
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeoligin-inspired synthetic lignans with selectivity for cell-type and bioactivity relevant for cardiovascular disease†‡
Thomas
Linder
 a,
Rongxia
Liu§
b,
Atanas G.
Atanasov¶
b,
Yuanfang
Li||
b,
Sophie
Geyrhofer
a,
Stefan
Schwaiger
c,
Hermann
Stuppner
c,
Michael
Schnürch
a,
Verena M.
Dirsch
*b and
Marko D.
Mihovilovic
*a
a,
Rongxia
Liu§
b,
Atanas G.
Atanasov¶
b,
Yuanfang
Li||
b,
Sophie
Geyrhofer
a,
Stefan
Schwaiger
c,
Hermann
Stuppner
c,
Michael
Schnürch
a,
Verena M.
Dirsch
*b and
Marko D.
Mihovilovic
*a
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163-OC, 1060 Vienna, Austria. E-mail: marko.mihovilovic@tuwien.ac.at
bDepartment of Pharmacognosy, University of Vienna, Althanstraße 14, 1090 Vienna, Austria. E-mail: verena.dirsch@univie.ac.at
cInstitute of Pharmacy/Pharmacognosy, Center for Molecular Biosciences Innsbruck, University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria
First published on 14th May 2019
Abstract
Recently, a natural compound leoligin, a furan-type lignan, was discovered as an interesting hit compound with an anti-inflammatory pharmacological activity profile. We developed a modular and stereoselective approach for the synthesis of the edelweiss-derived lignan leoligin and used the synthetic route to rapidly prepare leoligin analogs even on the gram scale. Proof of concept of this approach together with cell-based bio-assays gained structural analogs with increased selectivity towards vascular smooth muscle versus endothelial cell proliferation inhibition, a major benefit in fighting vascular neointima formation. In addition, we identified the structural features of leoligin analogs that define their ability to inhibit the pro-inflammatory NF-κB pathway. Results are discussed in the context of structural modification of these novel synthetic lignans.
Introduction
Cardiovascular disease (CVD) is the number one cause of death in the world.1 Atherosclerosis, along with hypertension, is the main cause of CVD.2 Initiation of atherosclerosis is associated with chronic injury of endothelial cells (ECs), followed by the accumulation of lipids and extracellular matrix in the intima, recruitment of monocytes that differentiate into pro-inflammatory macrophage foam cells, and exaggerated proliferation of vascular smooth muscle cells (VSMCs), resulting in neointima formation and arterial lumen loss.3Severe vascular lumen loss can be treated by cardiovascular interventions including coronary balloon angioplasty, stenting and coronary bypass surgery.4 Restenosis, the re-narrowing of surgically dilated arteries, appears to be a major problem of percutaneous transluminal coronary angioplasty (PTCA) or PTCA in combination with bare-metal stent implantation and occurs as a proliferative and pro-inflammatory response to injury resulting again in neointimal hyperplasia.5 An important stimulus for VSMC proliferation is the platelet-derived growth factor (PDGF). While VSMC proliferation promotes restenosis, re-lining of the vessel wall with ECs is desirable.6 Hence, re-endothelialization may lower the risk of thrombotic events that are increased by drug-eluting stents and bioresorbable scaffolds.4,7,8 Drug-eluting stents release anti-proliferative/immunosuppressant or cytotoxic drugs, such as rapamycin (sirolimus) or related substances and paclitaxel, respectively, to reduce local VSMC proliferation. These drugs, however, also inhibit EC proliferation.9–11 Thus, an ideal drug for restenosis treatment should be able to selectively inhibit VSMC proliferation.
