
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of a cross-layer linked G-octamer via conformational control: a stable G-quadruplex in H-bond competitive solvents†
Ying
He‡
a,
Yanbin
Zhang‡
b,
Lukasz
Wojtas
a,
Novruz G.
Akhmedov
c,
David
Thai
a,
Heng
Wang
a,
Xiaopeng
Li
a,
Hao
Guo
 *b and
Xiaodong
Shi
*a
*b and
Xiaodong
Shi
*a
aDepartment of Chemistry, University of South Florida, 4202 E. Fowler Avenue, Tampa, Florida 33620, USA. E-mail: xmshi@usf.edu
bDepartment of Chemistry, Fudan University, 2005 Songhu Road, Shanghai, 200438, People's Republic of China
cDepartment of Chemistry, West Virginia University, Morgantown, WV 26505, USA
First published on 6th March 2019
Abstract
Methanol soluble and stable guanosine octamers were successfully achieved via H-bond self-assembly. Through structural conformational design, we developed a new class of guanosine derivatives with modification on guanine (8-aryl) and ribose (2′,3′-isopropylidene). This unique design led to the formation of the first discrete G8-octamer with its structure characterized by single crystal X-ray diffraction, MS and NMR spectroscopy. The G8-octamer showed unique cation recognition properties, including the formation of a stable Rb+ templated G-quadruplex. Based on this observation, further modification on the 8-aryl moiety was performed to incorporate a cross-layer H-bond or covalent linkage. Similar G-octamers were obtained in both cases with structures confirmed by single crystal X-ray diffraction. Furthermore, the covalently linked G-quadruplex exhibited excellent stability even in MeOH and DMSO, suggesting a promising future for this new H-bond self-assembly system in biological and material applications.
Introduction
Non-covalent interactions enable construction of large structural motifs from small molecules.1–3 Molecular self-assembly, an important process typically driven by non-covalent interactions, is often dynamic and generally under thermodynamic control.4,5 With interest in the application of the self-assembly process to biomedical research, there is a growing demand with respect to preparing stable non-covalent assemblies in a biocompatible environment.6–8 However, supramolecular structures built through H-bonds are often studied in less polar aprotic solvents (such as CH2Cl2) to avoid the competition of H-bond interactions between substrates and solvents.9,10 Thus, the development of novel supramolecular systems which are stable in a polar protic solvent is highly desirable, though very challenging.11,12A G-quartet is an interesting supramolecular scaffold formed by H-bonds.13,14 As shown in Scheme 1, with an approximately 90-degree angle between the H-bond donor and acceptor, four guanine units are held together to form a G-quartet. Through ion–dipole interactions, alkali and alkaline earth metal cations can enhance the process by serving as the template to coordinate with central oxygen atoms.15–17 Stacking of G-quartets gives G-quadruplexes as bioactive building blocks found in DNA and RNA folding.18,19 In this case, the extent of G-quartet stacking in a G-quadruplex will be determined by the phosphate backbone, which is often associated with the formation of a counter folding subunit, such as the i-motif.20
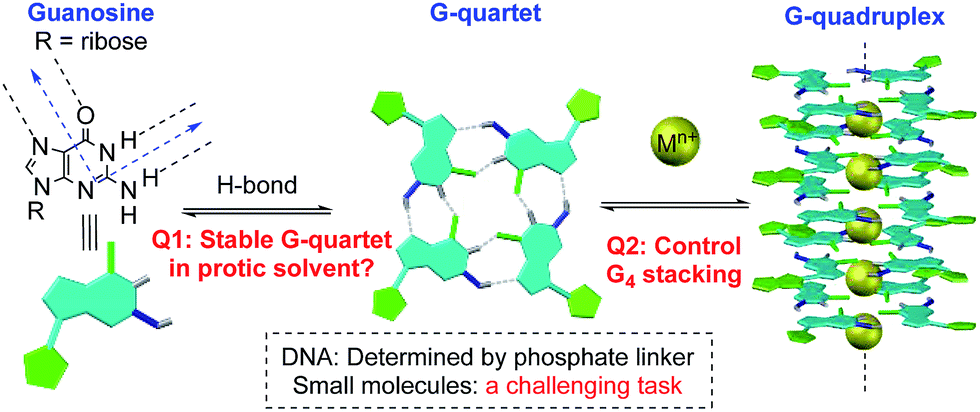 | ||
| Scheme 1 G-quadruplex formation: equilibrium and stability. | ||
Inspired by this unique H-bond assembly, researchers have been devoted to developing guanine derivatives to achieve controllable G-quadruplex formation from small molecules.21–23 Some interesting applications have been identified with various G-quartet assemblies, including lipophilic ion channels,24–26 supramolecular hydrogels,27–29 nanomaterials,30,31 potential targets for cancer therapy,32,33 and more.34–37 Although many examples of G-quartet formation through various modified guanine derivatives have been reported, studies on controlling G-quartet stacking molecularity are relatively rare.38,39 Factors to be taken into consideration include the cation concentration,39 the solvent,40 the anion,41 and so on.42 In many cases, mixtures of various “stacking isomers” (G8, G12 or G16) were observed, which highlights the significant challenges associated with controlling the vertical stacking.43–46 Moreover, the assembly of a specific and stable G-quadruplex in H-bond competitive solvents remains a challenging task.48 Herein, we report the construction of the first G-octamer with structures characterized by single crystal X-ray diffraction through monomer conformational design. Moreover, with this new system, stable G-quadruplexes were formed with significantly improved stability. Through the design of cross-layer H-bonds and covalent linkage, G-octamers were prepared with excellent stability in MeOH (no dissociation) and even in 50% DMSO, which offers a potential opportunity to extend the H-bond assembly system into biosystems for future applications.
Results and discussion
Design, synthesis and characterization of G-octamers
Ideally, a G4-tetramer would be the most concise target towards the construction of a simple and stable assembled structure. However, with a metal template in solution, further stacking of G4-tetramers leads to the mixture of G-quadruplex species.49 Thus, G4-tetramers are unfavorable for the formation of well-defined supermolecules.The simplest plausible G-quadruplex would be a G-octamer which is likely to adopt either top-to-top (T–T) or bottom-to-bottom (B–B) stacking patterns (Scheme 2A).50 In previous studies, Meijer et al. employed different concentrations of a guanosine derivative resulting in the formation of tunable G-octamers.41 Spada has reported G-octamer formation upon exposure to UV light through alkene isomerization.51 Wu presented several excellent examples of G-octamer formation through π–π interactions using NMR and MS studies.52 Hirao and coworkers applied Au(I)–Au(I) interaction between two G-quartet layers to achieve a G-octamer confirmed by NMR and CD spectra.53 However, to the best of our knowledge, no single crystal structure of a G-octamer has ever been reported, implying the challenging nature of preparing a stable and discrete G-octamer in a dynamic equilibrium.
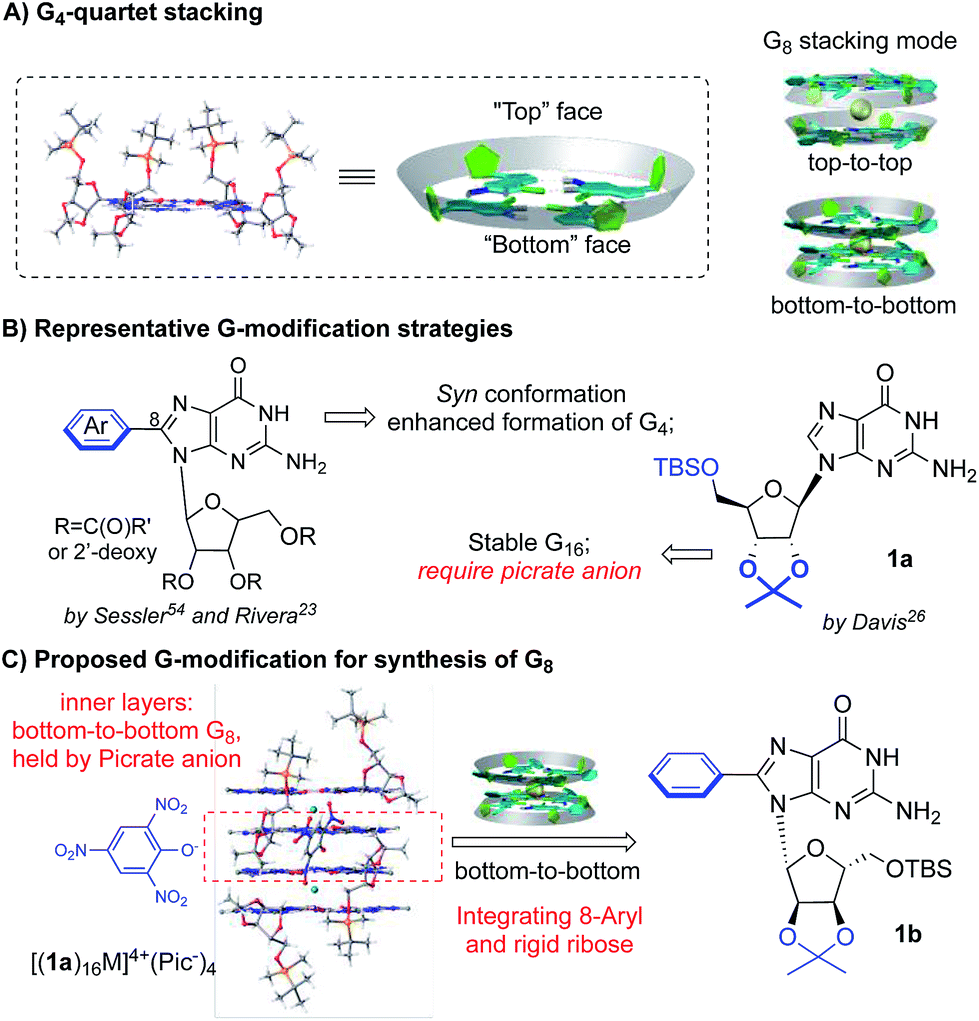 | ||
| Scheme 2 Achieving a stable G-octamer by controlling G-quartet stacking. | ||
To tackle this problem, we set out to design a G-derivative where the structure is rigid and predisposed for the conformation of a potential octamer structure so as to minimize the entropy cost involved in the self-assembly process. As shown in Scheme 2B, modification of guanosine often occurs on two positions: C-8 of purine and the hydroxyl of ribose. The Sessler group first reported on 8-aryl substituted guanosine in the formation of a G-quartet without templating cation in both solution and solid state.54
This seminal work initiated the concept of conformational control for G-quartet formation: the steric effect between the aryl substituent and protected ribose helped guanosine to adopt a syn conformation, preventing the ribbon formation and giving a tetramer as the dominant conformation. On the other hand, Davis's group developed lipophilic guanosine with bulky ribose to form a G-hexadecamer (Scheme 2B) both in solution and solid state (Mn+ = K+, Ba2+, Sr2+, and Pb2+).55 Notably, a picrate anion bridge played a crucial role: as revealed by single crystal X-ray diffraction (Scheme 2C), four picrate anions linked two G-octamers through H-bonds between anion and two inner G-quartets. The two G-octamers (from adjacent inner and outer G4) gave top-to-bottom stacking with ribose interdigitated between the adjacent layers. Interestingly, the two inner layers adopted bottom-to-bottom (B–B) stacking, which is more favorable than the T–T mode with the cation binding on the more “naked” convex face between the two layers. This result aroused our interest in developing a G-octamer through similar B–B stacking.
Considering the steric interaction between the C-8 substituent and ribose, we postulated that incorporation of C-8 aryl and the rigid ribose ring might provide a new system with steric hindrance between the G-quartet to force the G4 bowls to stack in a bottom-to-bottom manner, while obstructing ribose interdigitation at the top-face (Scheme 2C). To confirm this idea, compound 1b was designed, prepared and applied to assemble with various alkali and alkaline earth metal cations. 1H NMR spectra were obtained and selected regions of the 1H NMR spectra of these G-quadruplexes were compared with the G-hexadecamer from 1a as shown in Fig. 1.
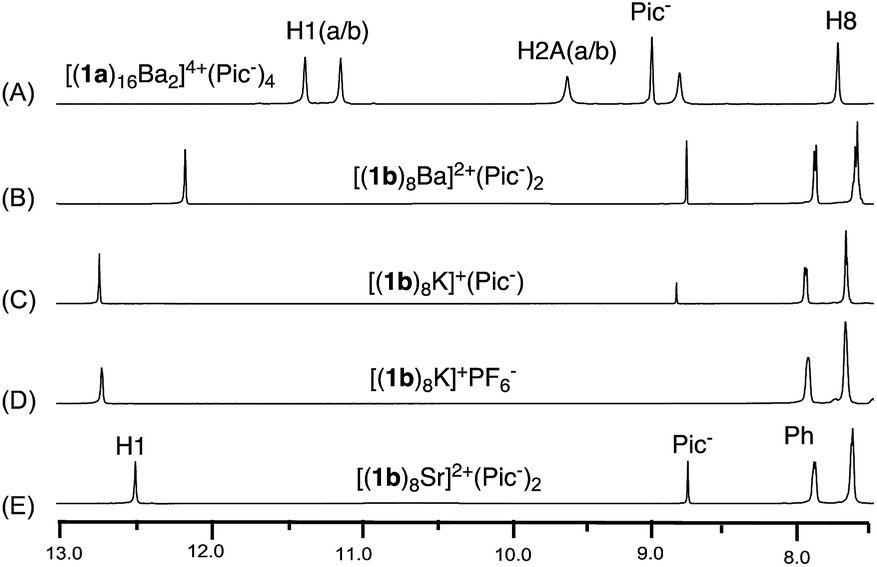 | ||
| Fig. 1 1H NMR spectra of G-quadruplexes (A) [(1a)16Ba2]4+·(Pic−)4; (B) [(1b)8Ba]2+·(Pic−)2; (C) [(1b)8K]+·(Pic−); (D) [(1b)8K]+·(PF6−); (E) [(1b)8Sr]2+·(Pic−)2 in CDCl3. | ||
As previously reported, treating 1a with alkali and alkaline earth metal salts (K+, Ba2+, Sr2+ and so on) gave two sets of signals in the 1H NMR spectra, corresponding to the inner and outer G-quartet.56 Conducting similar cation binding experiments with 1b in CDCl3 gave a single set of 1H NMR signals in all cases (M = K+, Ba2+, and Sr2+; A− = Picrate− or PF6−). Furthermore, ESI-MS demonstrated a clear doubly charged peak at m/z = 2123.01, corresponding to a mol. wt of 4246.68 for [(1b)8Ba]2+. The experimental and calculated isotope patterns further suggested an octameric composition. In addition, traveling wave ion mobility-mass spectrometry (TWIM-MS)57 confirmed no formation of stacking isomers, which excluded the formation of random aggregates in gas phase (see ESI† for details).
Finally, single crystal structures were obtained and unambiguously verified the G8-octamer formation with the proposed bottom-to-bottom stacking (Fig. 2A). The top view of the crystal structure (Fig. 2B) shows the G-quartet self-assembly in the tail to tail orientation. The five crystal structures obtained with ligand 1b include monovalent cations (K+ and Rb+) and divalent cations (Ba2+ and Sr2+). Picrate anion showed no clear binding with the G-quartet, consistent with what was observed in the 1H NMR spectra (see Fig. S2†). A complex with non-coordinated PF6− anion was also successfully obtained, [(1b)8K]+·(PF6−), confirming the “anion-free” binding mode of this new type of G8-quadruplex. The distances of the H-bond within the G4-quartet and between the G4 layers are compared in Table 1.
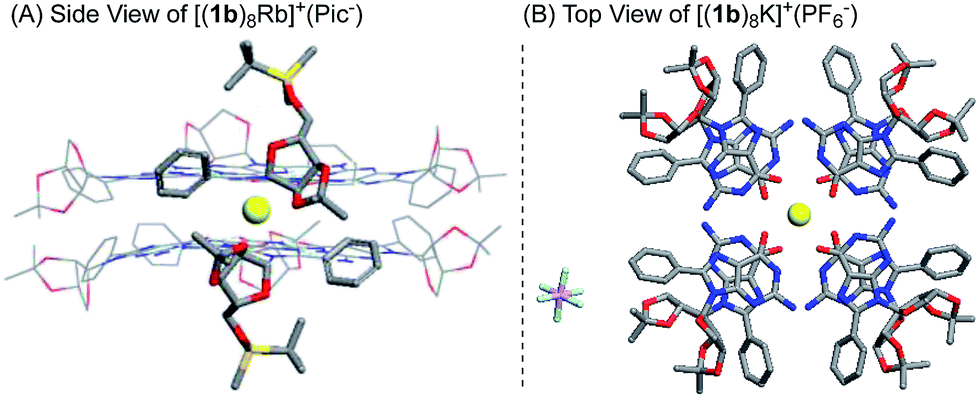 | ||
| Fig. 2 X-ray single crystal structure of the G8-octamer (A) [(1b)8Rb]+·(Pic−); (B) [(1b)8K]+·(PF6−). Similar structures for [(1b)8K]+·(Pic−), [(1b)8Ba]2+·(Pic−)2 and [(1b)8Sr]2+·(Pic−)2 were also obtained.§ | ||
| G-quadruplex | d(N1⋯O6) | d(N2⋯N7) | d(O–M) | d(G4–G4) | |
|---|---|---|---|---|---|
| a See ref. 46. | |||||
| [(1a)16Ba2]4+·(Pic−)4a | Inner | 2.92 ± 0.01 | 2.91 ± 0.07 | 2.75 ± 0.02 | 3.06 (i–o) |
| Outer | 2.86 ± 0.01 | 2.89 ± 0.04 | 2.79 ± 0.03 | 3.58 (i–i) | |
| [(1b)8K]+·(Pic−) | 2.82 ± 0.08 | 2.87 ± 0.04 | 2.72 ± 0.17 | 2.85 | |
| [(1b)8K]+·(PF6−) | 2.82 ± 0.04 | 2.89 ± 0.05 | 2.77 ± 0.03 | 2.96 | |
| [(1b)8Ba]2+·(Pic−)2 | 2.89 ± 0.04 | 2.89 ± 0.04 | 2.72 ± 0.05 | 2.89 | |
| [(1b)8Sr]2+·(Pic−)2 | 2.83 ± 0.03 | 2.86 ± 0.04 | 2.62 ± 0.05 | 2.75 | |
| [(1b)8Rb]+·(Pic−) | 2.85 ± 0.03 | 2.89 ± 0.03 | 2.81 ± 0.05 | 3.04 | |
According to these crystal structures, all G-octamers from 1b gave G-quartets with an (N1⋯O6) and (N2⋯N7) H-bond distance of around 2.9 Å, similar to that of the inner and outer layer in the G16-hexadecamer formed from 1a.46 These results indicated that both cations and the C-8 phenyl substituent had little influence on the H-bond in the G4-quartet. However, the size of the G8 was influenced by the average O–M distances of all these complexes from 1b, which might follow the trend where higher ionic potential (Z/r) resulted in shorter O–M distance (2.75–2.81 Å). Interestingly, in comparison with the G16-hexadecamer [(1a)16Ba2]4+·(Pic−)4, the G8-octamers [(1b)8Ba2]2+·(Pic−)2 gave a slightly shorter distance between the two G4 layers (2.89 Å vs. 3.06 Å and 3.58 Å). This result implied the stronger cation interaction of the (1b)4-quartet than the (1a)4-quartet. This improved cation interaction has been supported by G4-binding studies with Rb+ using the 1b ligand and led to the first crystal structures of Rb+ coordinated G-quadruplexes. Among all the G8 crystals, [(1b)8Rb]+·(Pic−) gave the longest O–M and G4–G4 distance due to its large radii58 and low valency. Very few examples of Rb coordinated G-quartets have been reported so far, indicating how challenging it is for guanosine to bind with Rb to form a discrete G-quadruplex.59 To the best of our knowledge, this is the first single crystal structure of a G-quadruplex containing Rb+, clearly suggesting the promising cation binding ability of guanosine derivative 1b.
Having successfully confirmed a new concise G8-quadruplex structure in solution (NMR), solid state (XRD) and gas phase (ESI-MS and TWIM-MS), we evaluated its stability in MeOH. Dissolving octamer [(1b)8K]+·(Pic−) in CD3OD gave a mixture of two sets of signals in NMR spectra, suggesting partial decomposition of this G8-octamer (vide infra). To obtain MeOH stable G-quadruplexes, further modification is still needed.
