. Al-Duaij, S. A. Macgregor and M. Pölleth, J. Am. Chem. Soc., 2006, 128, 4210 CrossRef CAS; (c) D. L. Davies, S. M. A. Donald, O. Al-Duaij, J. Fawcett, C. Little and S. A. Macgregor, Organometallics, 2006, 25, 5976 CrossRef CAS; (d) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC; (e) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (f) L. Li, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 3492 CrossRef CAS; (g) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS; (h) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC; (i) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed.
- (a) D. J. Tantillo, Applied Theoretical Organic Chemistry, World Scientific Publishing Europe Ltd, London, 2018 CrossRef; (b) S. M. Bachrach, Computational Organic Chemistry, John Wiley & Sons, Inc, Hoboken, New Jersey, 2014 CrossRef.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101 CrossRef CAS PubMed.
- (a) W. D. Jones, Acc. Chem. Res., 2003, 36, 140 CrossRef CAS PubMed; (b) F. Kakiuchi, Y. Matsuura, S. Kan and N. Chatani, J. Am. Chem. Soc., 2005, 127, 5936 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05142a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |