
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight triggers molecular shuttling in rotaxanes: control over proximity and charge recombination†
Maximilian
Wolf
a,
Ayumu
Ogawa
bc,
Mareike
Bechtold
ae,
Maxime
Vonesch
 c,
Jennifer A.
Wytko
c,
Koji
Oohora
b,
Stéphane
Campidelli
d,
Takashi
Hayashi
*b,
Dirk M.
Guldi
*a and
Jean
Weiss
*c
c,
Jennifer A.
Wytko
c,
Koji
Oohora
b,
Stéphane
Campidelli
d,
Takashi
Hayashi
*b,
Dirk M.
Guldi
*a and
Jean
Weiss
*c
aFriedrich-Alexander-Universität Erlangen-Nürnberg, Department of Chemistry and Pharmacy, Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: dirk.guldi@fau.de
bDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita 565-0871, Japan. E-mail: thayashi@chem.eng.osaka-u.ac.jp
cInstitut de Chimie de Strasbourg, UMR 7177 CNRS-Université de Strasbourg, 4, rue Blaise Pascal, 67000 Strasbourg, France. E-mail: jweiss@unistra.fr
dLICSEN, NIMBE, CEA, CNRS, Université Paris-Saclay, CEA Saclay, 91191 Gif-sur-Yvette Cedex, France
eInstitute of Inorganic Chemistry Karlsruhe Institute of Technology, Engesserstraße 15, D-76131 Karlsruhe, Germany
First published on 27th February 2019
Abstract
We present the synthesis of novel rotaxanes based on mechanically interlocked porphyrins and fullerene and their advanced investigations by means of photophysical measurements. To this end, a fullerene-capped dumbbell-type axle containing a central triazole was threaded through strapped (metallo)porphyrins—either a free-base or a zinc porphyrin. Femtosecond-resolved transient absorption measurements revealed charge-separation between the porphyrin and fullerene upon light excitation. Solvent polarity and solvent coordination effects induced molecular motion of the rotaxanes upon charge separation and enabled, for the first time, subtle control over the charge recombination by enabling and controlling the directionality of shuttling.
Introduction
The field of topologically sophisticated molecules and molecular motions has been the focus of renewed attention since the awarding of the Nobel Prize of chemistry to Sauvage, Stoddart, and Feringa.1 Following the first templated synthesis of a catenane2 up until the design of devices utilizing molecular shuttles,3 lessons were learned about both the synthetic methods available to make sophisticated molecular scaffolds,4,5 and the design of properties related to molecular motions. Molecular motions in catenanes and rotaxanes have been the focus of attention for decades6–10 and theoretical investigations correlated to experiments are progressively shining light on the dynamic behavior in rotaxane structures.11–13 Several methods, such as cation,14 anion15 or guest binding,16 pH changes,17 redox stimuli,18,19 solvents changes20,21 and light irradiation22–24 have been envisioned to trigger motion mostly in rotaxanes. In designing molecular shuttles powered by light, inspiration is taken from nature, which uses porphyrin derivatives in chloroplasts as light harvesters and electron donors.25Interest in interlocked photodyads has evolved over the years after initially focusing on the effects of the peculiar topology on the rate of charge separation (CS) and charge recombination.7 Combining porphyrins with C60, which is one of the best-known organic electron acceptors, has led to a plethora of electron donor–acceptor systems.26 In short, the interest in fullerene-based electron donor–acceptor systems arises from the small reorganization energy of fullerenes in electron transfer reactions. As a result, back electron transfer (BET) occurs in the Marcus inverted region and, thus, leads to long-lived charge-separated states (CSS).27 The drawback of simple, two-component, electron donor–acceptor components is that BET is usually fast. By introducing additional electron donors and/or acceptors, it is possible to achieve multi-step electron transfer reactions that lead to longer-lived charge-separated state (CSS).28 Many such systems have been reported.29,30 The disadvantage of large systems is that evaluating the deactivation pathways is non-trivial and undesired deactivation pathways are likely added as well.
These considerations led to the approach of constructing interlocked structures with a scaffold containing fullerenes and porphyrins.31 On many occasions, porphyrins and fullerenes have been utilized as stoppers in pseudorotaxanes or catenanes, but only in very few cases in rotaxanes. In none of the rotaxane cases does the photoactive porphyrin constitute the shuttle, which moves along the axle.32–34 The current work is a step-change as it describes the preparation and the properties of a rotaxane containing a strapped porphyrin as the ring and a fullerene stopper at the end of a mobile dumbbell. The photophysical properties of the two rotaxanes, that is, 1 and 1Zn, in which the dumbbell bears a fullerene derivative as electron acceptor and the rings respectively comprise a free-base porphyrin (H2P) or a zinc porphyrin (ZnP) as the photosensitizer, are compared. In the H2P-based rotaxane 1, the dumbbell is likely to glide freely within the ring. In the presence of zinc in, for example, the porphyrin-based rotaxane (1Zn), the position of the dumbbell is fixed by metal–ligand coordination and can be perturbed by the use of coordinating ligands. The variation in the lifetime of the charge-separated state provides a means of detecting the gliding of the dumbbell within the strapped porphyrin ring. This work provides, to the best of our knowledge, the first example of photoactive rotaxanes in which the directional or random motion of the shuttle is triggered by an intramolecular photoinduced charge transfer. Past work was mainly based on intermolecular rather than intramolecular charge transfer, either electrochemical or photochemical. As such, the use of light enables control over energy or charge transfer processes.6,35–38
Results and discussion
Synthesis
The rotaxanes studied in this work were prepared according to Scheme 1, using a recently reported tandem active metal templated copper alkyne–azide cyclization (CuAAC) reaction, in which zinc and copper promote the formation of dumbbells within a phenanthroline-strapped porphyrin.39 Azide stopper 2,40 fullerene derivative 3,41 and phenanthroline-strapped porphyrin 4Zn,42,43 were synthesized according to reported methods. Compounds 3, 4, and 4Zn also served as references for the photophysical studies. To prepare the free base rotaxane, first the copper(I)–phenanthroline complex of the strapped porphyrin 4Zn was generated in situ. Reaction of this complex with azide 2 and acetylene 3, followed by removal of both the copper and zinc ions using EDTA and then CF3CO2H, afforded free base rotaxane 1 in 48% yield. The removal of both metals from the rotaxane facilitated the purification step. The porphyrin was subsequently remetallated with zinc(II) acetate to provide rotaxane 1Zn. | ||
| Scheme 1 Synthesis of the studied compounds. | ||
Steady-state absorption and fluorescence
Absorption experiments were conducted with Zn-tetraphenylporphyrin (ZnTPP) as reference. To determine extinction coefficients, steady-state absorption spectra were measured in a concentration range from 10−8 to 10−6 M to prevent aggregation, on one hand, and to stay within the boundaries of the Lambert–Beer law, on the other hand (Fig. 1).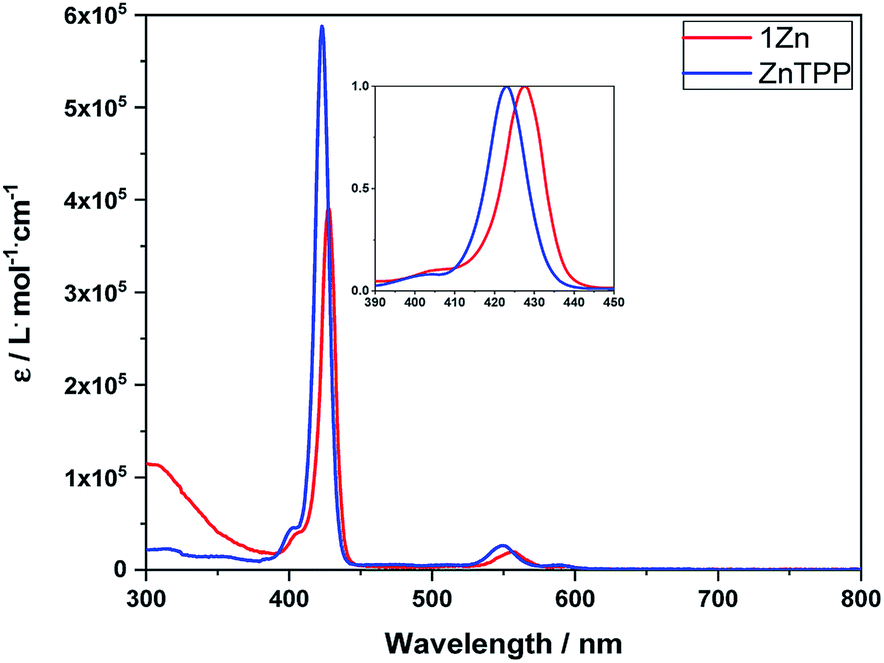 | ||
| Fig. 1 Steady-state absorption spectra of 1Zn (red) and ZnTPP (blue) in toluene. Inset: Normalized Soret absorption bands to highlight the red-shifted absorption. | ||
The absorption spectra of 1Zn in toluene reveal the typical porphyrin features including a Soret-band absorption at ∼420 nm and two Q-band absorptions at ∼550 and 590 nm. In addition, in the UV region below 400 nm, absorptions of the fullerene and the phenanthroline strap are discernable. Overall, all of the porphyrin absorption features are red-shifted by up to 5 nanometers in 1Zn relative to the ZnTPP reference. This shift is due to the complexation of triazole of the axle by the Zn-center leading to its penta-coordination.43–47 The extinction coefficient of the Soret-band is in the range of 105 M−1 cm−1. For 1, absorption patterns typical of H2TPP were observed in the visible region of the spectrum. Like what was seen for 1Zn, high energy absorptions (<400 nm) stem from C60 and the phenanthroline strap (see ESI,† Fig. S1).
Only negligible impact is seen in the absorption spectra of 1Zn in benzonitrile or in toluene, to which 500 equivalents of pyridine were added. Here, the additional red-shift is just 0.5 nm. Notably, in pyramidal, penta-coordinated ZnP-complexes, the Zn-center is known to be displaced out of the porphyrin plane towards the ligand.44–48 In turn, the Zn-center in 1Zn is not accessible from the top-side and strongly sterically encumbered from the bottom-side. The top-side is blocked by an overall steric hindrance stemming from the phenanthroline strap and the axle. The bottom-side lies within the inverted pyramid. Both effects result in very small binding constants for the hexa-coordination of the Zn-center.
In the steady-state fluorescence experiments, porphyrin fluorescence quantum yields of 0.0009, 0.0054, and 0.0014 for 1Zn in toluene, chlorobenzene, and benzonitrile, respectively, indicate a nearly quantitative quenching. In comparison to reference 4Zn, the quenching is as high as 0.97 in the aforementioned solvents. At this point, we infer the activation of non-radiative deactivation pathways, such as charge separation in 1Zn, which are absent in the ZnP reference. The same picture emerges for 1 (see ESI,† Table S1).
Electrochemistry and thermodynamic driving forces
Square wave voltammetry was performed on 1Zn in ortho-dichlorobenzene (o-DCB) with 0.1 M tetrabutylammonium perchlorate as supporting electrolyte. In the potential window of o-DCB, e.g. from −2.50 to 1 V vs. Ag/AgNO3, three fullerene reductions were discernable at −2.08, −1.52, and −1.16 V. In addition, the porphyrin was oxidized at +0.21 and 0.67 V and reduced at −1.69 V (see ESI,† Fig. S2).By combining the results from the steady-state spectroscopy and electrochemistry, the thermodynamic driving forces for charge transfer, e.g. charge separation and charge recombination, were calculated. The absorption and fluorescence data were used to derive the singlet excited state energy of 1Zn as 2.09 eV. The energy of the charge-separated state was calculated using the first porphyrin oxidation and the first fullerene reduction of 1Zn. Implicit in the difference between these values is the driving force for the charge separation from the excited state. The only correction required was for the dielectric constant of the medium as described in the literature;49 results are presented in Table 1. For the free base rotaxane 1, analogous calculations and results are listed in the ESI,† Fig. S3 and Table S2.
| Solvent | εS | ΔGCS in eV | ΔGCR-GS in eV | ΔGCR-3C60 in eV | ΔGCR-3ZnP in eV |
|---|---|---|---|---|---|
| a The dielectric continuum model, which was employed to calculate the driving forces, overestimates the influence of low polarity solvents. | |||||
| Tol | 2.38 | −0.16a | −1.93a | −0.43 | −0.4 |
| PhCl | 5.62 | −0.67 | −1.42 | 0.08 | 0.11 |
| PhCN | 25.2 | −0.96 | −1.13 | 0.37 | 0.4 |
The triplet excited state energies of both the Zn-porphyrin and the fullerene, with values of 1.53 or 1.50 eV, respectively,51 are higher in energy than the calculated energy of the ZnP˙+−C60˙− charge-separated state in 1Zn in chlorobenzene and benzonitrile. As such, charge recombination is expected to populate the ground state rather than a triplet excited state. In toluene, however, the triplet excited state of the fullerene is observed as a product of charge recombination (vide infra), as the charge-separated state is increased in energy. In 1, the charge-separated state is sufficiently high in energy to populate either the triplet excited state of C60 or that of H2P (see Table S2, ESI†), and accordingly, charge recombination results in these states as the predominant pathway of deactivation.
Transient absorption spectroscopy
Two different experimental setups were used to photoexcite the porphyrins at their Soret-band absorptions at either at 420 or 430 nm. First, time delays between 0 and 6000 ps and a time resolution of 150 fs were employed; second, time delays between <1 ns and 400 μs and a time resolution of ∼1 ns were used.In the ZnTPP reference, the second singlet excited state is formed instantaneously as demonstrated by its characteristic transient maxima at 450 and 580 nm as well as a broad absorption from 600 to 700 nm. All of the aforementioned were accompanied by bleaching of Q-band absorptions at 555 and 595 nm. Internal conversion (IC) with 5 × 1011 s−1 and intersystem crossing (ISC) with 5 × 108 s−1 to the first singlet excited state and the triplet excited state, respectively, constitute the deactivation pathways following the initial excitation. Spectral characteristics of the first singlet excited state are transient maxima at 450, 580, 620, 700 and 1260 nm, accompanied by transient minima at 555, 600 and 660 nm. The minima are due to ground state bleaching and stimulated emission. For the triplet excited state, the following features were observed: a maximum at 470 nm, ground state bleaching at 555 and 595 nm, and a broad and featureless absorption across the visible regions, with an 840 nm maximum.
Based on the thermodynamics of 1Zn, charge separation should afford the one-electron oxidized porphyrin (ZnP˙+) and the one-electron reduced fullerene (C60˙−), in addition to the deactivation cascade seen in the ZnTPP reference. Population of the second singlet excited state 1**ZnP is followed by IC to the first singlet excited state 1*ZnP, from where the further reaction steps occur. The corresponding excited state deactivation pathways are summarized in Fig. 2.
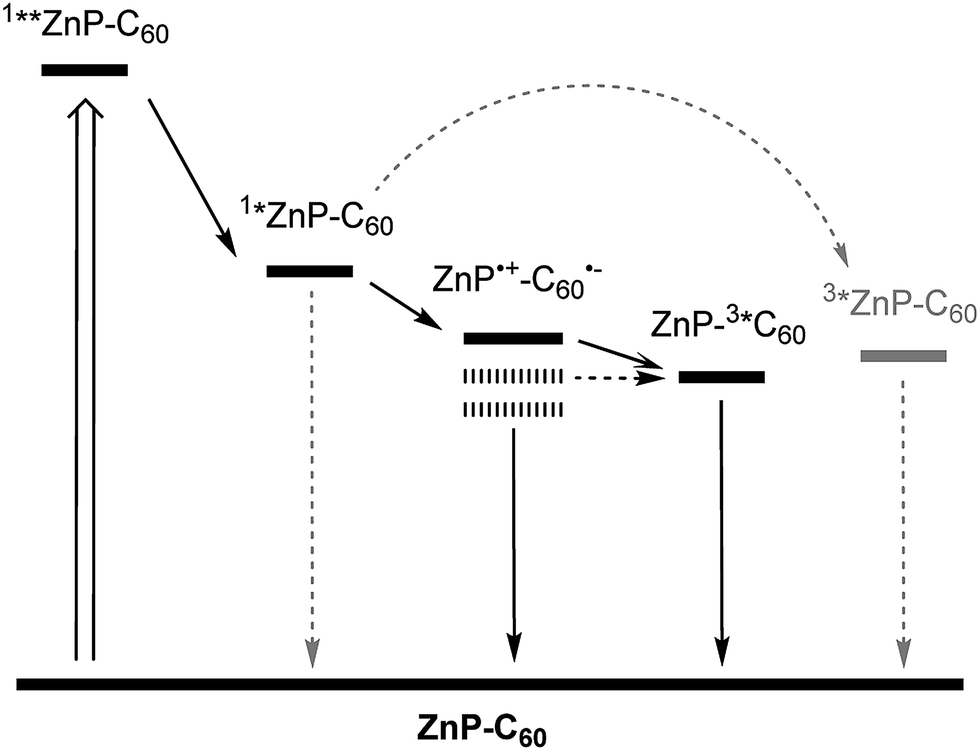 | ||
| Fig. 2 Summary of the deactivation pathways for photoexcited 1Zn in toluene (bold bars). Lower CSS energies for chlorobenzene and benzonitrile indicated by dashed bars. Minor pathways are indicated by dashed lines. | ||
Of relevance for the analysis are the fingerprint features of the one-electron oxidized porphyrin (ZnP˙+) and the one-electron reduced fullerene (C60˙−), with peaks at 410 and 1010 nm respectively,52–54 where neither contributions from the porphyrin nor the fullerene excited states render the kinetic analyses ambiguous. The analysis of the transient absorption spectra was conducted with global target analysis (GTA), taking into account the aforementioned spectral and kinetical observations and calculated energetics. GTA was performed via GloTarAn,55 a graphical user interface for the statistics package TIMP56 for R, which is especially designed for analyzing multiway spectroscopic data. Importantly, solvents of different polarity, i.e., toluene, chlorobenzene, and benzonitrile, were used. For the first two solvents, a four-species model was sufficient to fit the experimental data, whereas in benzonitrile a five-species model was necessary, which will be rationalized hereafter.
Even from the contour plots across the 400 to 1200 nm range at time delays up to 100 μs, it is evident that the 410 nm fingerprint of the one-electron oxidized porphyrin (ZnP˙+) and the 1010 nm fingerprint of the one-electron reduced fullerene (C60˙−) show vastly different lifetimes in apolar toluene (Fig. 3) and polar benzonitrile (Fig. 4). Concomitant with the decay of theZnP˙+ and C60˙− signatures at 410/450/650 and 1010 nm, respectively, is the population of the C60 triplet excited state in the form of the 700 nm fingerprint (also see ESI,† Fig. S4). Based on the intensity of this signal, the quantum yield of 3*C60 is estimated to be 20% in toluene, around 5% in chlorobenzene, and 0% in benzonitrile. The time absorption profiles at 410 and 1010 nm provided in Fig. 5 and 6 clearly depict the dynamics of the charge recombination processes. A clear monoexponential formation of the charge-separated state was observed—in all solvents and in all solvent combinations (see ESI,† Fig. S30–S39).
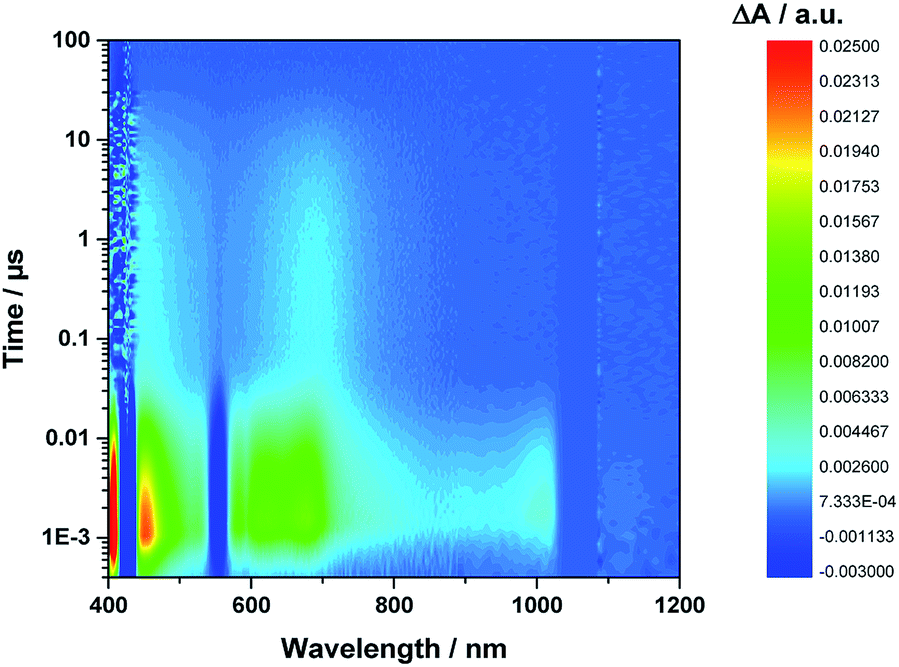 | ||
| Fig. 3 Contour plot of transient absorption measurements of 1Zn in toluene—please note the detector change at around 900 nm and the probe fundamental at around 1064 nm. | ||
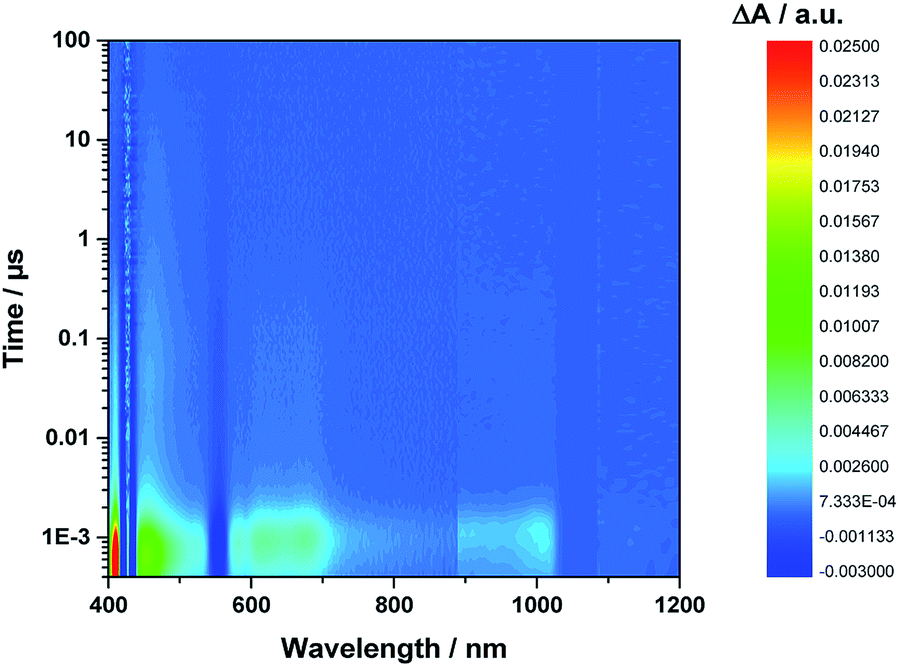 | ||
| Fig. 4 Contour plot of transient absorption measurements of 1Zn in benzonitrile—please note the detector change at around 900 nm and the probe fundamental at around 1064 nm. | ||
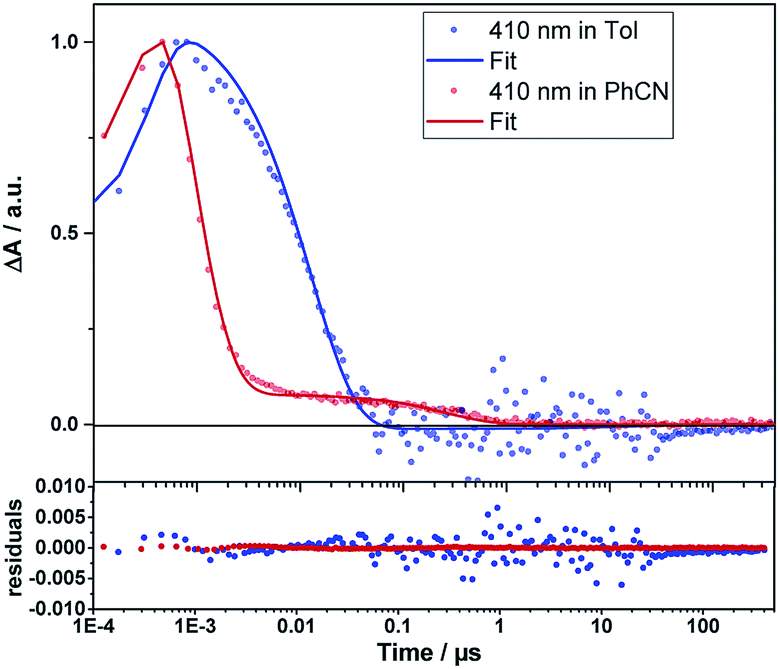 | ||
| Fig. 5 Time traces (dots) and the corresponding fits derived from GTA (lines) of the ZnP˙+ signal of 1Zn in toluene and benzonitrile. Bottom: fit residuals. | ||
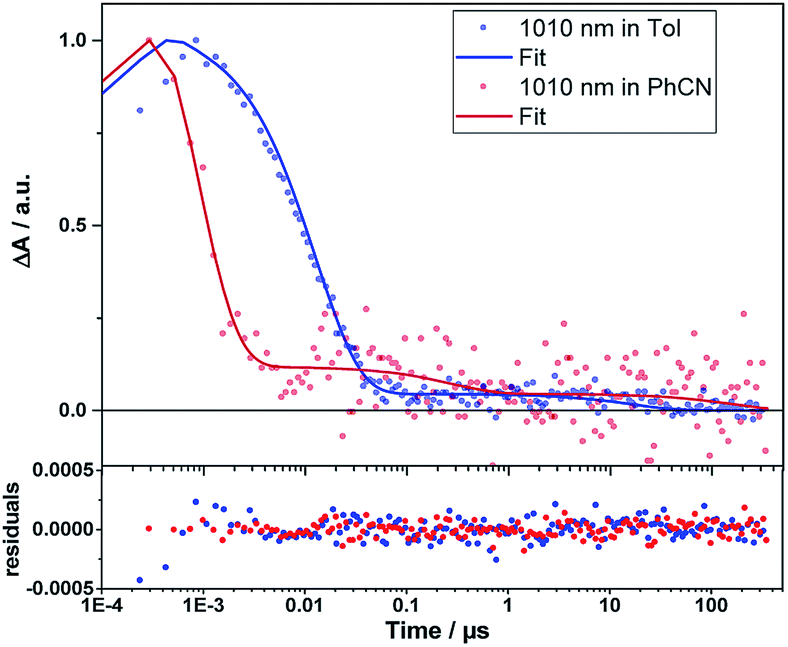 | ||
| Fig. 6 Time traces (dots) and the corresponding fits derived from GTA (lines) of the C60˙− signal of 1Zn in toluene and benzonitrile. Bottom: fit residuals. | ||
A closer look at the decay curves reveals that the charge-separated state (ZnP˙+−C60˙−) decay is biphasic in benzonitrile with a short-lived (<1 ns) and a long-lived (∼280 ns) component. In contrast, the decay is mono-phasic in toluene, with a lifetime that is longer than the short-lived component in benzonitrile. All the excited/charge-separated state lifetimes for 1Zn are listed in Table 2; those for 1 are provided in the ESI,† Table S3.
| Lifetimes of observed species | Toluene | Toluene + pyridine | Chlorobenzene | Chlorobenzene + pyridine | Benzonitrile |
|---|---|---|---|---|---|
| a State not observed. | |||||
| 1**ZnP–C60/ps | 0.44 | 0.59 | 1.14 | —a | 0.64 |
| 1*ZnP–C60/ps | 20.8 | 28.2 | 25.5 | 24.5 | 26.4 |
| ZnP˙+−C60˙−short/ns | 12.8 | 1.77 | 2.84 | 2.42 | 0.76 |
| ZnP˙+−C60˙−long/ns | —a | 9.42 | —a | 256 | 278 |
| ZnP–3*C60/μs | 16.4 | 12.7 | 13.3 | 14.2 | —a |
| 3*ZnP–C60/μs | —a | —a | 200 | 166 | 164 |
In other words, in toluene a short-lived charge-separated state is formed, which then deactivates via3C60 to the ground state, whereas in benzonitrile, an additional long-lived charge-separated state, accounting for approximately 10% of the amplitude of the fingerprints, is registered (also see ESI,† Fig. S5). A rationale for the short-lived component is based on the Marcus theory for electron transfer, in which lowering the energy of the charge-separated state in benzonitrile relative to toluene goes hand-in-hand with a faster deactivation. A similar consideration fails, however, to explain our observation of the long-lived component in benzonitrile.57 The coexistence of singlet and triplet CSS as a means to rationalize the short- and long-lived components is ruled out, as CS implies the conservation of spin.58 Alternatively, CS from the triplet excited states of ZnP or C60 to form a triplet CSS would be energetically feasible in benzonitrile (see Table 1). However, the population of the triplet excited states of either ZnP or C60 is negligible due to the highly efficient CS starting from the ZnP singlet excited state. Even an interconversion between singlet CSS and triplet CCS is unlikely. In short, any of the aforementioned pathways toward a triplet CSS are unlikely to play a role.
An alternative scenario, namely a change in electron donor–acceptor distance, must be considered. In particular, an increased spatial separation59 between electron donor and acceptor is likely induced by a movement of the porphyrin away from the fullerene via shuttling along the rotaxane's axle. Theoretical calculations reveal that the resulting difference in electron donor acceptor distance (edge to edge) is up to >8 Å (see ESI,† Fig. S9). Coordination of the Zn-center constitutes a bottleneck. For example, triazole of the axle is likely to form a coordination bond with the Zn-center, which, in turn, locks the position of the porphyrin close to the triazole—vide infra. Coordinating solvents such as benzonitrile are unlikely to trigger shuttling along the rotaxane axle because, in the ground state, simultaneous coordination of triazole and benzonitrile or even displacement of triazole by benzonitrile is highly unlikely.60 In stark contrast, oxidation of ZnP by means of charge separation results in a notable distortion of the ZnP scaffold, for which either S4-saddle or D2-ruffle geometries are reported.61,62 Oxidation also renders ZnP more electron deficient and more electrophilic.63 Distortion of the ZnP scaffold facilitates the bottom-side attack on the Zn-center. Both of these facts lead to a scenario in which de-coordination of the weak electron donating triazole and subsequent coordination of a more electron donating solvent molecule64 is favored. In other words, the ZnP shuttle becomes mobile along the axle only after its oxidation by means of a photoinduced charge transfer, and in the presence of a coordinating agent.65
The presence of 10−3 M pyridine in the toluene solution of 1Zn (5 × 10−5 M) induces the same effects that were discussed for benzonitrile. Most notably, two lifetimes were observed for 1Zn (see Fig. S10–S12, ESI†). However, the first lifetime, contributing to at least 90% of the signal amplitude, is drastically shortened, whereas the other remains comparable to that in toluene in the absence of pyridine.
This implies that, once the shuttle is mobile, the solvent polarity controls the directionality of the shuttling along the axle. In apolar toluene and in the presence of coordinating pyridine, the one-electron oxidized porphyrin (ZnP˙+) is drawn directionally towards the one-electron reduced fullerene (C60˙−). This leads to a drastically faster charge recombination of 1.7 ns as a major component and a nearly unchanged charge recombination of 9.42 ns as a minor component (indicating the fraction of 1Zn where the porphyrin remains anchored to the triazole). In polar benzonitrile, the solvent polarity is high enough to allow for random motion along the axle, i.e. decreasing and/or increasing the electron donor–acceptor distance. The largely decreased charge recombination of 278 ns cannot be explained by just the Marcus theory of charge transfer without considering an increased electron donor–acceptor separation. In contrast, the faster charge recombination of 0.76 ns is even shorter than that found in toluene when pyridine is present and is easily rationalized by the Marcus theory for charge transfer with dynamics that are placed in the inverted region of the Marcus parabola—see Table 1. Implicit are the same short distances between the one-electron oxidized porphyrin (ZnP˙+) and the one-electron reduced fullerene (C60˙−) as postulated in toluene.
To confirm our hypothesis, reference experiments were conducted with free base porphyrin rotaxane 1 (ESI,† Fig. S16–S29). Despite the fact that the driving forces for charge separation (see Table S2†) are up to 0.4 eV smaller in 1 than in 1Zn, charge separation (see Table S3†) is slowed down by only a few ps. Notably, the lack of a coordinating Zn-center in 1 leaves the shuttle free to move along the axle. A combination of π–π and charge transfer interactions, to name just a few, results in short electron donor–acceptor distances rather than large ones. In turn, charge separation and recombination are both accelerated.66 The triplet quantum yields correlate well with the underlying driving forces (Table S2, ESI†). However, solvent variation does not induce multiple CSS components in the excited state deactivation of 1. This is quite different from the observations for 1Zn. In other words, no triggered release of the porphyrin shuttle is possible in 1 due to its lack of a coordinating metal center.
Structural investigations
Considering the lifetime differences and the observation of a long-lived charge-separated state for 1Zn in the presence of coordinating solvents, the influence of the latter was investigated. The location of the dumbbell with respect to the porphyrin in the presence of a coordinating solvent (10% pyridine-d5 in benzene-d6) and of a non-coordinating solvent (benzene-d6) was deduced from 1D- and 2D-(1H-NMR) studies (see ESI,† Fig. S40–S53). Fig. 7 depicts the postulated structures in solution.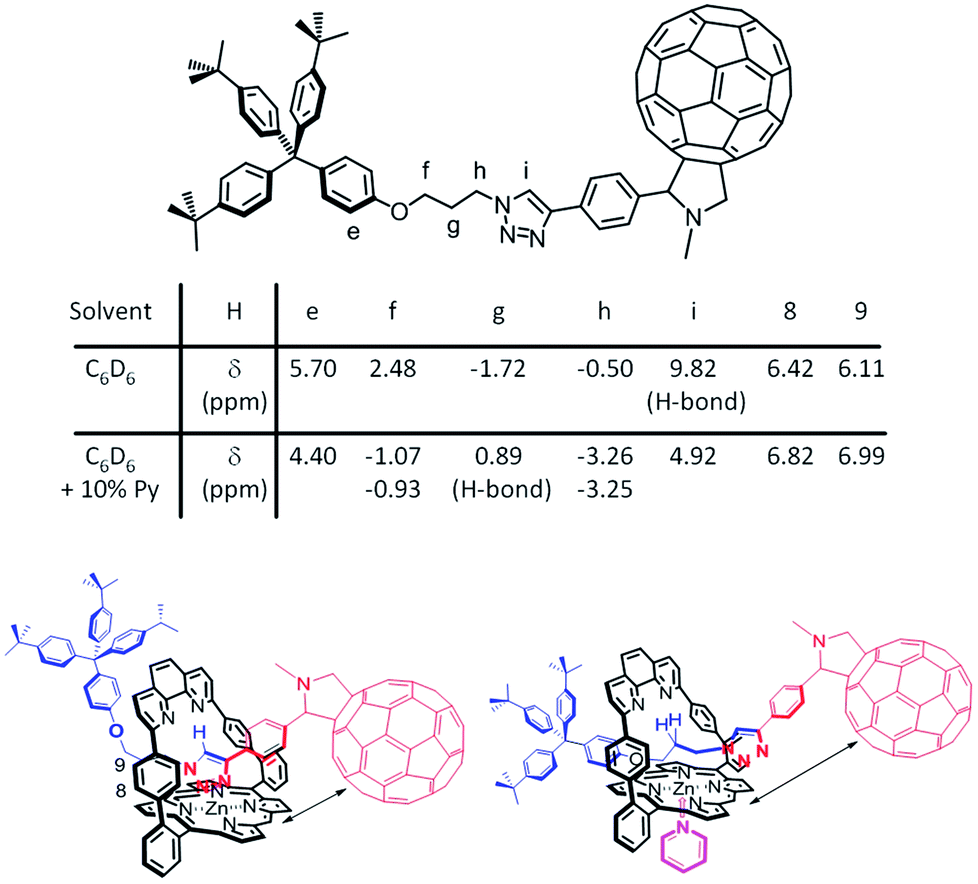 | ||
| Fig. 7 Comparison of the chemical shifts and position of the dumbbell of 1Zn in benzene-d6 (bottom left) and in benzene-d6 + 10% pyridine-d5 (bottom right). | ||
In benzene-d6, the triazole subunit of 1Zn is coordinated to the Zn-center and stabilized by a hydrogen bond of the triazole with the nitrogen atoms of the phenanthroline, as indicated by the downfield location of the triazole proton Hi at 9.82 ppm. In the presence of 10% pyridine-d5, which binds to the Zn-center opposite to the triazole, the triazole is relocated and sits outside of the phenanthroline pocket. Still, it is above the shielding cone of the porphyrin as seen by the upfield shift of Hi to 4.92 ppm. Deshielding of H8 and H9 corroborated that the triazole does sit outside of the phenanthroline pocket in pyridine-d5. In benzene-d6, these two protons are discernable at higher fields due to the influence of the triazole ring current.
Major differences of the chemical shifts also evolved for the alkyl protons. A coordinating solvent significantly affects the positioning of the dumbbell's propyl chain relative to the strapped porphyrin. Fig. 7 summarizes all the relevant chemical shifts stemming from the 1Zn protons in benzene-d6 and in a mixture of 10% pyridine-d5 in benzene-d6.
Compared to their chemical shifts in benzene-d6, the alkyl protons Hf and Hh are strongly shielded and appear as diastereotopic protons in the presence of pyridine-d5. In contrast, Hg protons are deshielded in pyridine-d5. Taking the aforementioned into account implies that in the presence of a coordinating solvent the propyl chain is positioned above the porphyrin with possible hydrogen bonding interactions between Hg and the phenanthroline nitrogen atoms. With respect to the position of the dumbbell, when a coordinating solvent is present, it is similar to what has been derived from the crystal structure (see ESI,† Fig. S54) for an analogue of 1Zn featuring two triaryl stoppers.
By correlating the observations made in the NMR studies with the results of the photophysics, the following conclusions are reached. In solution, shuttling in 1Zn is enabled by either coordinating solvents or a coordinating additive. Oxidation induces a distortion of the porphyrin scaffold and, in turn, facilitates easy access to the Zn-center. This consequently induces triazole-decoordination from the Zn-center as a starting point for directional or random shuttling in apolar or polar solvents, respectively.
Conclusions
In this work on rotaxanes comprising a strapped-porphyrin as a ring molecule and a dumbbell bearing one fullerene stopper, coordination forces are crucial for the excited state behavior. Distinct differences in the lifetime of the charge-separated state and the presence of more than a single lifetime were observed in 1Zn. Here, the coordinating properties and the polarity of the solvent have the strongest impact. For example, in apolar, non-coordinating toluene, a single lifetime was found for the charge-separated state, whereas in polar, coordinating PhCN, a short-lived and a long-lived charge-separated state evolved. In toluene and PhCl, these effects were also induced when a coordinating species, such as pyridine, was added to 1Zn.Depending on the solvent polarity, the shuttling is either directional or random. In apolar toluene and in the presence of pyridine, Coulombic interactions in the ZnP˙+−C60˙− radical ion pair dominate and, in turn, force a close proximity between the oxidized porphyrin and the reduced fullerene. This sets up the means for a directional molecular shuttling, which results in a drastically reduced lifetime of the charge-separated state. In highly polar benzonitrile, shuttling is random and allows the fullerene anion to move further away from the porphyrin radical cation in 1Zn. This enables the system to overcome the barrier of Coulombic attractions. As such, larger porphyrin-to-fullerene distances go hand-in-hand with a longer lifetime of the charge-separated state in a high-polarity coordinating solvent relative to that in either non-coordinating solvents or low-polarity solvents. Independent confirmation of our hypothesis came from the absence of a longer and/or a shorter-lived charge-separated state in the presence of a coordinating solvent in analogous experiments with 1, which features a strapped free-base porphyrin, without porphyrin–triazole interactions, rather than a zinc porphyrin.
Our results emphasize the importance of coordination interactions and their modulation in the excited state of a zinc-porphyrin rotaxane. To gain further insight, rotaxanes with longer, more rigid axles, which should enhance the effects on the distribution of lifetimes, are currently being designed and studied in our laboratories. Finally, a porphyrin with a metal center that binds pyridine more strongly than zinc, such as rhodium, could exhibit interesting behavior.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. V. thanks the French Ministry of Research for a PhD fellowship. This work was partially funded by JST-ANR grant ANR-14-JTIC-0002-01 (MECANO), JSPS KAKENHI (Grant Number JP15H05804 in Innovative Areas “Precisely Designed Catalysts with Customized Scaffolding”), Deutsche Forschungsgemeinschaft DFG (SFB953 Synthetic Carbon Allotropes), and the State of Bavaria (SolTech).Notes and references
- The Nobel Prize in Chemistry 2016, Nobel Media AB, 2018 Search PubMed.
- C. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS.
- S. Silvi, M. Venturi and A. Credi, J. Mater. Chem., 2009, 19, 2279 RSC.
- N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantoş and J. K. M. Sanders, Science, 2012, 338, 783–785 CrossRef CAS PubMed.
- J. J. Danon, A. Krüger, D. A. Leigh, J.-F. Lemonnier, A. J. Stephens, I. J. Vitorica-Yrezabal and S. L. Woltering, Science, 2017, 355, 159–162 CrossRef CAS PubMed.
- N. Armaroli, V. Balzani, J.-P. Collin, P. Gaviña, J.-P. Sauvage and B. Ventura, J. Am. Chem. Soc., 1999, 121, 4397–4408 CrossRef CAS.
- R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi and M. Venturi, Acc. Chem. Res., 2001, 34, 445–455 CrossRef CAS PubMed.
- S. Durot, V. Heitz, A. Sour and J.-P. Sauvage, Top. Curr. Chem., 2014, 354, 35–70 CrossRef CAS PubMed.
- C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS PubMed.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- G. Mancini, C. Zazza, M. Aschi and N. Sanna, Phys. Chem. Chem. Phys., 2011, 13, 2342–2349 RSC.
- N. K. Jena and N. A. Murugan, J. Phys. Chem. C, 2013, 117, 25059–25068 CrossRef CAS.
- S. S. Andersen, A. I. Share, B. L. C. Poulsen, M. Kørner, T. Duedal, C. R. Benson, S. W. Hansen, J. O. Jeppesen and A. H. Flood, J. Am. Chem. Soc., 2014, 136, 6373–6384 CrossRef CAS PubMed.
- A. V. Leontiev, C. J. Serpell, N. G. White and P. D. Beer, Chem. Sci., 2011, 2, 922 RSC.
- T. A. Barendt, I. Rašović, M. A. Lebedeva, G. A. Farrow, A. Auty, D. Chekulaev, I. V. Sazanovich, J. A. Weinstein, K. Porfyrakis and P. D. Beer, J. Am. Chem. Soc., 2018, 140, 1924–1936 CrossRef CAS PubMed.
- A. Tron, I. Pianet, A. Martinez-Cuezva, J. H. R. Tucker, L. Pisciottani, M. Alajarin, J. Berna and N. D. McClenaghan, Org. Lett., 2017, 19, 154–157 CrossRef CAS PubMed.
- S. Garaudée, S. Silvi, M. Venturi, A. Credi, A. H. Flood and J. F. Stoddart, ChemPhysChem, 2005, 6, 2145–2152 CrossRef PubMed.
- R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CAS.
- J. Baggerman, N. Haraszkiewicz, P. G. Wiering, G. Fioravanti, M. Marcaccio, F. Paolucci, E. R. Kay, D. A. Leigh and A. M. Brouwer, Chem.–Eur. J., 2013, 19, 5566–5577 CrossRef CAS PubMed.
- Z. Zhang, C. Han, G. Yu and F. Huang, Chem. Sci., 2012, 3, 3026 RSC.
- Y. Liu, C. Chipot, X. Shao and W. Cai, J. Phys. Chem. C, 2016, 120, 6287–6293 CrossRef CAS.
- A. C. Benniston and A. Harriman, Angew. Chem., Int. Ed. Engl., 1993, 32, 1459–1461 CrossRef.
- H. Murakami, A. Kawabuchi, K. Kotoo, M. Kunitake and N. Nakashima, J. Am. Chem. Soc., 1997, 119, 7605–7606 CrossRef CAS.
- J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611–619 CrossRef CAS.
- J. A. Wytko, R. Ruppert, C. Jeandon and J. Weiss, Chem. Commun., 2018, 54, 1550–1558 RSC.
- H. Imahori and S. Fukuzumi, Adv. Funct. Mater., 2004, 14, 525–536 CrossRef CAS.
- C. Luo, D. M. Guldi, H. Imahori, K. Tamaki and Y. Sakata, J. Am. Chem. Soc., 2000, 122, 6535–6551 CAS.
- J. D. Megiatto, A. Antoniuk-Pablant, B. D. Sherman, G. Kodis, M. Gervaldo, T. A. Moore, A. L. Moore and D. Gust, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 15578–15583 CAS.
- M. Wolf, A. Herrmann, A. Hirsch and D. M. Guldi, J. Am. Chem. Soc., 2017, 139, 11779–11788 CrossRef CAS PubMed.
- M. Wolf, C. Villegas, O. Trukhina, J. L. Delgado, T. Torres, N. Martín, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2017, 139, 17474–17483 CrossRef CAS PubMed.
