
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiiron oxo reactivity in a weak-field environment†‡
Elizabeth J.
Johnson‡
 ,
Claudia
Kleinlein‡
,
Rebecca A.
Musgrave
and
Theodore A.
Betley
*
,
Claudia
Kleinlein‡
,
Rebecca A.
Musgrave
and
Theodore A.
Betley
*
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA. E-mail: betley@chemistry.harvard.edu
First published on 9th May 2019
Abstract
Concomitant deprotonation and metalation of a dinucleating cofacial Pacman dipyrrin ligand platform tBudmxH2 with Fe2(Mes)4 results in formation of a diiron complex (tBudmx)Fe2(Mes)2. Treatment of (tBudmx)Fe2(Mes)2 with one equivalent of water yields the diiron μ-oxo complex (tBudmx)Fe2(μ-O) and free mesitylene. A two-electron oxidation of (tBudmx)Fe2(μ-O) gives rise to the diferric complex (tBudmx)Fe2(μ-O)Cl2, and one-electron reduction from this FeIIIFeIII state allows for isolation of a mixed-valent species [Cp2Co][(tBudmx)Fe2(μ-O)Cl2]. Both (tBudmx)Fe2(μ-O) and [Cp2Co][(tBudmx)Fe2(μ-O)Cl2] exhibit basic character at the bridging oxygen atom and can be protonated using weak acids to form bridging diferrous hydroxide species. The basicity of the diferrous oxo (tBudmx)Fe2(μ-O) is quantified through studies of the pKa of its conjugate acid, [(tBudmx)Fe2(μ-OH)]+, which is determined to be 15.3(6); interestingly, upon coordination of neutral solvent ligands to yield (tBudmx)Fe2(μ-O)(thf)2, the basicity is increased as observed through an increase in the pKa of the conjugate acid [(tBudmx)Fe2(μ-OH)(thf)2]+ to 26.8(6). In contrast, attempts to synthesize a diferric bridging hydroxide by two-electron oxidation of [(tBudmx)Fe2(μ-OH)(thf)2]+ resulted in isolation of (tBudmx)Fe2(μ-O)Cl2 with concomitant loss of a proton, consistent with the pKa of the conjugate acid [(tBudmx)Fe2(μ-OH)Cl2]+ determined computationally to be −1.8(6). The foregoing results highlight the intricate interplay between oxidation state and reactivity in diiron μ-oxo units.
Introduction
Diiron units featuring bridging oxygen ligands are important structural motifs in metalloenzymes.1–4 For example, methane monooxygenase, ribonucleotide reductase, and hemerythrin feature diiron units which are responsible for oxygen activation or transport of dioxygen to a variety of substrates.5,6 During these processes, a diiron unit which is bridged by one or two oxygen-based ligands passes through oxidation states ranging from FeIIFeII to FeIVFeIV. Synthetic model complexes featuring a μ-oxo or μ-hydroxo bridge have been prepared in order to create functional mimics of these enzymes.7–9 In particular, the interconversion between hydroxo- and oxo-bridged states has been of interest as it is proposed to play an important role during the catalytic cycle of metalloenzymes.10,11 While there are many synthetic examples of diferric Fe–O–Fe units,12–18 few mixed-valent FeII–O–FeIII complexes and FeII–O–FeII complexes have been reported.19–22 In contrast, a variety of model complexes for both diferric and diferrous hydroxides have been synthesized.9,23,24To better understand how the molecular oxidation state of a diiron core influences the acid–base properties of a bridging (hydr)oxo ligand, we explored the coordination and redox chemistry of a diiron unit in a cofacial dipyrrin Pacman unit. Herein, we report the synthesis and versatile reactivity of a family of diiron oxo and hydroxo complexes in three different molecular oxidation states. Reactions include acid–base chemistry as well as one and two electron redox chemistry, which demonstrate the intricate interplay between oxidation state, coordination environment, and reactivity.
Results and discussion
I. Synthesis of a diiron starting material