Cross-layer H-bonded G-octamers
To further improve the stability of the G-quadruplex, we sought to establish the interactions between the G-quartet layers. As highlighted in Fig. 2A, the 8-phenyl group in 1b adopted a tilted conformation and reached out from the G-quartet. This geometry provided an opportunity to further enhance the supramolecular structure by introducing new interactions between the two G-quartets.Notably, the Rivera group have reported the formation of an intralayer H-bond between carbonyl oxygen with N(2)H within the same G-quartet by using 2′-deoxy guanosine derivatives without rigid ribose functionalization.34,36–38 This work suggested the possibility of forming extra H-bonds by using both hydrogens of the N(2)–NH2 group. Inspired by this work, a carbonyl group was introduced at the meta-position of the 8-aryl position of 1b as illustrated in Fig. 3A. Based on this design, we hypothesized that N(2)–HA would form a H-bond with neighboring guanosine within the G-quartet, while the N(2)–HB could interact with the carbonyl group by forming a cross-layer H-bond.
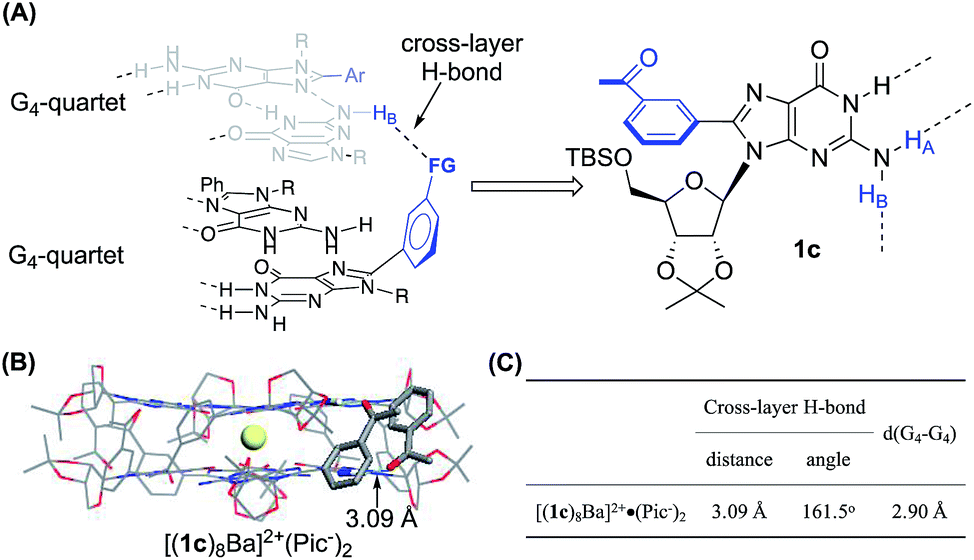 | ||
| Fig. 3 (A) General design of establishing a cross-layer H-bond; (B) single crystal structure and (C) H-bond information of [(1c)8Ba]2+·(Pic−)2.§ | ||
To confirm this idea, compound 1c was synthesized and applied to G-quadruplex construction upon interacting with metal cations. According to the 1H NMR spectra, treating 1c with Ba(Pic)2 led to the formation of a new G-quadruplex with one set of signals, similar to the G8-octamer obtained from 1b. Analysis of the NMR sample (in CDCl3) by ESI-MS gave a dominant, doubly charged peak with m/z at 2291.34, corresponding to [(1c)8Ba]2+ (mw = 4582.68).
Having confirmed the G8-octamer [(1b)8Ba]2+·(Pic−)2 formation, our next goal was to determine if there was a cross-layer H-bond as designed above. The 1H NMR spectra of [(1b)8Ba]2+·(Pic−)2 and [(1c)8Ba]2+·(Pic−)2 did not show peaks corresponding to N(2)H at room temperature. This is likely due to the rapid exchange between the two NH2 protons, even with the formation of a H-bond. Thus, the exchange rate of the two protons provides a direct indication of the H-bond strength in the G4-quartet. To explore the dynamic structure, variable temperature (VT) NMR experiments with [(1b)8Ba]2+·(Pic−)2 and [(1c)8Ba]2+·(Pic−)2 were performed and are summarized in Fig. 4.
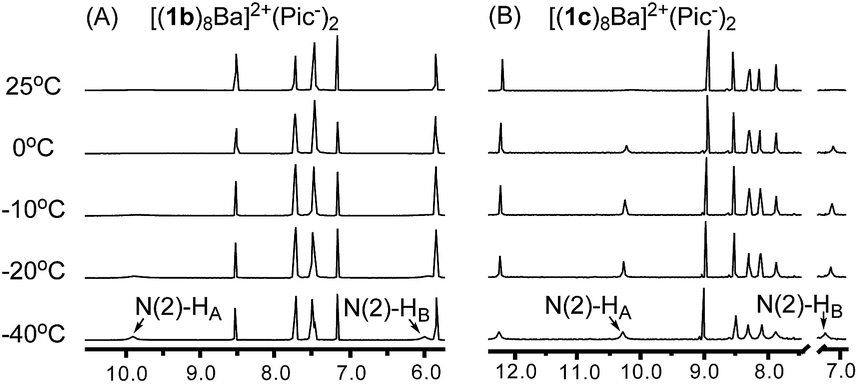 | ||
| Fig. 4 (A) VT NMR spectra of [(1b)8Ba]2+·(Pic−)2 and (B) VT NMR spectra of [(1c)8Ba]2+·(Pic−)2 confirmed the cross-layer H-bond design in establishing a cross-layer H-bond. | ||
For complex [(1c)8Ba]2+·(Pic−)2, the NH2 protons started appearing as broad peaks at 0 °C with the chemical shift at 10.28 ppm (HA) and 7.25 ppm (HB). In contrast, the VT NMR spectra of [(1b)8Ba]2+·(Pic−)2 did not show apparent peaks of the N(2)–H signals until further cooling the sample to −40 °C. The results indicated that there might be an extra H-bond in the 1c complex to lock the N(2)–NH2 from rapid exchange. Furthermore, a significantly downfield shifted chemical shift (7.25 ppm) was ascribed to the N(2)–HB proton in the (1c)8-octamer compared with the (1b)8-octamer (5.98 ppm). These observations provide clear evidence of the formation of a cross-layer H-bond in the (1c)8-octamer as designed.
Finally, the G8-octamer was verified by X-ray crystallography as shown in Fig. 3B. The crystal structure also confirmed the presence of the cross-layer interactions with a mean H-bond distance of 3.09 Å and a bond angle of 161.5°, suggesting a weak cross-layer H-bond present in solid state (Fig. 3C). This makes the structure a “self-assembled molecular-cuboid” purely constructed by H-bond linkage with all eight guanosine units. On the other hand, the distance of the two G-quartet layers (2.90 Å) in [(1c)8Ba]2+·(Pic−)2 remained similar to the 1b complex. Considering the specific “cage” size in the G8-octamer shown in Table 1, the cross-layer H-bond was not strong enough to generate extra enthalpy gain to balance the entropy cost caused by holding the two layers tighter.