Inflammation is an important contributor to the pathogenesis of atherosclerosis and restenosis.12 Nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) is a transcription factor that promotes the expression of pro-inflammatory mediators including cytokines such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6, adhesion molecules, and mediators involved in inflammation and tissue reorganization such as cyclooxygenase (COX)-2 or matrix metalloproteinases.13 Due to its central pro-inflammatory action, inhibition of NF-κB may be a promising pharmacological target in (neo)atherosclerosis and restenosis.13,14
Leoligin 1a (Fig. 1) is a furan-type lignan and a secondary metabolite found in the roots of edelweiss (Leontopodium nivale ssp. alpinum).15–17 Amongst other effects,18–21 it had previously been shown to inhibit intimal hyperplasia in a human saphenous vein organ culture model, as well as in a mouse model for vein graft disease.22 The observed effect is based on the inhibition of VSMC proliferation.22 Although not cytotoxic, leoligin also inhibited the proliferation of ECs. In addition, the potency of leoligin was not high enough (VSMC, IC50: 32.1 μM) for it to be considered a useful tool compound and to study its activities in cardiovascular disease. Since leoligin is exclusively present in the roots of Leontopodium ssp. at concentrations of 0.0155 and 0.0547 w% in cultivated edelweiss plants (L. nivale ssp. alpinum cv. ‘Helvetia’) and 0.005–0.010 w% in collected samples of the same species, the possibility of production in hairy root cultures was investigated.17 Since edelweiss is a strongly protected plant, a biosynthetic or purely synthetic pathway producing leoligin is required, where the latter one would also allow the preparation of leoligin derivatives.23
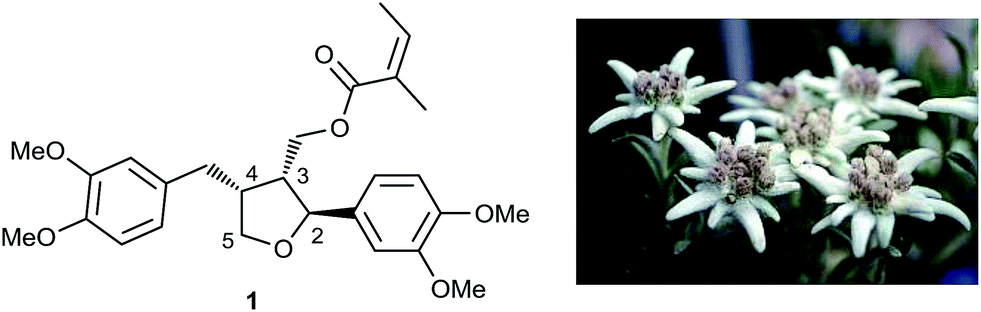 | ||
| Fig. 1 Leoligin, the major lignan isolated from L. nivale ssp. alpinum. | ||
Results and discussion
In the context of a multi-disciplinary project on the identification of natural products with anti-inflammatory activity,24 developing a fast and high-yield chemical synthesis method for leoligin was our first objective.25 With respect to intimal hyperplasia, the objectives were two-fold: (i) starting out from the natural hit molecule, a synthetic analog was sought with higher potency compared to leoligin, but also a better cell-selectivity with respect to inhibition of cell proliferation (VSMCs versus ECs); and (ii) since leoligin appeared to be a moderate NF-κB inhibitor (IC50: 19.7 μM), we set out to identify the structural features that define its anti-inflammatory action.For the preparation of synthetic leoligin analogs (and leoligin itself), it was important to develop a modular synthetic strategy, in order to gain access to a large variety of structural analogs with minimum effort. Recently, synthesis of a structure claimed to be leoligin was published.26 However, all steps were carried out in the absence of a chiral inducer and, additionally, diastereocontrol within the sequence seems highly questionable, in particular as spectral properties of the reported synthetic product do not match with those of the natural product isolate of leoligin (see ESI‡).
The stereoselective synthetic route developed by us is outlined in Scheme 1. Key steps of the synthetic strategy involve (i) establishing an enantiomerically pure status upon lipase mediated kinetic resolution; (ii) formation of a tetrahydrofuran ring system upon diastereoselective radical cyclization; (iii) regio- and stereo-selective arylation via hydroboration and subsequent Suzuki–Miyaura cross-coupling in a single-operation cascade and, finally, (iv) esterification employing a modified Mitsunobu protocol.
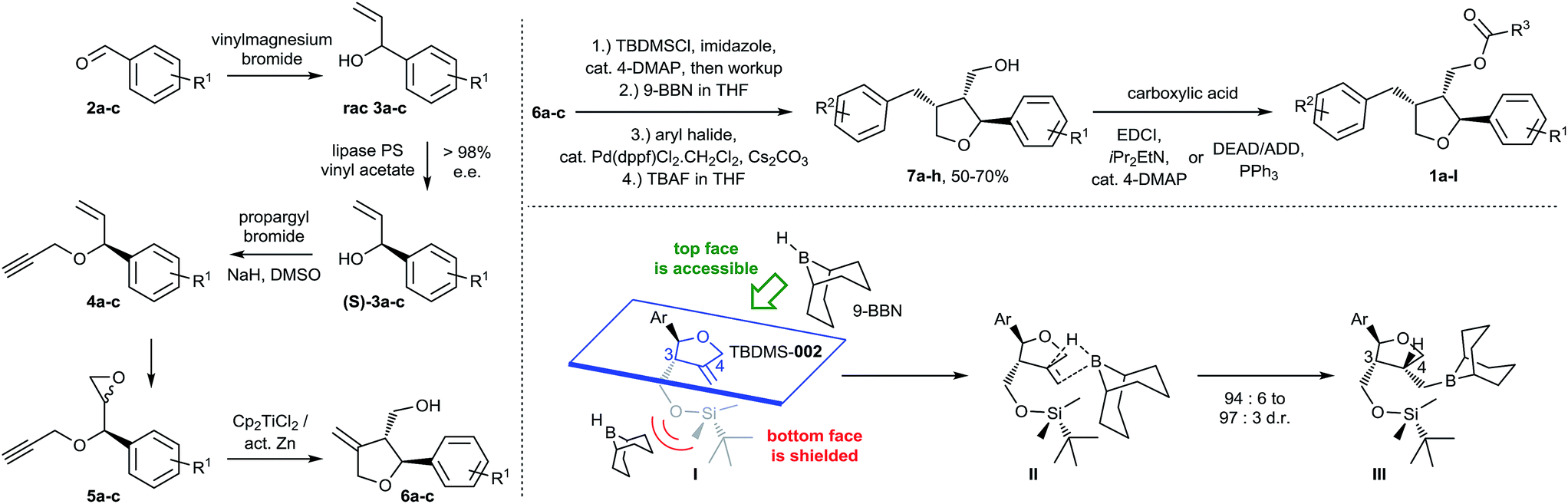 | ||
| Scheme 1 Synthesis of a library of furan-type lignans based on the leoligin hit-structure. | ||
The initial addition of vinylmagnesium bromide to benzaldehyde derivatives 2a–c is followed by the first key step, a lipase-catalyzed kinetic resolution27–31 of rac-3a–c employing Amano lipase PS. In this case, the stereoselectivity of the enzyme favors esterification of the undesired enantiomer, leaving behind enantiopure (>98% ee) alcohols (S)-3a–c. Etherification with propargylbromide gave intermediates 4a–c, paving the way for a standard m-CPBA epoxidation to 5a–c. The final cyclization to 6a–c was carried out using radical cyclization, which was previously applied by Roy and co-workers for the synthesis of structurally related racemic furan-type lignans (e.g. rac-dimethyl lariciresinol).32,33 After the individual steps were established, we successfully combined the transformation of 3 into 6 in a single operation, offering the advantage that neither 4 nor 5 had to be isolated but the crude materials could be subjected to the next steps, directly. This provided rapid access to variously substituted compounds 6a–c.