Formation of the diiron dipyrrin Pacman complex (tBudmx)Fe2(Mes)2 (1) proceeds cleanly from the reaction of the cofacial dipyrrin Pacman ligand tBudmxH2 with Fe2(Mes)4 (Mes = 2,4,6-Me3C6H2) in benzene at 65 °C for four hours (Scheme 1). The composition and purity of 1 were established by 1H NMR spectroscopy, 57Fe Mössbauer spectroscopy, and combustion analysis. X-ray diffraction studies on a single crystal of 1 obtained from a toluene solution layered with hexanes at −35 °C revealed two non-interacting iron sites residing in a trigonal-planar geometry (Fig. S-27†). 57Fe Mössbauer analysis of 1 confirmed the presence of a single iron environment (δ = 0.46 mm s−1, |ΔEQ| = 0.95 mm s−1) in line with other high-spin FeII complexes containing alkyl or aryl ligands (Fig. S-1†).25–27 Magnetometry data was consistent with the presence of two uncoupled S = 2 centers (Fig. S-23†).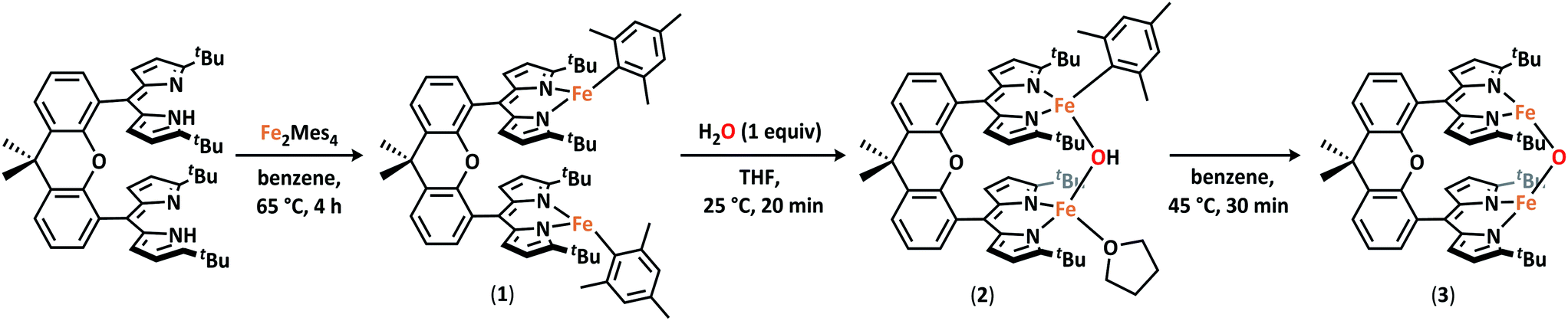 | ||
| Scheme 1 Synthesis of (tBudmx)Fe2(μ-O) (3). | ||
II. Isolation and characterization of a diferrous μ-oxo
Diiron complex 1 is a versatile starting material for the preparation of μ-hydroxo and μ-oxo diiron complexes. Addition of one equivalent of water to 1 results in complete consumption of the starting material and formation of a new, asymmetric species, as evidenced by the presence of 18 paramagnetically-shifted 1H NMR resonances along with the formation of free mesitylene.28 Cooling a concentrated diethyl ether solution of this product to −35 °C affords an orange crystalline material which was characterized by X-ray diffraction to unveil an asymmetric Pacman complex (tBudmx)Fe2(μ-OH)(Mes)(thf) (2) featuring a bridging hydroxide ligand (Scheme 1). One iron site remains bound to a mesitylene ligand with an Fe–O bond length of 2.045(3) Å while the second iron center is coordinated by a THF solvent molecule and exhibits a shorter Fe–O bond of 1.942(4) Å (Fig. S-28†). The asymmetric nature of this complex is furthermore reflected by the 57Fe Mössbauer spectrum, which contains two quadrupole doublets (δ = 0.80 mm s−1, |ΔEQ| = 3.23 mm s−1 and δ = 0.99 mm s−1, |ΔEQ| = 2.92 mm s−1) (Fig. S-2†). Solid-state Fourier-transform infrared (FTIR) spectra of 2 were collected to further confirm the identity of the bridging ligand as a μ-hydroxide (Fig. S-12†). Spectra of 2 show an O–H stretching band at 3638 cm−1 that shifts to 2685 cm−1 when D2O is used to prepare the analogous deuterated bridging hydroxide complex (tBudmx)Fe2(μ-OD)(Mes)(thf).While 2 is stable in ethereal solvents at room temperature, slow deprotonation of the bridging hydroxide ligand concomitant with release of the second mesitylene molecule is observed in non-coordinating solvents. Heating of 2 in benzene to 45 °C for 30 minutes leads to complete consumption of starting material and furnishes a new product (3) which is easily discernible by 1H NMR spectroscopy as a C2-symmetric species distinct from the starting material (Scheme 1).
Zero-field 57Fe Mössbauer analysis of 3 at 90 K (Fig. 2b, top) reveals the presence of a single iron environment with parameters (δ = 0.68 mm s−1, |ΔEQ| = 0.88 mm s−1) similar to other three-coordinate FeII dipyrrin complexes previously synthesized.29 The 57Fe Mössbauer spectrum contains 5% of an additional species with parameters corresponding to a four-coordinate high-spin FeII compound which we assign as a small amount of an unknown bridging hydroxide species. An X-ray diffraction study on single crystals grown from a concentrated diethyl ether solution revealed the major product 3 as a diiron complex bearing a bridging oxido ligand, (tBudmx)Fe2(μ-O) (Fig. 1a). A crystallographically imposed C2 axis renders the two halves of the complex equivalent. The Fe–O (1.7939(14) Å) bond length is substantially shorter than the Fe–O distances in 2 (2.045(3) Å, 1.942(4) Å) and in other FeII hydroxides, and is in line with previously reported diferrous μ-oxo species [LFe]2O (Fe–O 1.7503(4) Å, L = ArNC(tBu)CHC(tBu)NAr−, where Ar = 2,6-diisopropylphenyl;22 1.784(9) Å, L = PhBP3iPr;21 1.753(2) Å, L = PhBP3Ph (ref. 21)). The Fe–O–Fe angle in 3 (116.00(14)°) deviates significantly from linearity, which is more acute than previously reported diferrous oxo complexes (∠Fe–O–Fe 167.55(14)°, L = ArNC(tBu)CHC(tBu)NAr−;22 174.7(4)°, L = PhBP3iPr;21 147.7(3)°, L = PhBP3Ph (ref. 21)). The more acute Fe–O–Fe angle can be attributed to the restraints on the coordination chemistry imposed by the dinucleating ligand platform in 3.