With the confirmed cross-layer H-bonds, we evaluated the complex stability of [(1c)8K]+·(Pic−) in methanol. The results showed a similar stability to the complex formed with 1b (vide infra). Although the cross-layer H-bond approach could not boost G8-quadruplex stability in MeOH as anticipated, it provided an effective and novel approach to enhance G-quadruplex stability from monomer conformational design. Structural amendment was required to further improve the stability of the G8-octamer.
Cross-layer linkage through a covalent bond
To increase the stability to a new level, we turned to establishing a potential covalent linkage between the two G-quartets. By scrutinizing the crystal structure of [(1b)8Ba]2+·(Pic−)2, we found that the distance between the two meta position of the phenyl ring from each tetramer was 4.0 Å, a distance similar to three single bond lengths.58 According to the observation, the meta position of the two phenyl groups could serve as a reference site for constructing cross-layer covalent linkers. The guanosine dimer 1d and 1d′ were then prepared using the synthetic route summarized in Fig. 5A.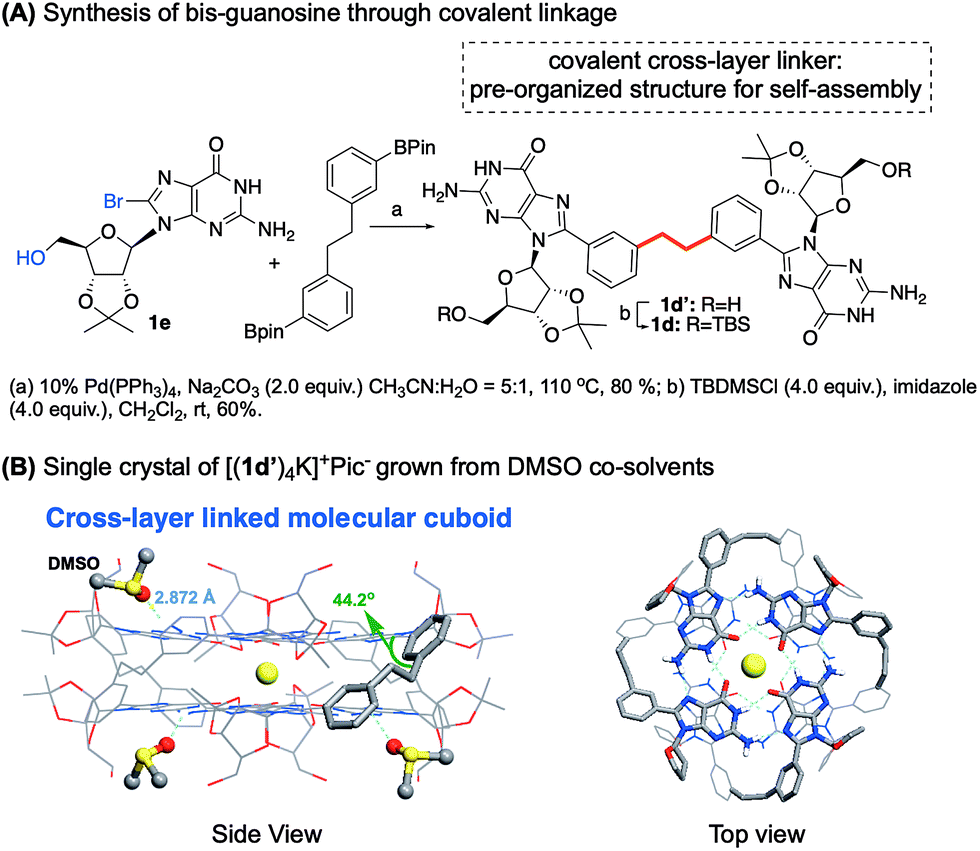 | ||
| Fig. 5 (A) Synthesis of bis-guanosine derivatives. (B) Single crystal structure of [(1d′)4K]+·(Pic−).§ | ||
Self-assembly of 1d with K+ and Ba2+ cations in CDCl3 gave a similar one-set of signals in the 1H NMR spectra, consistent with the formation of a G8-octamer. ESI-MS of complexes from 1d and Ba2+ gave a doubly charged peak at m/z = 2175.55 as the dominant signal, indicating a mol. wt of 4351.1 for the supermolecule as [(1d)4Ba]2+. An attempt to obtain a single crystal of the 1d complex failed initially, resulting in a rather thin, film-like solid formation. Fortunately, the single crystal structure was successfully obtained by switching the monomer to 1d′ using DMSO as a co-solvent, confirming the cross-layer covalent linked structure as proposed (Fig. 5B). Notably, for complex [(1d′)4Ba]2+·(Pic−)2, the dihedral angle between 8-aryl and guanine is 40.7 degrees, similar to the dihedral angles in complex [(1b)8Ba]2+·(Pic−)2 (42.5 degrees). Overall, through G-monomer conformational analysis, a series of G8-octamers was successfully prepared with functionalization at 8-phenyl (1b), a cross-layer H-bond linker (1c) and a covalent linker (1d).