Intermediate 6 was crucial for a strategy employing maximum flexibility towards focused compound library design, since it would allow access to diversely substituted benzylic groups in position three of the THF core upon sequential hydroboration and Suzuki–Miyaura cross-coupling. For that purpose, the hydroxymethyl group in 6 was protected, initially with an acetyl group, to test whether the ester required in the final products could be introduced already at this point. However, we observed an unsatisfactory diastereoselectivity of 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18 in favor of the desired 3,4-cis configuration. Since esterification with angelic acid would lead to a trigonal planar geometry of the resulting ester, it was believed that in this case also the steric demand of the ester would be insufficient to guarantee high 3,4-cis selectivity. Hence, we switched to silyl protecting groups with tetrahedral geometry and, indeed, enough steric shielding on the bottom face of the molecule was provided when employing the bulky TBDMS group to allow 3,4-cis diastereoselective hydroboration (∼95:5) with commonly used and bulky 9-borabicyclo[3.3.1]nonane (9-BBN) as the reagent. A more detailed explanation of the steric effects leading to good diastereoselectivity can be found in the ESI.‡ The corresponding intermediate could be converted in situ with aryl bromides or iodides via Suzuki–Miyaura coupling to the arylated products.34 Furthermore, even TBDMS deprotection could be carried out in situ affording products 7a–h in 94:6 to 97:3 d.r. (typically in 50 to 70% yield over 4 steps) without isolation of any intermediate. The introduced (hetero)aryl moieties cover a wide range of electron-rich and electron-deficient residues important for systematic biological evaluation (see ESI‡ for Experimental details).
:18 in favor of the desired 3,4-cis configuration. Since esterification with angelic acid would lead to a trigonal planar geometry of the resulting ester, it was believed that in this case also the steric demand of the ester would be insufficient to guarantee high 3,4-cis selectivity. Hence, we switched to silyl protecting groups with tetrahedral geometry and, indeed, enough steric shielding on the bottom face of the molecule was provided when employing the bulky TBDMS group to allow 3,4-cis diastereoselective hydroboration (∼95:5) with commonly used and bulky 9-borabicyclo[3.3.1]nonane (9-BBN) as the reagent. A more detailed explanation of the steric effects leading to good diastereoselectivity can be found in the ESI.‡ The corresponding intermediate could be converted in situ with aryl bromides or iodides via Suzuki–Miyaura coupling to the arylated products.34 Furthermore, even TBDMS deprotection could be carried out in situ affording products 7a–h in 94:6 to 97:3 d.r. (typically in 50 to 70% yield over 4 steps) without isolation of any intermediate. The introduced (hetero)aryl moieties cover a wide range of electron-rich and electron-deficient residues important for systematic biological evaluation (see ESI‡ for Experimental details).
The synthesis of leoligin and its derivatives was completed by simple esterification to 1a–l with various saturated, unsaturated and (hetero)aromatic acyl residues, either via a standard Steglich reaction35 or via a Mitsunobu protocol (employing the more easily removable ADDP reagent).36,37 The latter had to be applied for the preparation of the parent leoligin and all other compounds containing the angelic acid ester moiety to avoid double bond isomerization which occurred otherwise upon EDCI-mediated acid activation and reversible Michael addition by the 4-DMAP acylation catalyst.38
Overall, our synthetic route was successfully employed to obtain leoligin on a gram-scale with matching NMR signals and physical properties compared with the natural compound isolate (see ESI‡), hence, unambiguously confirming the relative and absolute stereochemistry of the natural compound by total synthesis. Moreover, we could rapidly synthesize tailor made analogs to probe the structure–activity relationship within more elaborate pharmacological profiling. In these examples, modifications were made at three different positions: (i) variation of the substitution pattern of the aryl group in position 2; (ii) variation of the substitution pattern of the benzylic arene in position 4; and (iii) variation of the ester functionality.