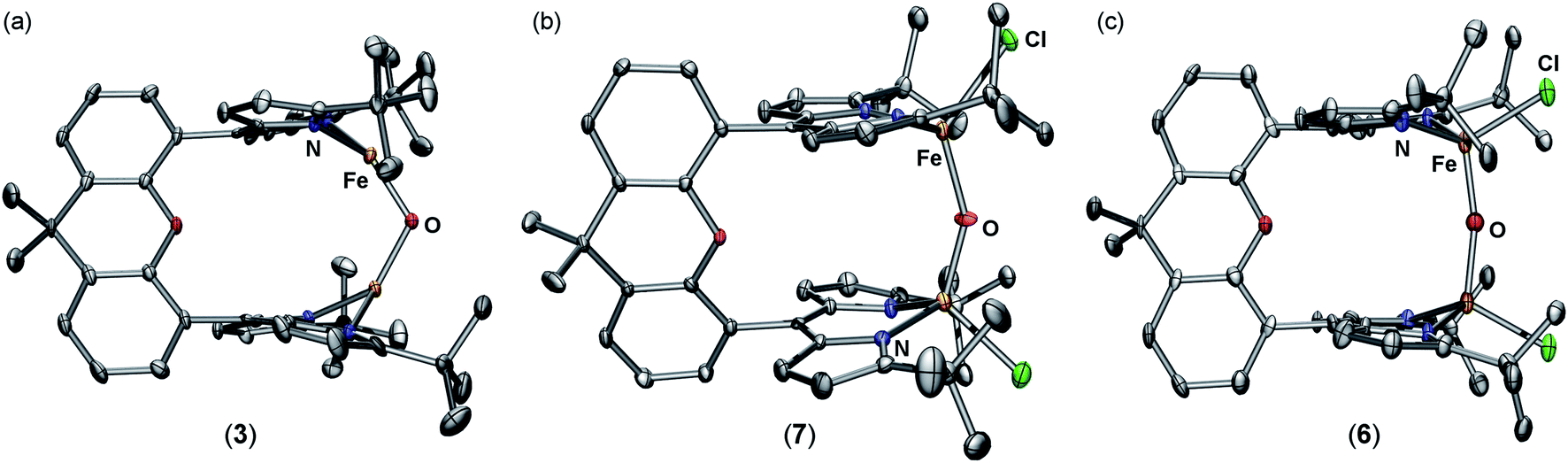 | ||
| Fig. 1 Solid-state molecular structures for (a) (tBudmx)Fe2(μ-O) (3), (b) [Cp2Co][(tBudmx)Fe2(μ-O)Cl2] (7), and (c) (tBudmx)Fe2(μ-O)Cl2 (6) with thermal ellipsoids at 50% probability level. Color scheme: Fe, orange; N, blue; O, red; Cl, green. Hydrogens, solvent molecules, and counterion in 7 are omitted for clarity. | ||
III. Reactivity and basicity of the diferrous μ-oxo
With 3 in hand, we began examining its reactivity profile. Treatment of 3 with one equivalent of a weak acid such as lutidinium tetraphenylborate in THF results in quantitative formation of a diiron μ-hydroxide cation [(tBudmx)Fe2(μ-OH)(thf)2][BPh4] (4) (Scheme 2). Given the observed reactivity of 3 with acids, we sought to quantify the basicity of 3 by establishing the pKa value of its conjugate acid. Following the precedent set by Smith et al., we attempted to use a combined approach of experiment and calculation to justify the reactivity of 3 with various weak acids.30 Due to the limited solubility of 3 and many of the suitable acids in non-coordinating solvents, we selected THF as the solvent for this study. Experimentally probing the protonation of 3 proved to be challenging, as the anticipated conjugate acid product, [(tBudmx)Fe2(μ-OH)]+ (4a), is not stable to reaction conditions, as in many cases it reacts with the generated conjugate base, leading to multiple species observed by 1H NMR spectroscopy. Consequently, efforts focused on replicating the theoretical procedure used by Smith et al. to computationally determine the pKa value of 4a. Using density functional theory (see ESI for details†), the acid–base reaction shown in Scheme 3 was considered, in which HA is a weak acid with reported pKa value in THF.31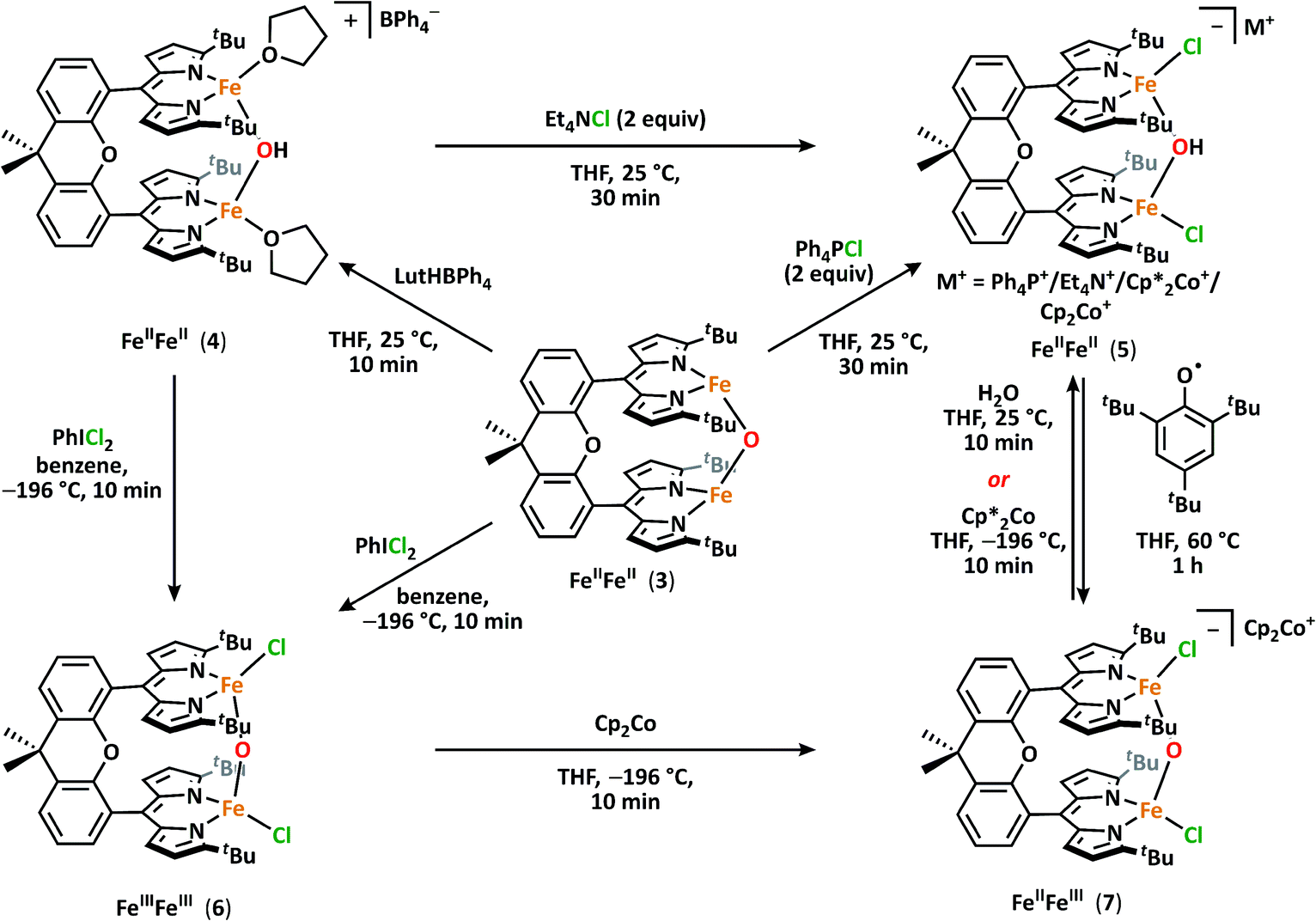 | ||
| Scheme 2 Interconversion between diiron hydroxide and oxide species. | ||
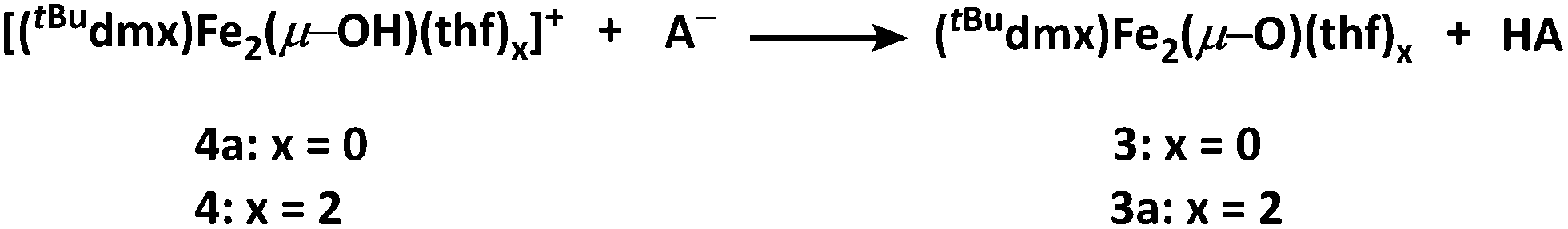 | ||
| Scheme 3 General acid–base reaction studied. | ||
Since these reactions were performed in THF, we examined the stability of 3 under these reaction conditions and discovered that solvents can bind the open coordination site at each metal center to begin to form (tBudmx)Fe2(μ-O)(thf)2 (3a) within minutes, as evidenced by 57Fe Mössbauer spectroscopy (Fig. S-4†). Thus, we modelled the acid–base reaction for both 3 and for 3a (Scheme 3), the latter being the suspected active metal species during these reactions. The pKa value calculated for 4a (which is three-coordinate at iron) was 15.3(6), and upon coordination of two solvent molecules to form [(tBudmx)Fe2(μ-OH)(thf)2]+ (4) increased to 26.8(6). We hypothesize that the coordination of a fourth ligand to each iron center results in the bridging oxo moiety adopting greater O2− character, thus rendering the oxo ligand more basic. Computationally, we observe that the Fe–O covalency in 3a decreases upon addition of the solvent ligands to 3, reflected in a diminished Mayer bond order32,33 (3, 0.811; 3a, 0.798), thus resulting in enhanced oxide character and basicity in 3a (Table S-6†). Several examples of metal oxo and nitrido complexes featuring enhanced reactivity upon coordination of additional neutral ligands34–36 or increased basicity due to coordination of anionic ligands37–40 have been studied. However, to our knowledge, no synthetic examples have been reported in which the change in basicity of a metal oxo species has been quantified upon coordination of neutral donors. In this case, we remarkably observe an increase in the basicity of a diiron bridging oxo complex by ten orders of magnitude upon coordination of the neutral donor THF.
To further demonstrate the basicity of these diiron(II) μ-oxo complexes, we examined the reactivity of 3 upon coordination of anionic ligands. Stirring 3 in the presence of excess chloride sources such as tetraphenylphosphonium chloride in THF affords [Ph4P][(tBudmx)Fe2(μ-OH)Cl2] (5a) in high yield with no observable intermediates as ascertained by 1H NMR (Scheme 2). The origin of the hydroxide proton in this product is unknown and protonation could not be prevented even in silylated glassware and freshly dried solvent. We propose that upon coordination of chloride ligands and formation of an anticipated dianionic iron complex, the basicity of the bridging oxo increases further, resulting in instantaneous protonation. Attempts to determine the pKa value of the [(tBudmx)Fe2(μ-OH)Cl2]− (the conjugate acid of the anticipated dianionic complex [(tBudmx)Fe2(μ-O)Cl2]2−) were not successful; however, based on the observed reactivity we hypothesize that this species would be more basic than 3a and thus feature a pKa > 26.8. The assignments of 4 and 5 as diiron μ-hydroxides were confirmed by X-ray diffraction studies (Fig. 2a and S-32†), 57Fe Mössbauer spectroscopy (Fig. 2b, bottom; S-6†), and FTIR spectroscopy (Fig. S-14 and S-15†). One notable observation was that the O–H (ν = 3548 cm−1) and O–D (ν = 2587 cm−1) frequencies for 4 and its deuterated analogue [(tBudmx)Fe2(μ-OD)(thf)2][BPh4] in the FTIR spectrum are lower than are often observed for such stretches,29 which we hypothesize is due to a hydrogen-bonding interaction between the bridging hydroxide and a THF solvent molecule (OH–OTHF 1.899(3) Å), as observed in the solid-state. Magnetometry studies indicate that 4 features antiferromagnetic coupling of two S = 2 iron centers with an exchange coupling J = −13.3 cm−1, which is in line with the expected values for diiron bridging hydroxide complexes (Fig. S-25†).10
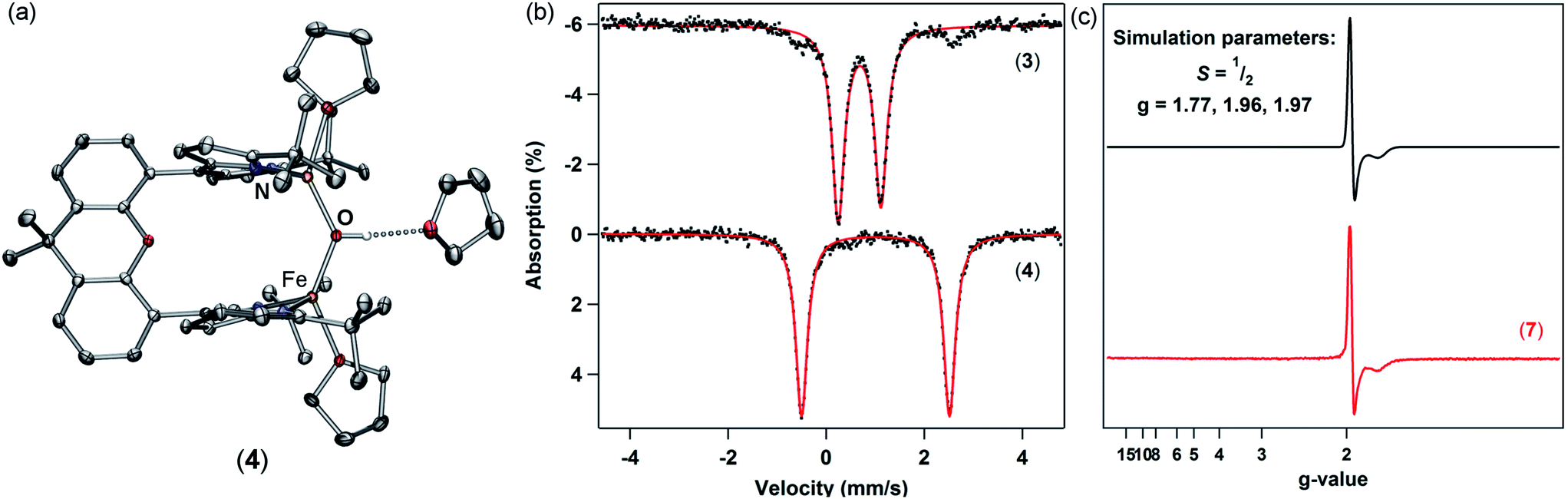 | ||
| Fig. 2 (a) Solid-state molecular structure for [(tBudmx)Fe2(μ-OH)(thf)2][BPh4] (4) with thermal ellipsoids at 50% probability level. Color scheme: Fe, orange; N, blue; O, red. All hydrogen atoms (except the μ-hydroxo proton), solvent molecules (except the hydrogen-bonded THF), and the counterion are omitted for clarity. (b) Zero-field 57Fe Mössbauer spectra of 3 (top) and 4 (bottom) collected at 90 K: δ = 0.68 mm s−1, |ΔEQ| = 0.88 mm s−1 for 3 and δ = 1.01 mm s−1, |ΔEQ| = 3.02 mm s−1 for 4. (c) EPR spectrum of 7 (X-band, 9.3298 GHz) in a 2-methyltetrahydrofuran glass at 4 K (red) and simulated spectrum (black). | ||
IV. Oxidation of diferrous μ-oxo
Having established the basic nature of 3, we were interested in studying its redox chemistry. Particularly, we sought to address how the acid/base properties of the oxo complexes were influenced by the oxidation states of the metal centers. Addition of one equivalent of iodobenzene dichloride to 3 in benzene resulted in an immediate color change to dark red and a new paramagnetically shifted 1H NMR spectrum. 57Fe Mössbauer analysis of the reaction mixture revealed a single iron-containing species with isomer shift and quadrupole splitting values supporting the assignment of high-spin FeIII (δ = 0.30 mm s−1, |ΔEQ| = 1.38 mm s−1) indicating formation of (tBudmx)Fe2(μ-O)Cl2 (6) (Fig. S-7†). Single crystals of 6 were grown by slow diffusion of diethyl ether into a saturated THF solution of 6 at −35 °C (Fig. 1c). The solid state molecular structure of 6 features a crystallographically imposed C2 axis that renders both iron sites equivalent. The iron–oxygen distance of 1.7734(10) Å is in agreement with the plethora of crystallographically characterized diferric μ-oxo compounds, and the Fe–O–Fe angle of 167.1(3)° only slightly deviates from linearity.41Complex 6 can similarly be accessed by a two-electron oxidation of the diferrous hydroxide 4 using iodobenzene dichloride (Scheme 2). Oxidation of the hydroxide 4 does not furnish the anticipated cationic diferric bridging hydroxide species, rather the reaction product was spectroscopically identical to 6. Loss of the hydroxide proton in 4 was confirmed using IR spectroscopy. Unfortunately, we were not able to identify the fate of the proton in this reaction; however, the putative diferric bridging hydroxide intermediate [(tBudmx)Fe2(μ-OH)Cl2]+ (6a) was found to be quite acidic with a computationally determined pKa of −1.8(6), which we propose is the driving force for facile loss of the proton concomitant with oxidation to the diferric state.