- D. I. Schuster, J. Org. Chem., 2013, 78, 6811–6841 CrossRef CAS PubMed.
- K. Li, D. I. Schuster, D. M. Guldi, M. Á. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 3388–3389 CrossRef CAS PubMed.
- A. S. D. Sandanayaka, N. Watanabe, K.-I. Ikeshita, Y. Araki, N. Kihara, Y. Furusho, O. Ito and T. Takata, J. Phys. Chem. B, 2005, 109, 2516–2525 CAS.
- A. S. D. Sandanayaka, H. Sasabe, Y. Araki, N. Kihara, Y. Furusho, T. Takata and O. Ito, J. Phys. Chem. A, 2010, 114, 5242–5250 CrossRef CAS PubMed.
- A. Mateo-Alonso, C. Ehli, G. M. A. Rahman, D. M. Guldi, G. Fioravanti, M. Marcaccio, F. Paolucci and M. Prato, Angew. Chem., Int. Ed., 2007, 46, 3521–3525 CrossRef CAS PubMed.
- A. S. D. Sandanayaka, H. Sasabe, Y. Araki, N. Kihara, Y. Furusho, T. Takata and O. Ito, Aust. J. Chem., 2006, 59, 186 CrossRef CAS.
- M. R. Panman, P. Bodis, D. J. Shaw, B. H. Bakker, A. C. Newton, E. R. Kay, D. A. Leigh, W. J. Buma, A. M. Brouwer and S. Woutersen, Phys. Chem. Chem. Phys., 2012, 14, 1865–1875 RSC.
- B. Delavaux-Nicot, H. Ben Aziza, I. Nierengarten, T. Minh Nguyet Trinh, E. Meichsner, M. Chessé, M. Holler, R. Abidi, E. Maisonhaute and J.-F. Nierengarten, Chem.–Eur. J., 2018, 24, 133–140 CrossRef CAS PubMed.
- Y. Miyazaki, C. Kahlfuss, A. Ogawa, T. Matsumoto, J. A. Wytko, K. Oohora, T. Hayashi and J. Weiss, Chem.–Eur. J., 2017, 23, 13579–13582 CrossRef CAS PubMed.
- H. Zheng, W. Zhou, J. Lv, X. Yin, Y. Li, H. Liu and Y. Li, Chem.–Eur. J., 2009, 15, 13253–13262 CrossRef CAS PubMed.
- K.-H. Le Ho, I. Hijazi, L. Rivier, C. Gautier, B. Jousselme, G. de Miguel, C. Romero-Nieto, D. M. Guldi, B. Heinrich, B. Donnio and S. Campidelli, Chem.–Eur. J., 2013, 19, 11374–11381 CrossRef PubMed.
- J. A. Wytko, E. Graf and J. Weiss, J. Org. Chem., 1992, 57, 1015–1018 CrossRef CAS.
- J. Froidevaux, P. Ochsenbein, M. Bonin, K. Schenk, P. Maltese, J.-P. Gisselbrecht and J. Weiss, J. Am. Chem. Soc., 1997, 119, 12362–12363 CrossRef CAS.
- C. H. Kirksey, P. Hambright and C. B. Storm, Inorg. Chem., 1969, 8, 2141–2144 CrossRef CAS.
- M. Nappa and J. S. Valentine, J. Am. Chem. Soc., 1978, 100, 5075–5080 CrossRef CAS.
- C. B. Storm, A. H. Turner and M. B. Swann, Inorg. Chem., 1984, 23, 2743–2746 CrossRef CAS.
- G. Szintay and A. Horváth, Inorg. Chim. Acta, 2000, 310, 175–182 CrossRef CAS.
- M. A. Bobrik and F. A. Walker, Inorg. Chem., 1980, 19, 3383–3390 CrossRef CAS.
- D. M. Guldi, C. Luo, M. Prato, A. Troisi, F. Zerbetto, M. Scheloske, E. Dietel, W. Bauer and A. Hirsh, J. Am. Chem. Soc., 2001, 123, 9166–9167 CrossRef CAS PubMed.
- A. A. Maryott and E. R. Smith, Table of dielectric constants of pure liquids, National Bureau of Standards, Washington D.C., 1951 Search PubMed.
- D. M. Guldi, C. Luo, M. Prato, E. Dietel and A. Hirsch, Chem. Commun., 2000, 373–374 RSC.
- J. Fajer, D. C. Borg, A. Forman, D. Dolphin and R. H. Felton, J. Am. Chem. Soc., 1970, 92, 3451–3459 CrossRef CAS PubMed.
- N. Carnieri and A. Harriman, Inorg. Chim. Acta, 1982, 62, 103–107 CrossRef CAS.
- C. A. Reed and R. D. Bolskar, Chem. Rev., 2000, 100, 1075–1120 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Software, 2012, 49, 3 Search PubMed.
- K. M. Mullen and I. H. M. van Stokkum, J. Stat. Software, 2007, 18, 3 Search PubMed.
- At room temperature porphyrin–fullerene conformations are indistinguishable. Instead, all possible conformers of C60 relative to the porphyrin (e.g. cis or trans) coexist in solution.
- J. T. Buck, A. M. Boudreau, A. DeCarmine, R. W. Wilson, J. Hampsey and T. Mani, Chem, 2018, 5, 138–155 Search PubMed.
- M. Kuss-Petermann and O. S. Wenger, J. Am. Chem. Soc., 2016, 138, 1349–1358 CrossRef CAS PubMed.
- L. Favereau, A. Cnossen, J. B. Kelber, J. Q. Gong, R. M. Oetterli, J. Cremers, L. M. Herz and H. L. Anderson, J. Am. Chem. Soc., 2015, 137, 14256–14259 CrossRef CAS PubMed.
- H. Song, N. P. Rath, C. A. Reed and W. R. Scheidt, Inorg. Chem., 1989, 28, 1839–1847 CrossRef CAS.
- R. W. Scheidt, JBIC, J. Biol. Inorg. Chem., 2001, 6, 727–732 Search PubMed.
- A. S. Hinman and B. J. Pavelich, J. Electroanal. Chem., 1989, 269, 53–61 CrossRef CAS.
- N. S. Lebedeva, A. I. Vyugin, N. A. Pavlycheva, E. V. Parfenyuk and O. I. Davidova, Thermochim. Acta, 2002, 390, 179–186 CrossRef CAS.
- Pure non-coordinating solvents such as toluene or chlorobenzene disable the shuttling (for results in chlorobenzene see ESI,† Fig. S6–S8).
- The latter is further accelerated by the fact that the triplet excited states of C60 and/or H2P evolve as products of charge recombination.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional data and graphs. CCDC 1864783. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05328f |
| This journal is © The Royal Society of Chemistry 2019 |