In the first round, we screened a set of compounds with different ester functionalities for NF-κB and VSMC inhibitory action at a single dose (20 μM), which is close to the IC50 of leoligin for NF-κB (19.7 μM) and VSMC (32.1 μM) inhibition. For this, we used HEK-293/NF-κB-luc cells containing a NF-κB-driven luciferase reporter gene and PDGF-activated rat aortic VSMCs, respectively, to identify promising candidates, which were then subjected to dose–response studies (see ESI‡ for details on all cell-based assays). We discovered that the NF-κB-inhibitory activity is very susceptible to the acyl moiety; for instance, compound 1b (Fig. 2), which differs from leoligin only in the shift of one methyl group on the α,β-unsaturated system, had a much more suppressive effect on NF-κB activation, whereas VSMC proliferation inhibition remained unaffected. It was hypothesized that this might be due to the significantly reduced Michael–acceptor properties (which were problematic already in the esterification reactions) of the modified ester due to increased steric shielding of the β-carbon. Hence, saturated alkyl esters should show similar NF-κB inhibition, which was initially supported by compound 1c, which showed a similarly strong inhibition to 1b but still no effect on VSMCs. Increasing the steric bulk in the ester functionality led to an even higher activity as in compounds 1d (IC50 = 2.2 μM) and 1e (IC50 = 1.6 μM), the latter one reaching the potency of the positive control parthenolide (IC50 = 1.7 μM), a sesquiterpene lactone from feverfew of which the orally bioavailable analog, dimethylamino-parthenolide, reached clinical trials.39 Besides the improved potency for NF-κB inhibition it is of additional importance that compound 1d also shows VSMC proliferation inhibition for the first time, even though not very pronounced (IC50 = 24.3 μM). This effect is further increased in compound 1e (IC50 = 10.3 μM). Other ester modifications did not further increase the potency for VSMC proliferation inhibition. It has to be mentioned that all tested compounds so far also impaired EC proliferation. Hence, further investigation on compounds with increased selectivity for inhibiting VSMC proliferation versus ECs was necessary.
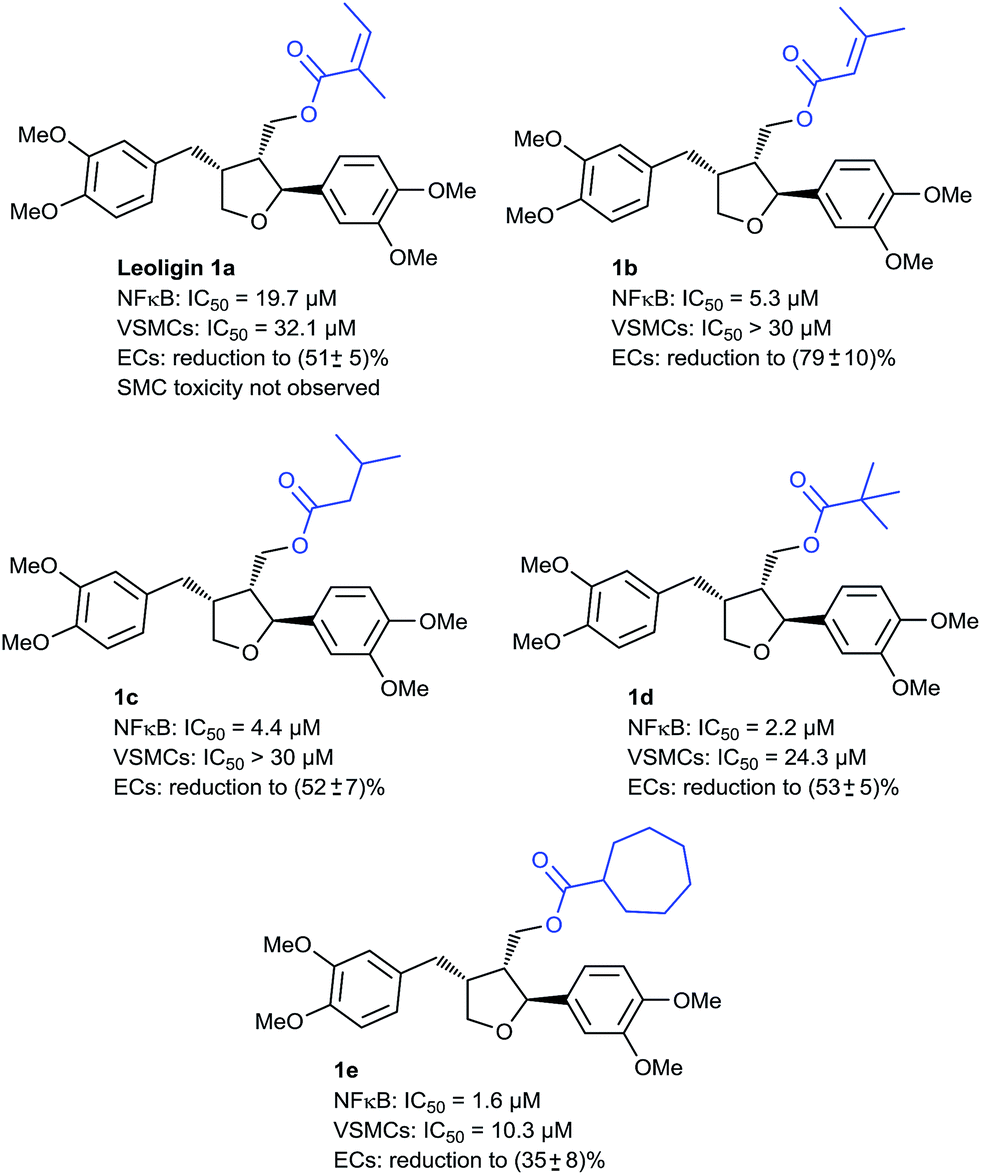 | ||
| Fig. 2 Development of leoligin analogs as NF-κB inhibitors. NF-κB inhibition is expressed by their IC50 values. Inhibition of EC proliferation is given as the reduction in cell number ± standard error of the mean at 30 μM concentration with respect to the negative control (100%). | ||
Again, we first screened at a single dose (30 μM), close to the IC50 of leoligin (32.1 μM) for inhibition of VSMC proliferation.