Herein, we observe a dramatic decrease in the basicity upon oxidation to the diferric state from a diferrous μ-oxo. This trend can be explained by consideration of the Fe–O covalency, in which greater covalency results in decreased basicity of the bridging oxo. Increased covalency upon oxidation is supported by a combination of computational and magnetometry studies; specifically, increased Mayer bond orders (from 0.811 in diferrous 3 to 0.947 in diferric 6, Table S-6†), enhanced orbital overlap in the unrestricted corresponding orbitals in 6 compared to 3 (Tables S-8 and S-9†), and an increased exchange coupling constant upon oxidation to the diferric state (Table 1, Fig. S-24 and S-26†) are observed. Taking into account the previous comparison between 3 and 3a, the covalency of the Fe–O interactions are not governed solely by the geometry of the linkage, but rather are largely dictated by the electrophilicity of the iron sites within the Fe2(μ-O) unit. As such, the oxo moiety is more basic in 3a than 3 due to THF ligation diminishing the iron electrophilicity, and is more basic in 3 than 6 due to lower iron electrophilicity in the more reduced molecular oxidation state.
| Complex | Fe–O distances (Å) | Fe–O–Fe angle (°) | Fe–Fe distance (Å) | pKa of conjugate acid | J calc (cm−1) | J exp (cm−1) | Reference |
|---|---|---|---|---|---|---|---|
| a Based on geometry-optimized structure. b L = (2,6-diisopropylphenyl)NC(tBu)CHC(tBu)(2,6-diisopropylphenyl). c L = PhBP3iPr. d L = PhBP3Ph. e L = 2,2′-(2-methyl-2-(pyridine-2-yl)propane-1,3-diyl)bis-(azanediyl)bis(methylene)diphenol. f L = 4-((1-methyl-1H-imidazol-2-yl)methyl)-1-thia-4,7-diazacyclononane. | |||||||
| 3 | 1.7939(14) | 116.00(14) | 3.0425(10) | 15.3(6) | −65.9 | −53.2 | This work |
| 3a | 1.858, 1.859 | 126.281 | 3.315 | 26.8(6) | −69.3 | — | This work |
| 6 | 1.7734(10) | 167.1(3) | 3.5244(17) | −1.8(6) | −96.4 | −122 | This work |
| (FeII)2(μ-O)b | 1.7503(4) | 167.55(14) | 3.4831(4) | — | ∼−200 to −250 | — | 22 |
| (FeII)2(μ-O)c | 1.784(9) | 174.7(4) | 3.573(9) | — | — | — | 21 |
| (FeII)2(μ-O)d | 1.753(2) | 147.7(3) | 3.367(3) | — | — | — | 21 |
| (FeIII)2(μ-O)e | 1.8194(16), 1.8156(16) | 143.71(10) | 3.4542(7) | 21.3(1) | — | −87.5 | 10 |
| (FeIII)2(μ-O)f | 1.791(2), 1.803(2) | 168.47(13) | 3.5763(6) | 6.1(3) | — | — | 43 |
The foregoing results should be compared with the analysis of the relationship between the Fe–O–Fe angle, Fe–O covalency, and basicity for diiron(III) oxo complexes put forth by Solomon and coworkers.3,42 In Solomon's studies, enhanced basicity is observed with a more acute Fe–O–Fe angle due to a reduction in the Fe–O orbital overlap, thereby resulting in pronounced electron density on the bridging oxide ligand when examining an isovalent series.3,42 Further comparison with other diiron μ-oxo complexes corroborates this relationship, as a diiron(III) μ-oxo synthesized by Houser and coworkers10 (∠Fe–O–Fe 143.71(10)°, pKa 21.3(1)) is several orders of magnitude more basic than 6 (∠Fe–O–Fe 167.1(3)°, pKa –1.8(6)) (Table 1); whereas an example by Grapperhaus and coworkers43 with similar core metrics to 6 (∠Fe–O–Fe 168.47(13)°, pKa 6.1(3)) is less basic than the Houser example. The Grapperhaus example is more basic than 6, which can be attributed to differences in coordination geometry; the tetrahedral iron sites in 6 are influenced more by the enhanced covalency to the oxo moiety (due to the increased iron electrophilicity) than the octahedrally coordinated iron sites in the Grapperhaus example.43
V. Synthesis and characterization of a mixed-valent diiron μ-oxo
Further exploration of the redox chemistry of the diiron μ-oxo complexes studied herein was pursued. A cyclic voltammogram of 6 measured in THF revealed a quasi-reversible reduction event at −861 mV versus [Cp2Fe]+/0 (Fig. S-20–S-22†). The peak-to-peak separation for this event varies as a function of scan rate, possibly indicating slow electron transfer rate constants. At slower scan rates, i.e. 0.02 V s−1, the peak current for oxidation decreases and the redox event becomes irreversible. Nevertheless, these data suggest that a mixed-valent μ-oxo complex is chemically accessible.The redox chemistry of 6 was further explored by using chemical reductants. Cp2Co (exhibiting a reduction potential of −1.33 mV versus [Cp2Fe]+/0 in dichloromethane44) was employed to reduce 6. Treatment of 6 with one equivalent of cobaltocene in THF for ten minutes affords [Cp2Co][(tBudmx)Fe2(μ-O)Cl2] (7). A frozen solution EPR spectrum collected at 4 K displays a pseudo-axial signal with features at g = 1.97, and 1.77 consistent with an S = 1/2 spin state (Fig. 2c). Antiferromagnetic coupling of high-spin ferric (S = 5/2) and ferrous (S = 2) centers in 7 explains the doublet spin state and supports the formation of a mixed-valent diiron bridging oxo complex. X-ray diffraction studies on single crystals of 7 obtained from a concentrated THF solution at 70 °C unveiled two crystallographically distinct iron sites (Fig. 1b); two distinct sites were further observed by 57Fe Mössbauer spectroscopy, the parameters of which support one center being more consistent with a high spin FeII and the other as a high spin FeIII center (Fig. S-8 and S-9†). The Fe–O bond distances in 7 (1.8034(15) Å, 1.8928(15) Å) are longer than those in both diferrous 3 and diferric 6, and the difference of 0.09 Å indicates a potentially localized nature. The Fe–O–Fe angle of 148.98(10)° resides in between the fairly linear Fe–O–Fe vector in the diferric state (167.1(3)°) and the significantly more acute Fe–O–Fe angle of the diferrous complex (116.00(14)°).