G-quadruplex H-bond stability in MeOH
As discussed above, our intrinsic motivation in exploring these different G-quadruplexes was to develop H-bonded guanosine self-assembly that could survive in protic solvents (H-bond competitive). With all these different G16 and G8 quadruplexes prepared, we dissolved them in CD3OD to compare the 1H NMR spectra. As shown in Fig. 6A, N(1)–H and N(2)–H protons did not appear in 1H NMR spectra with CD3OD as the solvent due to the H/D exchange. Thus, evaluation of the 1H NMR spectra will mainly be focused on the non-exchangeable aromatic protons and ribose protons. Dissolving the G16-hexadecamer [(1a)16K4]4+·(Pic−)4 in CD3OD gave only one set of signals, identical to the 1a monomer in CD3OD. The result suggested complete dissociation of the G16-hexadecamer to 1a monomer in MeOH. Interestingly, the G8-octamer [(1b)8K]+·(Pic−) and [(1c)8K]+·(Pic−) in CD3OD gave two sets of signals, indicating the existence of dissociated monomer and possible oligomers or a G-quadruplex in the solution phase. Although the exact structures of the guanosine species in these two cases are not determined at this moment, the fact that H-bonded guanosine complexes were formed with 1b and 1c clearly suggests the improved H-binding ability of these two monomers over 1a. Surprisingly, when dissolving the G8-octamer [(1d)4K]+·(Pic−) in CD3OD, only one set of signals was observed. Notably, in this case, N(1)–H gave a broad signal at 12.95 ppm, clearly suggesting the formation of a G-quadruplex through a H-bond. NMR solvent signal suppression was applied for the G-quadruplex [(1d)4K]+·(Pic−) in CD3OH. The peak at 12.9 ppm clearly showed up and was confirmed to be H1 of G in the G-quadruplex (see detailed NMR spectra in Fig. S7†). Thus, with monomer 1d, a G-quadruplex remains intact in protic solvent CD3OD. Impressively, this G-quadruplex did not dissociate even at elevated temperature in CD3OD, with the N(1)H peak remaining at 60 °C (see Fig. S6† for VT NMR spectra). This observation indicated that there was a high kinetic barrier to break the 1d G-quartet for H/D exchange, which highlighted the stability of [(1d)4K]+·(Pic−) in a H-bond competitive system. Injecting an MeOH solution of [(1d)4Ba]2+·(Pic−)2 complex into ESI-MS gave a dominant double charged peak with m/z = 2175.55, corresponding to [(1d)4Ba]2+ (Fig. 6B). It is noteworthy that TWIM-MS of this G-quadruplex in methanol solution was recorded as a single band (m/z = 2175.55) with drift time at 14.33 ms, which is in agreement with the size of the G8-octamer (see detailed discussion in Table S1†). To the best of our knowledge, this is one of the few stable G-quadruplex systems from small molecule self-assembly to survive in a H-bond competitive environment.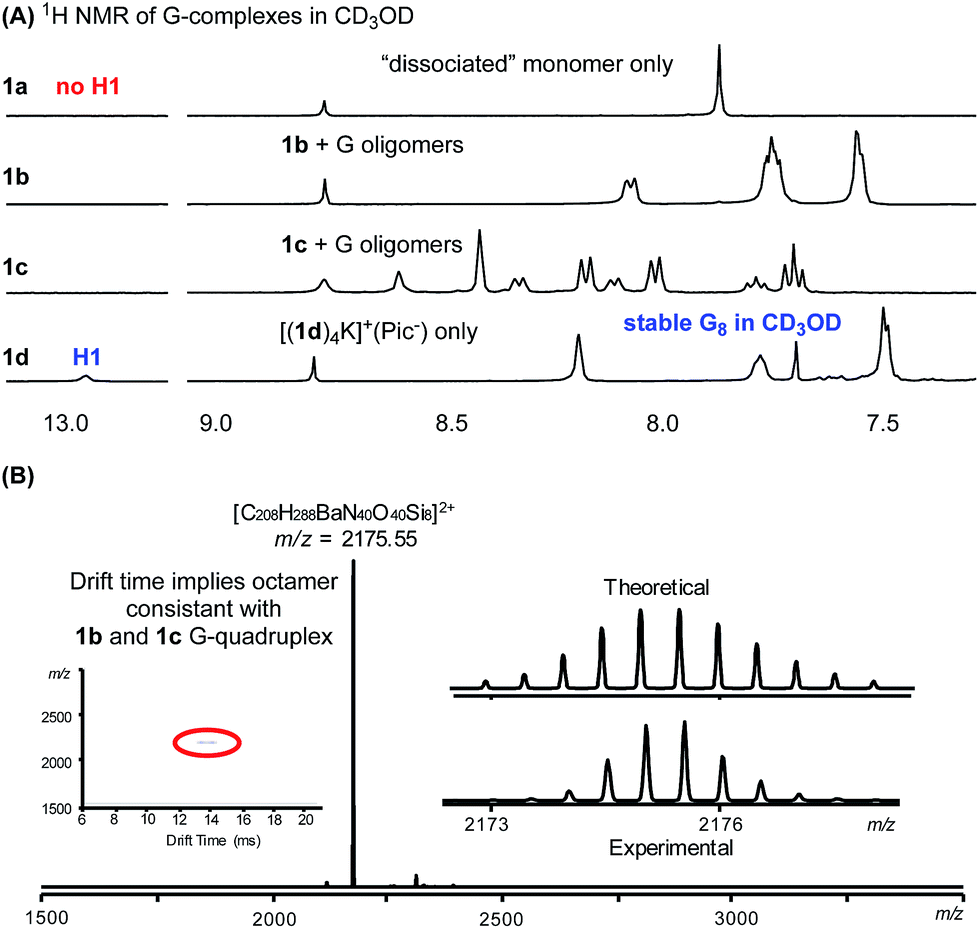 | ||
| Fig. 6 (A) 1H NMR spectra showing G-quadruplex stability in CD3OD. (B) MS of [(1d)4Ba]2+·(Pic−)2 in MeOH. | ||
Evaluating G-quadruplex stability
With the success in maintaining G-quadruplex stability in protic solvent MeOH, we sought to evaluate whether a similar stability trend exists with polar aprotic solvents. DMSO is a strong polar solvent, which can disrupt the H-bond in G-quartets and cause the decomposition of G-quadruplexes. To evaluate how the incorporation of the phenyl group and cross-layer interaction impact on thermodynamic stability, G-quadruplexes were treated with CDCl3/DMSO-d6 solvent mixture. A summary of 1H NMR spectra from these experiments is shown in Fig. 7.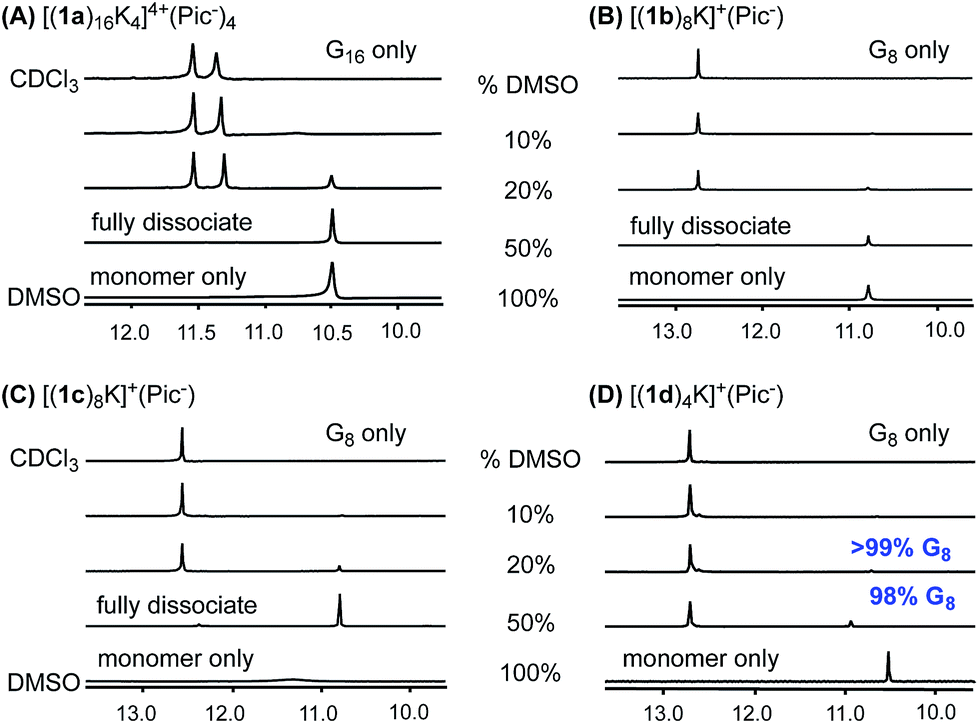 | ||
Fig. 7 DMSO-d6 titration: 1H NMR spectra of (A) [(1a)16K4]4+·(Pic−)4, (B) [(1b)8K]+·(Pic−), (C) [(1c)8K]+·(Pic−), (D) [(1d)4K]+·(Pic−) in CDCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO-d6 with DMSO-d6 fractions of 10%, 20% and 50%. :DMSO-d6 with DMSO-d6 fractions of 10%, 20% and 50%. | ||
As shown in the 1H NMR spectra, G-quadruplexes from 1a, 1b and 1c started to dissociate in mixed solvents containing 20% DMSO-d6. Compared with the reported G16-hexadecamer from 1a,46 the G8-octamer formed by 1b and 1c showed comparable stability in 20% DMSO-d6. Eventually, all three G-quadruplexes gave complete dissociation in 50% DMSO-d6 solution with only one set of signals corresponding to the monomer. In contrast, [(1d)4K]+·(Pic−) showed significantly improved stability, with only 2% complex dissociation in 50% DMSO-d6. This result demonstrates the significantly enhanced stability of G-quadruplexes constructed by 1d.
To quantify the thermodynamic stability of G-quadruplexes,47 VT NMR experiments were carried out for pure complexes of [(1a)16K4]4+·(Pic−)4, [(1b)8K]+·(Pic−), [(1c)8K]+·(Pic−), and [(1d)4K]+·(Pic−) in CDCl3/DMSO-d6 with a fraction of 20% DMSO-d6. The values of complex dissociation enthalpy and entropy for each G-quadruplex were calculated from van't Hoff plots and are compared in Table S6 (see detailed discussion in the ESI†). For complex [(1d)4K]+·(Pic−), no significant increase in monomer concentration was observed with the increase in temperature. This might be attributed to the high kinetic barrier for G-quadruplex [(1d)4K]+·(Pic−) dissociation. To confirm this hypothesis, a NOESY experiment at 50 °C was performed for [(1b)8K]+·(Pic−), [(1c)8K]+·(Pic−) and [(1d)4K]+·(Pic−) (Fig. S14–S16†). The results suggested that kinetic exchange between the complex and monomer for [(1d)4K]+·(Pic−) was too slow to be recorded by NMR spectroscopy.
In addition to DMSO titration, the stability of G8 and G16 complexes could also be evaluated using tandem-MS by increasing the operating voltage. The cation fragments of G-quadruplexes were separated and treated with increasing voltage. The operating voltage for G-quadruplex cation fragment decomposition are summarized in Table 2.
It is noteworthy that [(1a)16Ba2]4+·(Pic−)4 only showed a doubly charged peak at m/z = 2123.31 corresponding to [(1a)8Ba]2+, indicating that the picrate bridge dissociated under the MS conditions. Further comparison of all the cation fragments of the G-quadruplexes revealed a clear stability trend as [(1d)4Ba]2+ > [(1c)8Ba]2+ > [(1b)8Ba]2+ > [(1a)8Ba]2+. Overall, the covalent linking strategy significantly helped to stabilize the G8-octamer, both in H-bond competitive solvents and gas phase.
Conclusions
In summary, with modification on both guanine (8-aryl) and ribose (sterically hindered 2′,3′ position), a stable G8-octamer was formed with its structure characterized by single crystal X-ray diffraction for the first time. Through the analysis of cross-layer interactions, a covalently tethered 8-aryl guanosine dimer was designed and prepared for supramolecular assembly. The expected G8-octamer was confirmed by X-ray, MS and NMR spectroscopy with significantly improved stability in MeOH and 1:1 DMSO/CDCl3 mixture. To the best of our knowledge, this is the first example of discrete G-quadruplexes formed from small molecules with enhanced stability in a protic solvent (MeOH) and a polar aprotic solvent (DMSO). Meanwhile, formation of the stable G8-octamer with a concise and well-defined bottom-to-bottom stacking mode provides a novel supramolecular platform. Incorporation of this new system into material and biological applications is expected and currently undergoing in our group.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the NSF (CHE-1665122), the NIH (1R01GM120240-01), and NSFC (21629201) for financial support.Notes and references
- M. Fujita, Structure and Bonding, Springer, Berlin, 2000, pp. 177–201 Search PubMed.
- D. S. Lawrence, T. Jiang and M. Levett, Chem. Rev., 1995, 95, 2229–2260 CrossRef CAS.
- D. P. Craig and D. P. Mellor, Structure and Bonding, Springer, Berlin, 1976, pp. 1–48 Search PubMed.
- M. Fujita, Chem. Soc. Rev., 1998, 27, 417–425 RSC.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS PubMed.
- P. Y. W. Dankers and E. W. Meijer, Bull. Chem. Soc. Jpn., 2007, 80, 2047–2073 CrossRef CAS.
- H.-W. Jun, S. E. Paramonov and J. D. Hartgerink, Soft Matter, 2006, 2, 177–181 RSC.
- P. Xin, P. Zhu, P. Su, J.-L. Hou and Z.-T. Li, J. Am. Chem. Soc., 2014, 136, 13078–13081 CrossRef CAS PubMed.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed.
- J. L. Sessler, C. M. Lawrence and J. Jayawickramarajah, Chem. Soc. Rev., 2007, 36, 314–325 RSC.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2004, 126, 11408–11409 CrossRef CAS PubMed.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed.
- J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296–313 RSC.
- G. P. Spada and G. Gottarelli, Synlett, 2004, 4, 596–602 Search PubMed.
- E. Bouhoutsos-Brown, C. L. Marshall and T. J. Pinnavaia, J. Am. Chem. Soc., 1982, 104, 6576–6584 CrossRef CAS.
- J. P. N. V. Hud, Quadruplex Nucleic Acids, The Royal Society of Chemistry, Cambridge, 2006, pp. 100–130 Search PubMed.
- J. A. Walmsley, R. G. Barr, E. Bouhoutsos-Brown and T. J. Pinnavaia, J. Phys. Chem., 1984, 88, 2599–2605 CrossRef CAS.
- G. Biffi, M. Di Antonio, D. Tannahill and S. Balasubramanian, Nat. Chem., 2013, 6, 75 CrossRef PubMed.
- J. N. Parkinson, Quadruplex Nucleic Acids, The Royal Society of Chemistry, Cambridge, 2006, pp. 1–30 Search PubMed.
- M. Zeraati, D. B. Langley, P. Schofield, A. L. Moye, R. Rouet, W. E. Hughes, T. M. Bryan, M. E. Dinger and D. Christ, Nat. Chem., 2018, 10, 631–637 CrossRef CAS PubMed.
- I. C. M. Kwan, Y.-M. She and G. Wu, Can. J. Chem., 2011, 89, 835–844 CrossRef CAS.
- M. a. D. C. Rivera-Sánchez, M. García-Arriaga, G. Hobley, A. V. Morales-de-Echegaray and J. M. Rivera, ACS Omega, 2017, 2, 6619–6627 CrossRef PubMed.
- M. d. C. Rivera-Sánchez, I. Andújar-de-Sanctis, M. García-Arriaga, V. Gubala, G. Hobley and J. M. Rivera, J. Am. Chem. Soc., 2009, 131, 10403–10405 CrossRef PubMed.
- R. N. Das, Y. P. Kumar, O. M. Schütte, C. Steinem and J. Dash, J. Am. Chem. Soc., 2015, 137, 34–37 CrossRef CAS PubMed.
- S. L. Forman, J. C. Fettinger, S. Pieraccini, G. Gottarelli and J. T. Davis, J. Am. Chem. Soc., 2000, 122, 4060–4067 CrossRef CAS.
- M. S. Kaucher, W. A. Harrell and J. T. Davis, J. Am. Chem. Soc., 2006, 128, 38–39 CrossRef CAS PubMed.
- T. Bhattacharyya, P. Saha and J. Dash, ACS Omega, 2018, 3, 2230–2241 CrossRef CAS.
- G. M. Peters and J. T. Davis, Chem. Soc. Rev., 2016, 45, 3188–3206 RSC.
- G. M. Peters, L. P. Skala and J. T. Davis, J. Am. Chem. Soc., 2016, 138, 134–139 CrossRef CAS PubMed.
- M. García-Iglesias, T. Torres and D. González-Rodríguez, Chem. Commun., 2016, 52, 9446–9449 RSC.
- D. González-Rodríguez, P. G. A. Janssen, R. Martín-Rapún, I. D. De Cat, S. De Feyter, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 4710–4719 CrossRef PubMed.
- H. Han and L. H. Hurley, Trends Pharmacol. Sci., 2000, 21, 136–142 CrossRef CAS PubMed.
- F. W. B. Van Leeuwen, W. Verboom, X. Shi, J. T. Davis and D. N. Reinhoudt, J. Am. Chem. Soc., 2004, 126, 16575–16581 CrossRef CAS PubMed.
- J. E. Betancourt, C. Subramani, J. L. Serrano-Velez, E. Rosa-Molinar, V. M. Rotello and J. M. Rivera, Chem. Commun., 2010, 46, 8537–8539 RSC.
- S. Martic, G. Wu and S. Wang, Inorg. Chem., 2008, 47, 8315–8323 CrossRef CAS PubMed.
- J. M. Rivera, M. Martín-Hidalgo and J. C. Rivera-Ríos, Org. Biomol. Chem., 2012, 10, 7562–7565 RSC.
- J. M. Rivera and D. Silva-Brenes, Org. Lett., 2013, 15, 2350–2353 CrossRef CAS PubMed.
- M. Martín-Hidalgo and J. M. Rivera, Chem. Commun., 2011, 47, 12485–12487 RSC.
- K. B. Sutyak, P. Y. Zavalij, M. L. Robinson and J. T. Davis, Chem. Commun., 2016, 52, 11112–11115 RSC.
- J. E. Betancourt, M. Martín-Hidalgo, V. Gubala and J. M. Rivera, J. Am. Chem. Soc., 2009, 131, 3186–3188 CrossRef CAS PubMed.
- D. González-Rodríguez, J. L. J. van Dongen, M. Lutz, A. L. Spek, A. P. H. J. Schenning and E. W. Meijer, Nat. Chem., 2009, 1, 151–155 CrossRef PubMed.
- J. E. Betancourt and J. M. Rivera, J. Am. Chem. Soc., 2009, 131, 16666–16668 CrossRef CAS PubMed.
- J. E. Betancourt and J. M. Rivera, Org. Lett., 2008, 10, 2287–2290 CrossRef CAS PubMed.
- V. Gubala, J. E. Betancourt and J. M. Rivera, Org. Lett., 2004, 6, 4735–4738 CrossRef CAS PubMed.
- S. B. Zimmerman, J. Mol. Biol., 1976, 106, 663–672 CrossRef CAS PubMed.
- X. Shi, K. M. Mullaugh, J. C. Fettinger, Y. Jiang, S. A. Hofstadler and J. T. Davis, J. Am. Chem. Soc., 2003, 125, 10830–10841 CrossRef CAS PubMed.
- E. Fadaei, M. Martín-Arroyo, M. Tafazzoli and D. González-Rodríguez, Org. Lett., 2017, 19, 460–463 CrossRef CAS PubMed.
- M. García-Arriaga, G. Hobley and J. M. Rivera, J. Am. Chem. Soc., 2008, 130, 10492–10493 CrossRef PubMed.
- A. L. Marlow, E. Mezzina, G. P. Spada, S. Masiero, J. T. Davis and G. Gottarelli, J. Org. Chem., 1999, 64, 5116–5123 CrossRef CAS.
- M. García-Arriaga, G. Hobley and J. M. Rivera, J. Org. Chem., 2016, 81, 6026–6035 CrossRef PubMed.
- S. Lena, P. Neviani, S. Masiero, S. Pieraccini and G. P. Spada, Angew. Chem., Int. Ed., 2010, 49, 3657–3660 CrossRef CAS PubMed.
- S. Marti, X. Liu, S. Wang and G. Wu, Chem.–Eur. J., 2008, 14, 1196–1204 CrossRef PubMed.
- X. Meng, T. Moriuchi, M. Kawahata, K. Yamaguchi and T. Hirao, Chem. Commun., 2011, 47, 4682–4864 RSC.
- J. L. Sessler, M. Sathiosatham, K. Doerr, V. Lynch and K. A. Abboud, Angew. Chem., Int. Ed., 2000, 39, 1300–1303 CrossRef CAS PubMed.
- X. Shi, J. C. Fettinger and J. T. Davis, Angew. Chem., Int. Ed., 2001, 40, 2827–2831 CrossRef CAS PubMed.
- X. Shi, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc., 2001, 123, 6738–6739 CrossRef CAS PubMed.
- H. Wang, X. Qian, K. Wang, M. Su, W.-W. Haoyang, X. Jiang, R. Brzozowski, M. Wang, X. Gao, Y. Li, B. Xu, P. Eswara, X.-Q. Hao, W. Gong, J.-L. Hou, J. Cai and X. Li, Nat. Commun., 2018, 9, 1815 CrossRef PubMed.
- J. G. Speight, Lange's Handbook of Chemistry, McGraw-Hill, New York, 16th edn, 2005, pp. 1–150 Search PubMed.
- R. Ida and G. Wu, Chem. Commun., 2005, 4294–4296 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, NMR spectra, ESI-MS spectra and crystallographic data. CCDC 1871565–1871570 and 1871754. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00190e |
| ‡ Equal contribution. |
| § All the structures reported in this article have been deposited with the Cambridge Crystallographic Data Centre. The accession numbers for [(1b)8Ba]2+·(Pic−)2; [(1b)8K]+·(PF6−); [(1b)8K]+·(Pic−); [(1b)8Sr]2+·(Pic−)2; [(1b)8Rb]+·(Pic−); [(1c)8Ba]2+·(Pic−)2; [(1d′)4K]+·(Pic−) reported in this paper are: 1871565, 1871566, 1871567, 1871568, 1871569, 1871570, 1871754, correspondingly. |
| This journal is © The Royal Society of Chemistry 2019 |