Here it turned out that modifications in the benzylic arene were crucial for obtaining the desired activity. In an initial screening it was observed that p-tert-butyl bearing alcoholic compound 7b was already much more active on these cells compared to leoligin, while being inactive on NF-κB (Fig. 3).
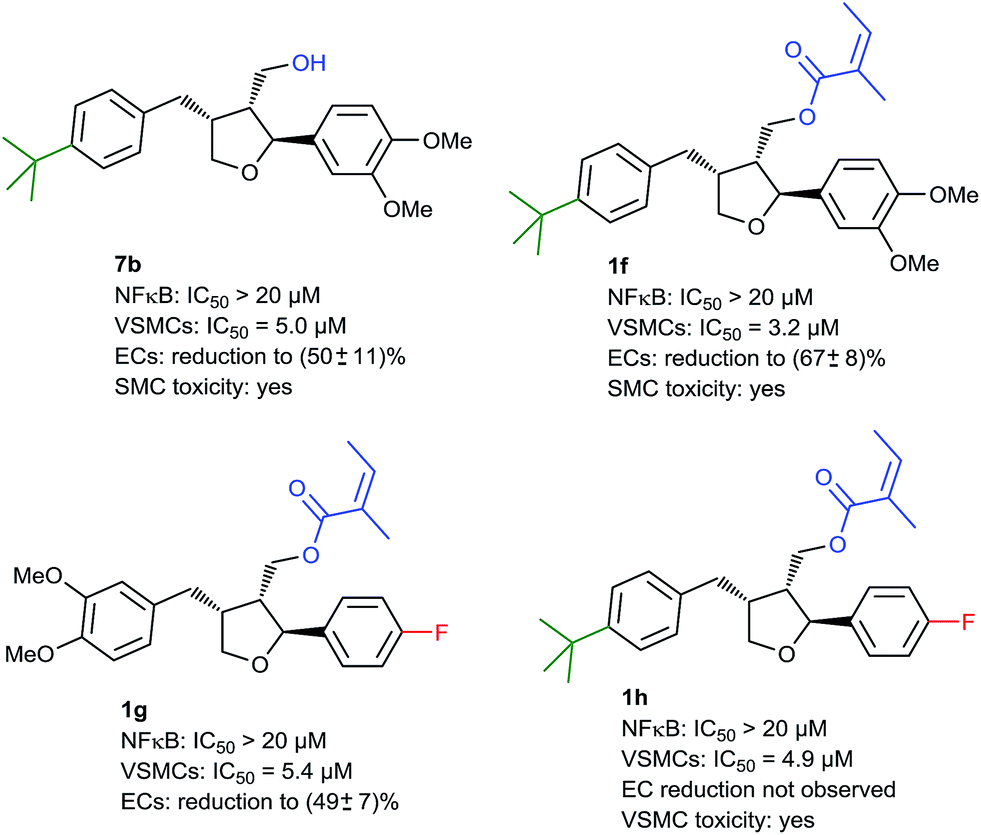 | ||
| Fig. 3 Development of leoligin analogs as inhibitors of VSMC proliferation. Inhibition of VSMC proliferation is expressed by their IC50 values. Inhibition of EC proliferation is given as the reduction in the number of cells ± standard error of the mean at 30 μM concentration with respect to the negative control (100%). VSMC toxicity was also tested (via LDH release) at 30 μM concentration; for simplicity, significant positive toxicity (p < 0.1 in one-tailed t-test) is described qualitatively with respect to the negative control. | ||
This was interesting because the analog leoligin alcohol (dimethyl lariciresinol) and most of the screened compounds devoid of an acyl group did not show any appreciable VSMC proliferation inhibition (data not shown). Consequently, attaching the angelic acid scaffold also present in leoligin should result in even higher potency. Indeed, we found that the corresponding compound 1f was about ten times more active than the natural product (IC50: 3.2 μM), however 1f also showed undesired VSMC toxicity (Fig. 3).