VI. Reactivity of the mixed-valent diiron μ-oxo
Upon synthesizing the mixed-valent oxo complex 7via the method previously described, 1H NMR and 57Fe Mössbauer spectroscopies reveal the formation of 7 together will a small amount of hydroxide 5. While 7 is stable at 70 °C for 12 hours and does not react with weak C–H bonds (such as those in 1,4-cyclohexadiene), this mixed valent species is protolytically sensitive. Addition of one equivalent of water to 7 leads to immediate consumption of 7 and quantitative formation of [Cp2Co][(tBudmx)Fe2(μ-OH)Cl2] (5d).45 We were not able to unequivocally determine the origin of the reducing equivalent which reduces a potential mixed-valent hydroxide intermediate species to diferrous 5, but the hydroxide generated upon proton transfer to 7 could act as a potential reductant. Alternatively, 7 can be converted to [Cp2*Co][(tBudmx}Fe2(μ-O)Cl2] (5c) by addition of one equivalent of decamethylcobaltocene. These reactions demonstrate the intricate interplay between oxidation and protonation state.Lastly, we were able to evaluate the strength of the O–H bond in [Et4N][(tBudmx)Fe2(μ-OH)Cl2] (5b) by addition of 2,4,6-tri-tert-butylphenoxyl radical as a hydrogen atom acceptor. Slow generation of 7 is observed upon addition of 2,4,6-tri-tert-butylphenoxyl radical to 5 (Fig. S-19†), suggesting that the BDEs of the O–H bond of 5 and 2,4,6-tri-tert-butylphenol are comparable.46
Conclusions
The Pacman ligand platform tBudmxH2 was selected as a template for the synthesis of dinuclear iron complexes. Reaction of tBudmxH2 with Fe2(Mes)4 affords a diiron complex 1 that is an excellent starting material for the introduction of bridging oxygen atoms. Our results indicate that the tBudmxH2 ligand serves as a preorganized scaffold to support a diiron core but is sufficiently flexible to allow for moderate structural rearrangements.Importantly, we demonstrate that this ligand platform supports a diiron core in multiple oxidation states. As such, this system provided us with the unique opportunity to compare the reactivity of a series of diiron μ-oxo complexes in a similar coordination environment. We showcase that the diiron species have a distinct inherent preference for oxo or hydroxide bridge formation, similar to bimetallic iron enzymes in nature.1–4 While a diferrous, a mixed-valent, and a diferric μ-oxo complex could be isolated, oxidized bridging hydroxide complexes remained elusive. The acidity of the bridging hydroxide increases substantially upon oxidation (pKa of 6a = −1.8(6)), which precludes isolation of a diferric bridging hydroxide. Similarly, the basicity of the bridging oxygen atom increases upon reduction (pKa of 3a = 15.3(6)) and even further upon coordination of neutral (pKa of 4 = 26.8(6)) or anionic ligands (pKa of 5 is expected to be greater than 26.8).
The primary influence of changing the Fe–O–Fe bonding and, thus, its attendant covalency is the relative electrophilicity of the Fe sites within that unit. Whereas solvation of the [Fe2(μ-O)] core results in a substantial increase of the oxo basicity relative to 3, anation leads to oxo protonation without a discernible intermediate. Solvation and anation electrostatically reinforce the iron centers, diminishing the Fe–O interaction and increasing oxo basicity. Oxidation of diferrous 3 to diferric 6, on the other hand, leads to an increased covalent interaction as the iron sites are now more electrophilic following oxidation. Consequently, the oxo moiety is far less basic in the higher oxidation state complex.
Finally, we were able to isolate a rare mixed-valent diiron μ-oxo complex and observed a decomposition pathway distinct from previously reported mechanisms. Rapid disproportionation of a mixed-valent diiron oxo complex to a diferrous hydroxide together with a diferric μ-oxo complex has been described in the literature.10 In our system, decomposition of the mixed-valent complex proceeds cleanly to diferrous hydroxide 5 without the formation of a diferric species. We believe that the reactivity showcased by the diiron complexes presented herein complements current mechanistic understanding and work is underway to elucidate the reactivity of these complexes with dioxygen.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the NIH (GM-115815), the Dreyfus Foundation (Teacher-Scholar Award to T. A. B.), and Harvard University. E. J. J. is grateful for an NSF Predoctoral Fellowship; C. K. was supported by the Jacques-Emile Dubois Graduate Student Dissertation Fellowship Fund; and R. A. M. is grateful for postdoctoral support from the European Union's Horizon 2020 Research and Innovation Programme under grant agreement No. 752684. The authors would like to thank Diana Iovan for helpful discussions regarding calculations.Notes and references
-
L. Que and A. E. True, Dinuclear Iron- and Manganese-Oxo Sites in Biology, in Progress in Inorganic Chemistry, John Wiley & Sons, Inc., 2007, pp. 97–200, DOI:10.1002/9780470166390.ch3
.