When screening compounds with different substitution patterns on the C2-aryl ring, compound 1g in which the 3,4-dimethoxy groups were replaced with p-fluorine showed an activity profile in favor of VSMC proliferation inhibition, and we expected a synergistic effect by combining these two modifications as in compound 1h. This did not turn out to be the case, but surprisingly, no effect on EC proliferation of 1h was observed at 30 μM (contrary to 7b, 1f and leoligin), which was precisely the desired profile (Fig. 3). To our dismay, however, a subsequent cytotoxicity study measuring cellular lactate dehydrogenase (LDH) release showed that this compound was significantly toxic to VSMCs (at a concentration of 30 μM) indicating that necrosis and/or apoptosis had occurred. Thus, this issue had to be addressed next.
Since we had shown that the desired VSMC/EC selectivity profile can be achieved by modifications of the aryl moieties, a second series of such compounds was synthesized and subjected to an activity screening. Since VSMC toxicity always occurred in the presence of a tert-butyl group on the benzylic arene, this motif was now avoided. In this screening, compound 1i (Fig. 4) showed some preference for the inhibition of VSMC proliferation vs. the inhibition of NF-κB activity while toxicity was not encountered, although the overall potency was considerably lower. To improve the potency, it was envisioned to combine the structural variations of compounds 1i and previously discussed compound 1g, with the aim to attain a potent compound without VSMC toxicity. The corresponding compound 1j was synthesized and indeed it proved to be the most active analog, so far (VSMC: IC50: 1.9 μM), while no inhibition of NF-κB or EC proliferation was observed, and no cytotoxicity was found. Compounds with similar modifications on the two arene rings were tested as well (e.g.1k and 1l, Fig. 4), showing the same activity profile and establishing this interesting chemotype as a lead for future optimizations. Although an IC50 of 1.9 μM does not match the potency of compounds in use, such as paclitaxel that reaches an IC50 of 108 nM in our system, 1j showed higher selectivity towards VSMCs versus ECs,40 which was one important goal of this study.
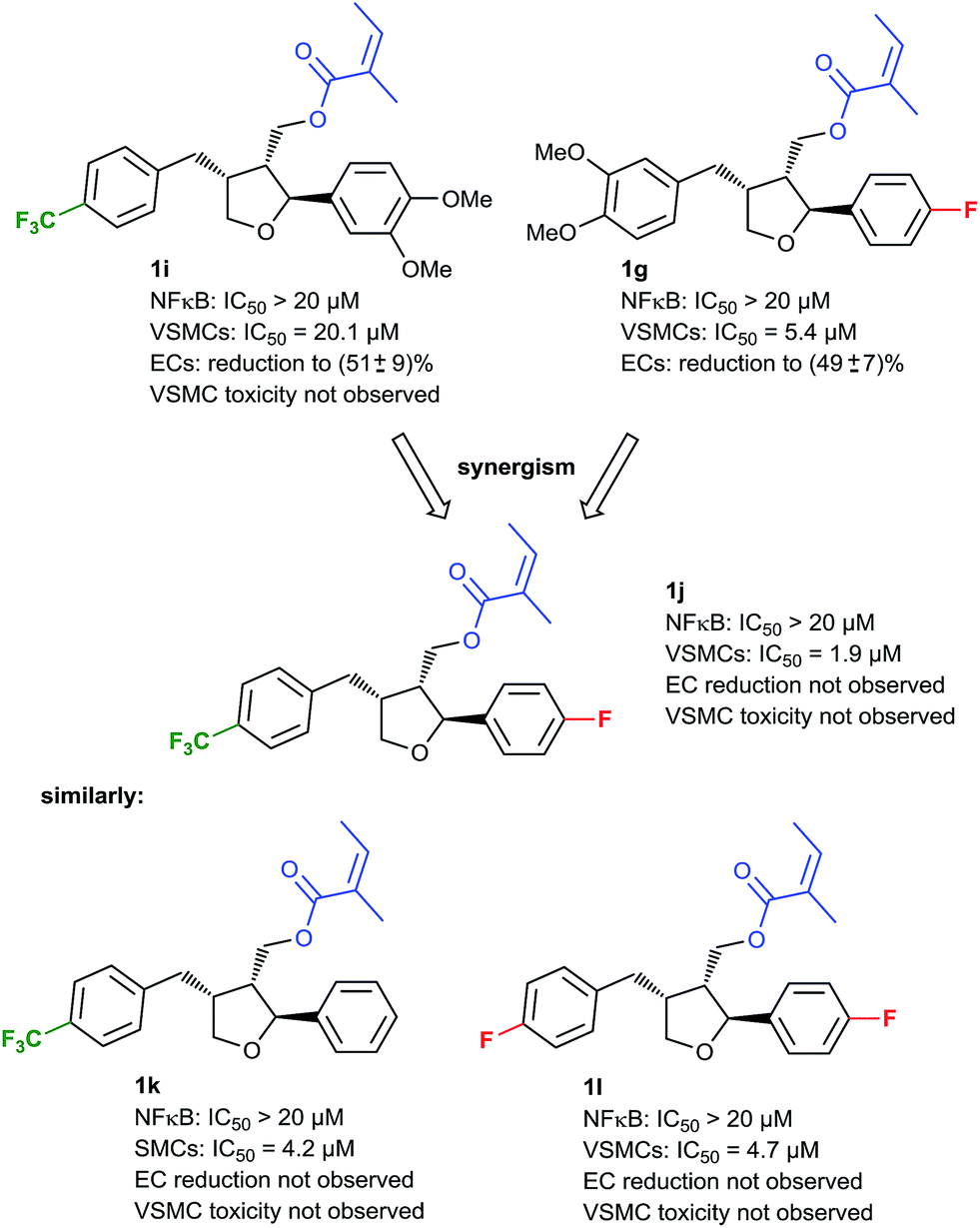 | ||
| Fig. 4 Further development of NF-κB- and VSMC-selective inhibitors. | ||
Conclusions
In conclusion, we succeeded in establishing the first total synthesis of leoligin in a stereoselective fashion on a gram-scale. Furthermore, we could show that, based on chemistry that allowed mid- and late-stage incorporation to replace moieties pertinent to the tetrahydrofuran core of leoligin, the phenotype-based activity profile of this natural product can be individualized and selective tool compounds for investigations of its pharmacology can be obtained. Specifically, compounds which selectively inhibit VSMC proliferation or suppress the NF-κB pathway were obtained by a rational design approach. This allows further elaboration of this natural product hit compound towards an interesting pharmacological tool with potential as a lead towards drug development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported in part by the Austrian Science Fund (FWF): S10710 and S10704 (NFN ‘Drugs from Nature Targeting Inflammation’). The contributions of Diyana Ogurlu and Natalie Maier-Revoredo to technical work are gratefully acknowledged.Notes and references
- C. D. Mathers and D. Loncar, PLoS Med., 2006, 3, e442 CrossRef PubMed.
- I. F. Charo and R. Taub, Nat. Rev. Drug Discovery, 2011, 10, 365 CrossRef CAS PubMed.
- K. Wadey, J. Lopes, M. Bendeck and S. George, Cardiovasc. Res., 2018, 114, 601–610 CrossRef CAS PubMed.
- R. A. Byrne, G. W. Stone, J. Ormiston and A. Kastrati, Lancet, 2017, 390, 781–792 CrossRef.
- B. Mills, T. Robb and D. F. Larson, Perfusion, 2012, 27, 520–528 CrossRef CAS PubMed.
- B. E. Claessen, J. P. S. Henriques, F. A. Jaffer, R. Mehran, J. J. Piek and G. D. Dangas, J. Am. Coll. Cardiol., 2014, 7, 1081–1092 Search PubMed.
- A. Colombo, F. Giannini and C. Briguori, J. Am. Coll. Cardiol., 2017, 70, 607–619 CrossRef PubMed.
- Y. Sotomi, Y. Onuma, C. Collet, E. Tenekecioglu, R. Virmani, S. Kleiman Neal and W. Serruys Patrick, Circ. Res., 2017, 120, 1341–1352 CrossRef CAS PubMed.
- A. I. McDonald and M. L. Iruela-Arispe, Vasc. Pharmacol., 2015, 72, 9–15 CrossRef CAS PubMed.
- K. Korybalska, E. Kawka, A. Breborowicz and J. Witowski, J. Physiol. Pharmacol., 2017, 68, 397–405 CAS.
- D.-H. Lee and J. M. de la Torre Hernandez, Eur. Cardiol., 2018, 13, 54–59 CrossRef PubMed.
- K. Toutouzas, A. Colombo and C. Stefanadis, Eur. Heart J., 2004, 25, 1679–1687 CrossRef CAS PubMed.
- J. Dabek, A. Kulach and Z. Gasior, Pharmacol. Rep., 2010, 62, 778–783 CrossRef CAS PubMed.
- B. Pamukcu, G. Y. H. Lip and E. Shantsila, Thromb. Res., 2011, 128, 117–123 CrossRef CAS PubMed.
- M. J. Dobner, E. P. Ellmerer, S. Schwaiger, O. Batsugkh, S. Narantuya, M. Stütz and H. Stuppner, Helv. Chim. Acta, 2003, 86, 733–738 CrossRef CAS.
- M. J. Dobner, S. Sosa, S. Schwaiger, G. Altinier, R. Della Loggia, N. C. Kaneider and H. Stuppner, Planta Med., 2004, 70, 502–508 CrossRef CAS PubMed.
- C. Wawrosch, S. Schwaiger, H. Stuppner and B. Kopp, Fitoterapia, 2014, 97, 219–223 CrossRef CAS PubMed.
- L. Wang, A. Ladurner, S. Latkolik, S. Schwaiger, T. Linder, J. Hosek, V. Palme, N. Schilcher, O. Polansky, E. H. Heiss, H. Stangl, M. D. Mihovilovic, H. Stuppner, V. M. Dirsch and A. G. Atanasov, J. Nat. Prod., 2016, 79, 1651–1657 CrossRef CAS PubMed.
- B. Scharinger, B. Messner, A. Tuerkcan, D. Schuster, A. Vuorinen, F. Pitterl, K. Heinz, K. Arnhard, G. Laufer, M. Grimm, H. Stuppner, H. Oberacher, P. Eller, A. Ritsch and D. Bernhard, J. Mol. Cell. Cardiol., 2016, 99, 35–46 CrossRef CAS PubMed.
- K. Duwensee, S. Schwaiger, I. Tancevski, K. Eller, M. van Eck, P. Markt, T. Linder, U. Stanzl, A. Ritsch, J. R. Patsch, D. Schuster, H. Stuppner, D. Bernhard and P. Eller, Atherosclerosis, 2011, 219, 109–115 CrossRef CAS PubMed.
- S. Schwaiger, M. Adams, C. Seger, E. P. Ellmerer, R. Bauer and H. Stuppner, Planta Med., 2004, 70, 978–985 CrossRef CAS PubMed.
- U. Reisinger, S. Schwaiger, I. Zeller, B. Messner, R. Stigler, D. Wiedemann, T. Mayr, C. Seger, T. Schachner, V. M. Dirsch, A. M. Vollmar, J. O. Bonatti, H. Stuppner, G. Laufer and D. Bernhard, Cardiovasc. Res., 2009, 82, 542–549 CrossRef CAS PubMed.
- A. G. Atanasov, B. Waltenberger, E.-M. Pferschy-Wenzig, T. Linder, C. Wawrosch, P. Uhrin, V. Temml, L. Wang, S. Schwaiger, E. H. Heiss, J. M. Rollinger, D. Schuster, J. M. Breuss, V. Bochkov, M. D. Mihovilovic, B. Kopp, R. Bauer, V. M. Dirsch and H. Stuppner, Biotechnol. Adv., 2015, 33, 1582–1614 CrossRef CAS PubMed.
- B. Waltenberger, A. G. Atanasov, E. H. Heiss, D. Bernhard, J. M. Rollinger, J. M. Breuss, D. Schuster, R. Bauer, B. Kopp, C. Franz, V. Bochkov, M. D. Mihovilovic, V. M. Dirsch and H. Stuppner, Monatsh. Chem., 2016, 147, 479–491 CrossRef CAS PubMed.
- T. Linder, M. Schnürch and M. D. Mihovilovic, Targets Heterocycl. Syst., 2015, 19, 274–298 CAS.
- Y. Xia, L. Zou, J. Zhang and W. Zhang, J. Chem. Res., 2018, 42, 350–353 CrossRef CAS.
- W. S. Qayed, A. S. Aboraia, H. M. Abdel-Rahman and A. F. Youssef, J. Chem. Pharm. Res., 2015, 7, 311–322 CAS.
- U. Bornscheuer and R. Kazlauskas, Hydrolases in Organic Synthesis: Regio- or Stereoselective Biotransformations, Wiley, 1999 Search PubMed.
- A. Ghanem and H. Y. Aboul-Enein, Chirality, 2005, 17, 1–15 CrossRef CAS PubMed.
- P. Chen and P. Xiang, Tetrahedron Lett., 2011, 52, 5758–5760 CrossRef CAS.
- X. Fu and K. L. Parkin, J. Am. Oil Chem. Soc., 2004, 81, 45 CrossRef CAS.
- P. K. Mandal, G. Maiti and S. C. Roy, J. Org. Chem., 1998, 63, 2829–2834 CrossRef CAS.
- T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986–997 CrossRef CAS.
- C.-H. Chen, Y.-K. Chen and C.-K. Sha, Org. Lett., 2010, 12, 1377–1379 CrossRef CAS PubMed.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed., 1978, 17, 522–524 CrossRef.
- O. Mitsunobu, Synthesis, 1981, 1981, 1–28 CrossRef.
- K. C. K. Swamy, N. N. B. Kumar, E. Balaraman and K. V. P. P. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef CAS PubMed.
- X. Liang, G. Grue-Sørensen, A. K. Petersen and T. Högberg, Synlett, 2012, 23, 2647–2652 CrossRef CAS.
- A. Ghantous, A. Sinjab, Z. Herceg and N. Darwiche, Drug Discovery Today, 2013, 18, 894–905 CrossRef CAS PubMed.
- R. Liu, E. H. Heiss, N. Sider, A. Schinkovitz, B. Gröblacher, D. Guo, F. Bucar, R. Bauer, V. M. Dirsch and A. G. Atanasov, Mol. Nutr. Food Res., 2015, 59, 843–852 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Johannes Fröhlich on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures, NMR-spectra of all compounds, and description of pharmacological experiments. See DOI: 10.1039/c9sc00446g |
| § Current affiliation: School of Pharmacy, Yantai University, 264005, Yantai, PR China. |
| ¶ Current affiliation: Institute of Genetics and Animal Breeding of the Polish Academy of Sciences, 05-552, Jastrzebiec, Poland. |
| || Current affiliation: Institute for Cardiovascular Prevention (IPEK), Ludwig-Maximilians-University, Munich, Germany. |
| This journal is © The Royal Society of Chemistry 2019 |