- B. J. Wallar and J. D. Lipscomb, Chem. Rev., 1996, 96, 2625–2658 CrossRef CAS PubMed
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y.-S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–350 CrossRef CAS PubMed
- J. B. Vincent, G. L. Olivier-Lilley and B. A. Averill, Chem. Rev., 1990, 90, 1447–1467 CrossRef CAS
- H. Thomann, M. Bernardo, J. M. McCormick, S. Pulver, K. K. Andersson, J. D. Lipscomb and E. I. Solomon, J. Am. Chem. Soc., 1993, 115, 8881–8882 CrossRef CAS
- V. J. DeRose, K. E. Liu, D. M. Kurtz, B. M. Hoffman and S. J. Lippard, J. Am. Chem. Soc., 1993, 115, 6440–6441 CrossRef CAS
- S. Friedle, E. Reisner and S. J. Lippard, Chem. Soc. Rev., 2010, 39, 2768–2779 RSC
- U. Bossek, H. Hummel, T. Weyhermüller, E. Bill and K. Wieghardt, Angew. Chem., Int. Ed., 1996, 34, 2642–2645 CrossRef
- J. A. R. Hartman, R. L. Rardin, P. Chaudhuri, K. Pohl, K. Wieghardt, B. Nuber, J. Weiss, G. C. Papaefthymiou, R. B. Frankel and S. J. Lippard, J. Am. Chem. Soc., 1987, 109, 7387–7396 CrossRef CAS
- A. Jozwiuk, A. L. Ingram, D. R. Powell, B. Moubaraki, N. F. Chilton, K. S. Murray and R. P. Houser, Dalton Trans., 2014, 43, 9740–9753 RSC
- W. H. Armstrong and S. J. Lippard, J. Am. Chem. Soc., 1984, 106, 4632–4633 CrossRef CAS
- K. S. Murray, Coord. Chem. Rev., 1974, 12, 1–35 CrossRef CAS
- D. M. Kurtz, Chem. Rev., 1990, 90, 585–606 CrossRef CAS
- E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987–1012 CrossRef CAS PubMed
- R. Shakya, D. R. Powell and R. P. Houser, Eur. J. Inorg. Chem., 2009, 2009, 5319–5327 CrossRef
- W. H. Armstrong, A. Spool, G. C. Papaefthymiou, R. B. Frankel and S. J. Lippard, J. Am. Chem. Soc., 1984, 106, 3653–3667 CrossRef CAS
- R. E. Norman, S. Yan, L. Que, G. Backes, J. Ling, J. Sanders-Loehr, J. H. Zhang and C. J. O'Connor, J. Am. Chem. Soc., 1990, 112, 1554–1562 CrossRef CAS
- J. Rosenthal, T. D. Luckett, J. M. Hodgkiss and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 6546–6547 CrossRef CAS PubMed
- F. Arena, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Chem. Soc., Chem. Commun., 1986, 1369–1371 RSC
- S. C. Payne and K. S. Hagen, J. Am. Chem. Soc., 2000, 122, 6399–6410 CrossRef CAS
- C. T. Saouma, C. C. Lu, M. W. Day and J. C. Peters, Chem. Sci., 2013, 4, 4042–4051 RSC
- N. A. Eckert, S. Stoian, J. M. Smith, E. L. Bominaar, E. Münck and P. L. Holland, J. Am. Chem. Soc., 2005, 127, 9344–9345 CrossRef CAS PubMed
- J. D. Cohen, S. Payne, K. S. Hagen and J. Sanders-Loehr, J. Am. Chem. Soc., 1997, 119, 2960–2961 CrossRef CAS
- S. K. Ghosh and S. P. Rath, J. Am. Chem. Soc., 2010, 132, 17983–17985 CrossRef CAS
- S. L. Daifuku, J. L. Kneebone, B. E. R. Snyder and M. L. Neidig, J. Am. Chem. Soc., 2015, 137, 11432–11444 CrossRef CAS PubMed
- A. M. Tondreau, C. Milsmann, A. D. Patrick, H. M. Hoyt, E. Lobkovsky, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 15046–15059 CrossRef CAS PubMed
- H. Andres, E. L. Bominaar, J. M. Smith, N. A. Eckert, P. L. Holland and E. Münck, J. Am. Chem. Soc., 2002, 124, 3012–3025 CrossRef CAS PubMed
- R. Karlsson, J. Chem. Eng. Data, 1973, 18, 290–292 CrossRef CAS
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS PubMed
- I. Nieto, F. Ding, R. P. Bontchev, H. Wang and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 2716–2717 CrossRef CAS PubMed
- F. Ding, J. M. Smith and H. Wang, J. Org. Chem., 2009, 74, 2679–2691 CrossRef CAS PubMed
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS
- I. Mayer, Int. J. Quantum Chem., 1984, 26, 151–154 CrossRef CAS
- W.-L. Man, W. W. Y. Lam, S.-M. Yiu, T.-C. Lau and S.-M. Peng, J. Am. Chem. Soc., 2004, 126, 15336–15337 CrossRef CAS PubMed
- N. S. Venkataramanan and S. Rajagopal, Tetrahedron, 2006, 62, 5645–5651 CrossRef CAS
- R. E. Cowley, N. A. Eckert, S. Vaddadi, T. M. Figg, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2011, 133, 9796–9811 CrossRef CAS PubMed
- M. T. Green, J. H. Dawson and H. B. Gray, Science, 2004, 304, 1653–1656 CrossRef CAS PubMed
- C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que, S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef CAS PubMed
- K. A. Prokop, S. P. de Visser and D. P. Goldberg, Angew. Chem., Int. Ed., 2010, 49, 5091–5095 CrossRef CAS PubMed
- K. L. Stone, L. M. Hoffart, R. K. Behan, C. Krebs and M. T. Green, J. Am. Chem. Soc., 2006, 128, 6147–6153 CrossRef CAS PubMed
- R. M. Theisen, J. Shearer, W. Kaminsky and J. A. Kovacs, Inorg. Chem., 2004, 43, 7682–7690 CrossRef CAS PubMed
- C. A. Brown, G. J. Remar, R. L. Musselman and E. I. Solomon, Inorg. Chem., 1995, 34, 688–717 CrossRef CAS
- J. Cui, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2010, 49, 10427–10435 CrossRef CAS PubMed
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS
- J. M. Carroll and J. R. Norton, J. Am. Chem. Soc., 1992, 114, 8744–8745 CrossRef CAS
-
Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press LLC, Boca Raton, FL, 2003 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1554783–1554789. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00605b |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |