
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of a flexible electrochemiluminescence platform for sweat detection†
Miao-Miao
Chen
a,
Shi-Bo
Cheng
a,
Kailun
Ji
b,
Jingwen
Gao
b,
Yan-Ling
Liu
a,
Wei
Wen
 b,
Xiuhua
Zhang
*b,
Shengfu
Wang
b and
Wei-Hua
Huang
*a
b,
Xiuhua
Zhang
*b,
Shengfu
Wang
b and
Wei-Hua
Huang
*a
aLaboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: whhuang@whu.edu.cn
bHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Ministry of Education, Key Laboratory for the Synthesis and Application of Organic Functional Molecules, College of Chemistry and Chemical Engineering, Hubei University, Wuhan 430062, China. E-mail: zhangxh@hubu.edu.cn
First published on 22nd May 2019
Abstract
Flexible and wearable chemical sensors show great capability and potential in retrieving physiologically related chemical or biochemical information from elastic and curvilinear living bodies. However, so far, no flexible electrochemiluminescence (ECL) device has been reported, though ECL measurements have been extensively investigated and widely applied in many fields. Herein, we for the first time designed and fabricated a flexible ECL sensor by immobilizing highly luminescent nanospheres on Au nanotube (Au NT) networks, and subsequently coating an elastic molecularly imprinted polymer (MIP) thereon. The as-prepared flexible ECL platform displayed successive and desirable mechanical compliance while generating a very stable ECL signal during deformation, facilitating highly selective detection of physiologically relevant chemicals from bodies. On-body wearable sampling and subsequent detection of lactate and urea from sweat showed the ECL performance of this sensor displaying desirable fidelity, reusability and high stability against disturbance. This work successfully incorporated the ECL sensing model into a flexible and wearable device, therefore providing a promising new path for non-invasively monitoring the products of metabolism for health care and biomedical investigations.
Introduction
Wearable sensors are nowadays playing an attractive role in maintaining an optimal health status and assessing physical performance of the human body.1–3 Following the many physical wearable devices that have been fabricated for sensing vital signs (e.g., skin temperature,4,5 pressure,6,7 and electrocardiogram8,9), flexible chemical sensors have more recently been developed to dynamically evaluate the wellness of the wearer at a deeper, molecular level.10,11 Electrochemical (EC) sensors have shown the advantages of involving easy-to-manage equipment and rapid response, and have been developed in order to detect specific chemical constituents and hence gain pivotal insights into the human body.12,13 On the one hand, recent research has focused on mechanotransduction detection by developing stretchable EC sensors to monitor chemical messengers released from mechanical deformation of cells and tissues.14–16 On the other hand, wearable EC sensors hold considerable promise in non-invasively retrieving health metrics by reliably detecting metabolites in bio-fluids such as tears,17,18 saliva,19,20 and especially sweat.21–25 So far, two primary classes of wearable EC devices for monitoring sweat have been developed: ion-selective sensors and enzymatic biosensors. The operation of the former is based on measurable changes in potential with changes in electrolyte levels (e.g., H+, Na+, K+, Ca2+) in secreted sweat.21,26–28 The latter sensors have been developed for the purpose of gleaning non-electroactive biomarkers (e.g., glucose, lactate, alcohol) by amperometric detection of products from enzyme-catalyzed reactions of analytes.21,29,30 However, despite the considerable success achieved by the use of enzymatic biosensors for wearable applications, several inevitable problems with labile enzymes or intricate immobilization of enzyme-like materials (e.g., Pt metal, Pt–Pb alloy, and transition-metal oxides) on the electrode still exist during the healthcare-monitoring procedure.29Electrochemiluminescence (ECL) is a process in which light is emitted from excited species electrogenerated at the electrode surface during an exergonic electron transfer reaction.31–34 ECL does not need an external light source, and has received considerable attention and extensive application due to its free light source, accurate potential control, simple operation processes and high sensitivity.35–39 When coupled with the immunoassay or molecular imprinting technique, the use of ECL sensors has made great strides in the sensitive and selective detection of diverse analytes including protein, DNA, and other target molecules.40–43 Especially when compared with EC sensors, ECL sensors exhibit excellent sensitivity for non-electroactive target molecules while eliminating possible interference from the electro-active compounds. In this regard, it can be predicted that flexible ECL devices have great potential in wearable technology innovation. However, despite the progress in the fabrication of flexible electroluminescent devices for wearable lighting or other applications,44,45 designing and fabricating wearable ECL sensors capable of retrieving physiologically relevant information has not been reported.
Considering these circumstances, we developed a flexible and wearable ECL platform for sensitive and selective detection of lactate and urea in human sweat. This platform was achieved by immobilizing highly luminescent nanospheres (HLNs) on a PDMS-supported Au nanotube (Au NT) electrode and subsequently coating a molecularly imprinted polymer (MIP) film thereon (Fig. 1). Here, the Ru–PEI@SiO2 HLNs with improved light emission performance was synthesized by covalently bonding [Ru(dcbpy)3]2+ with PEI and doping the complex into silica nanospheres. Au NTs with high aspect ratios were distributed randomly on PDMS, which furnished a bendable form to adhere well with the epidermis. Moreover, the MIP film with target imprinting molecules enabled the as-prepared MIP-ECL platform to display specific recognition sites and a predetermined selectivity during the elution and detection process. These processes allowed, for the first time, the detection of excreted metabolites (lactate and urea) in human sweat samples as well as on-body investigation by a flexible ECL platform, therefore indicating its promising potential toward medical and clinical diagnosis and also broadening the detection models and targets in flexible sensors.
 | ||
| Fig. 1 Schematic illustration showing (a) the synthesis of Ru–PEI@SiO2 HLNs and (b) fabrication of a flexible MIP-ECL sensor for the epidermal analyte detection. (i) Galvanic conversion of Ag NWs/PDMS to an Au NT/PDMS electrode. (ii) Immobilization of HLNs on an Au NT electrode. (iii) UV-vis light-induced polymerization to form a target-imprinted MIP layer on HLNs/Au NTs. (iv) Elution of flexible MIP-ECL sensors. (v) Epidermal analyte detection. The corresponding ECL signals are displayed above the bottom three graphs, respectively. | ||
Results and discussion
Characterization and ECL behaviour of Ru–PEI@SiO2 nanospheres
It has been reported that the ECL intensity can be obviously enhanced by shortening the distance between the luminophore and co-reactant to improve the electron transfer efficiency.46–48 As depicted schematically in Fig. 1a, the multicarboxyl [Ru(dcbpy)3]2+ and amino-rich PEI was covalently bonded together through an amide reaction. The Ru–PEI complex was then closely doped into silica to produce Ru–PEI@SiO2 HLNs. The as-prepared HLNs were spherically shaped with some polymer coatings (Fig. 2a, the spherical Ru@SiO2 nanoparticles without PEI are shown in Fig. S1† for comparison), and monodispersed with an average diameter of 24.7 ± 2.4 nm (mean ± SD, n = 395, Fig. 2b). The zeta potential of the HLNs was measured to be 48.7 mV (Fig. 2c), indicating that the nanospheres were positively charged. This result suggested that the Ru–PEI complex was assembled into silica successfully and that many amino groups were exposed.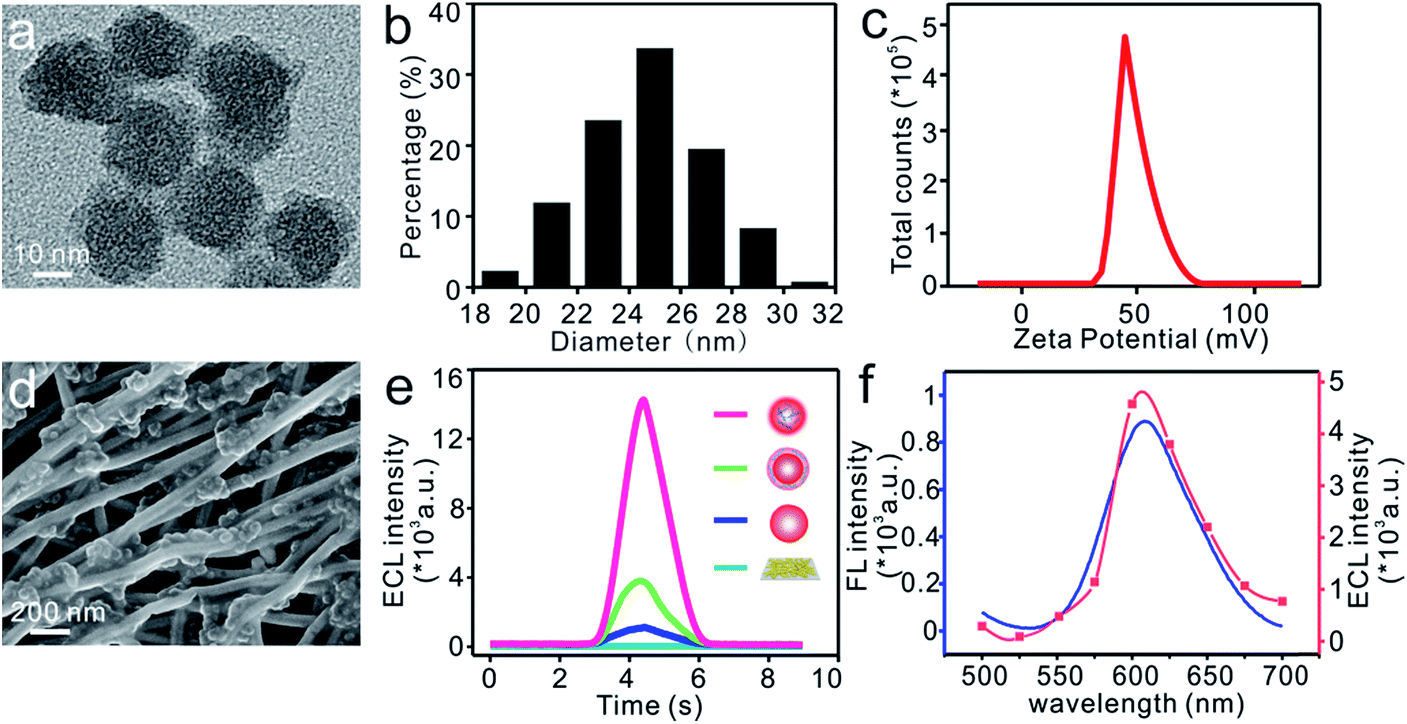 | ||
| Fig. 2 (a) HRTEM image, (b) size distribution and (c) zeta potential of Ru–PEI@SiO2 nanospheres. (d) SEM image of the Ru–PEI@SiO2/Au NT/PDMS sensor. (e) ECL spectra of bare Au NTs/PDMS (blue), Ru@SiO2/Au NTs/PDMS (dark blue), Ru@SiO2@PEI/Au NTs/PDMS (green) and HLNs/Au NTs/PDMS (red) each in 600 μL of 0.1 M PBS (pH 7.4). (f) Fluorescence spectrum of Ru–PEI@SiO2 nanospheres (blue) in methanol and ECL spectrum of Ru–PEI@SiO2/Au NTs (red) in 0.1 M PBS (pH 7.4) through a series of optical filters: 500 nm, 525 nm, 550 nm, 575 nm, 600 nm, 625 nm, 650 nm, 675 nm, and 700 nm. | ||
The flexible ECL platform was prepared by immobilization of the HLNs on a well-established stretchable electrode14 composed of high-aspect-ratio Au NTs randomly distributed on an elastic PDMS substrate (Fig. S2a†), which is denoted as HLNs/Au NTs/PDMS. SEM results (Fig. 2d) showed that the nanospheres intimately adhered to the surface or intersection of Au NTs, benefiting from the interaction of the amino group on the HLNs with the Au NTs. To demonstrate the enhanced ECL emission of the HLNs, the ECL performances of different materials immobilized on electrodes were tested and compared (Fig. 2e) in 600 μL of 0.1 M PBS (pH 7.4) without anything else added. Hardly any ECL emission was observed when Au NTs/PDMS was employed as a control (Fig. 2e, blue curve). The peak ECL intensity of Ru@SiO2/Au NTs/PDMS (nanospheres without PEI) was about 1087 a.u. (Fig. 2e, dark blue curve), generated from an annihilation reaction.49 When encompassing the Ru@SiO2 nanosphere surface with PEI via electrostatic interactions,50 Ru@SiO2@PEI/Au NTs/PDMS showed an apparent increased ECL intensity (3776 a.u., Fig. 2e, green curve). This result suggested that PEI with amine groups could work as a co-reactant and indeed enhance the ECL of Ru(II). As expected, the HLNs/Au NTs/PDMS yielded the highest ECL intensity (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 333 a.u., Fig. 2e, red curve) when the co-reactant PEI was covalently bonded to Ru(II) and then encapsulated into nanospheres. Based on the previous reports,48 the corresponding ECL mechanism is herein proposed as follows:
333 a.u., Fig. 2e, red curve) when the co-reactant PEI was covalently bonded to Ru(II) and then encapsulated into nanospheres. Based on the previous reports,48 the corresponding ECL mechanism is herein proposed as follows:
| Ru(II)–PEI@SiO2 − 2e → Ru(III)–PEI˙+@SiO2 | (1) |
| Ru(III)–PEI˙+@SiO2 → Ru(III)–PEI˙@SiO2 + H+ | (2) |
| Ru(III)–PEI˙@SiO2 → Ru(II)*–PEI@SiO2 + products | (3) |
| Ru(II)*–PEI@SiO2 → Ru(II)–PEI@SiO2 + hν | (4) |
When the electrochemical oxidation of Ru(II)–PEI@SiO2 occurred on the Au NT electrode, Ru(II) began to be oxidized to yield Ru(III) at a potential of about 1.1 V (Fig. S3a†), while the secondary amine in the coreactant was oxidized to a radical cation. After deprotonation, intramolecular electron-transfer annihilation occurred, generating an excited state that eventually decayed to the ground state, with this decay accompanied by emission of light. The marked improvement of the ECL emission of HLNs/Au NTs/PDMS was due to the greatly enhanced electron-transfer efficiency inside closely packed nanospheres. The Ru–PEI complex was comprised of the luminophore [Ru(dcbpy)3]2+ and coreactant PEI in one molecule. This design greatly shortened the electron-transfer path and lowered energy loss, thereby making the ECL reaction proceed more effectively. Furthermore, closely packing the Ru–PEI complex into nanospheres increased the immobilized amounts of luminophore and co-reactant on the Au NTs, which further endowed the flexible electrode with improved ECL performance. Based on this ECL signal, the molar ratio of Ru(II) to PEI was optimized to be 5:2 (Fig. S3b†). Adding a greater relative amount of PEI resulted in too much of it outside of the nanospheres (shown in Fig. S3c†), which could impede the electron transfer, causing the rapidly decreased signal. Moreover, the modified amount of HLNs was optimized to be 10 μL on a 0.5 cm × 0.5 cm electrode to achieve the maximum ECL intensity (Fig. S3d†).
The photo-physical properties of the HLNs were characterized with fluorescence and ECL spectra. As shown in Fig. 2f, an obvious ECL signal with a maximum emission peak at 606 nm was observed, very close to the fluorescence emission peak at 610 nm (the optimal excitation was at 474 nm, and the shift of 4 nm might have resulted from the effect of solvents51). This result led to a more comprehensive understanding of the mechanism from the final excited state Ru(II)*–PEI to the ground state. Taken together, immobilization of HLNs on an Au NT-based electrode resulted in a flexible ECL platform demonstrating excellent ECL performance without the need for any extra coreactant.
Characterization and ECL behavior of the flexible MIP-ECL sensor
To endow the HLN/Au NT electrode with high selectivity for non-invasive sweat analysis, we prepared an elastic MIP polymer on the flexible electrode. A mixture including the lactate or urea template molecule, functional monomer of MAA, EDMA cross-linker and AIBN initiators was coated on the HLN/Au NT surface. MAA strongly interacted with the template molecules due to its hydrogen-bond donor and acceptor characteristics to form the pre-polymerization complex.52,53 Free radical polymerization was then photochemically initiated by applying AIBN,54 and EDMA was used to fix MAA around the template molecules to form a highly cross-linked rigid polymer even after the removal of templates. Afterwards, the template molecule was removed by elution, leaving specific recognition sites for target detection, which contributed to the high adsorption efficiency and selectivity for analytes.55 Inspection of the photographs taken of the as-prepared MIP-ECL sensor in the bent state under sunlight and UV-light irradiation (Fig. 3a) showed that the introduction of elastic MIP layer very well maintained the flexibility of HLN/Au NT electrode and the excellent light-emission performance of the HLNs. | ||
| Fig. 3 (a) Acquired optical photographs of the bending characteristics of the flexible MIP-ECL sensor exposed to natural light or ultraviolet-light irradiation (λ = 365 nm). (b, c) SEM micrographs of the (b) cross-sectional structure and (c) surface morphology of the flexible MIP-ECL sensor after cyclic bending. (d) ECL signals of MIP/HLNs/Au NTs in 600 μL 0.1 M PBS (pH 7.4) before elution (green), after elution (red), and then during rebinding (blue). (e) ECL response for increasing concentrations of lactate from top to bottom, including concentrations of 0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, 7.5, 10, 12.5, 15, 17.5, and 20 mM. (f) Calibration plot for determining lactate concentration. | ||
The cross-sectional structure and surface morphology of the flexible MIP-ECL sensor after cyclic bending was characterized using SEM. As shown in Fig. 3b and c, the nanospheres were firmly adhered to the Au NT network, and the cross-section (Fig. 3b) as well as surface morphology (Fig. 3c) of the MIP layer did not change (the SEM images of the MIP-ECL sensor before cyclic bending are not shown here). These results suggested a high stability of the structure and morphology of the whole sensor during cyclic bending treatment.
To demonstrate the capability of the flexible MIP-ECL sensor in the detection of analyte, lactate was first chosen as the target molecule, and the corresponding ECL and CV responses were recorded during its elution and incubation process. When the lactate-imprinted MIP was polymerized on the sensor surface, there was a very weak current signal (Fig. S4a,† green curve) and almost no ECL response was observed (Fig. 3d, green curve). This result indicated that the insulated polymer layer largely blocked the electron transfer path and permitted neither the redox reaction nor any resulting ECL emission from occurring. When the imprinted lactate molecules were eluted from the MIP layer in the desorption process, the proposed sensor exhibited a markedly enhanced ECL intensity (Fig. 3d, red curve) accompanied by a significant current response (Fig. S4a,† red curve). This result was ascribed to the elution treatments having formed cavities inside the MIP polymer with memory of the shape, size and functional groups of the lactate targets. These cavities might have served as channels to facilitate electron transfer, thereby improving the efficiency and probability of Ru(II) oxidation and reinforcing ECL intensity. Subsequently, the sensor was immersed in a solution of 17.5 mM lactate to rebind templates, and both the ECL intensity (Fig. 3d, blue curve) and current signal (Fig. S4a,† blue curve) subsequently declined. This result was attributed to the cavities, serving as recognition and binding sites, having been re-occupied by the target molecules, which blocked the electron transfer channel, leading to the decreased signal output. As a control, ECL performance with a flexible NIP-ECL sensor (surface appearance shown in Fig. S4b†) showed some variability, caused by nonspecific absorption of analyte on the NIP layer (Fig. S4c†). Moreover, the experimental parameters including elution time (6 min, Fig. S5a†) and incubation time (6 min, Fig. S5b†) were optimized.
The formation of sweat lactate is a function of eccrine gland energy metabolism, and its content in human sweat usually varies from 3.7 mM to maximal levels of 25 mM during exercise.56 During physical activity, lactate is produced by glycogen in the eccrine sweat glands when the human body instigates the more efficient anaerobic metabolism for supplying the body with energy.57,58 Accordingly, sweat lactate is a widely recognized biomarker for exercise intensity and muscular exertion, and higher lactate levels correspond to increased muscular exertion during periods of anaerobic exercise. Due to the importance of this relationship, lactate samples at physiologically relevant and lower concentrations were tested with the developed lactate-imprinted ECL sensor. A stepwise diminishing ECL intensity was observed as the concentration of lactate was increased (Fig. 3e). Two highly linear segments were observed when plotting the ΔI/I0 signal as a function of target lactate concentration (Fig. 3f). The concentrations for the first segment ranged from 50 μM to 1.0 mM with a linear equation ΔI/I0 = 0.1267c + 0.0741 (R2 = 0.9955). The concentrations for the second segment ranged from 2.5 mM to 20.0 mM and the linear equation was expressed as ΔI/I0 = 0.0388c + 0.1124 (R2 = 0.9973). This flexible MIP-ECL platform was also tested with another metabolite, namely urea, and here too exhibited a well-defined concentration dependence with a highly linear response between 45 μM and 19.5 mM urea (Fig. S6†), and the linear equation was expressed as ΔI/I0 = 0.0443c + 0.0564 (R2 = 0.9980). The detection limits for lactate and urea were calculated to be 16.7 and 15.0 μM (S/N = 3), obviously lower than those obtained from previously reported wearable and flexible electrochemical sensors.25,56
Selectivity, stability and flexibility of the MIP-ECL sensor
Human sweat commonly contains a wide variety of metabolites and electrolytes that can potentially affect the test results.59 Major sweat electrolytes (NaCl, KCl, CaCl2) or biomolecules, e.g., glucose (Glu), uric acid (UA), urea, ascorbic acid (AA), were added, each at a concentration much higher than the physiologically relevant concentration, into respective solutions of 2.5 mM lactate to assess the selectivity of the MIP-ECL platform. As shown in Fig. 4a, the responses of the samples were hardly affected by these added species, indicating a favourable tolerance of the flexible MIP-ECL sensor to the interferences and ensuring the fidelity of the sensor readings in real sample analysis. In addition, we recorded the ECL signals by performing continuous scanning for 20 cycles apiece after the elution and incubation process. A negligible fluctuation of ECL intensity was observed (Fig. 4b), revealing the excellent stability of the device.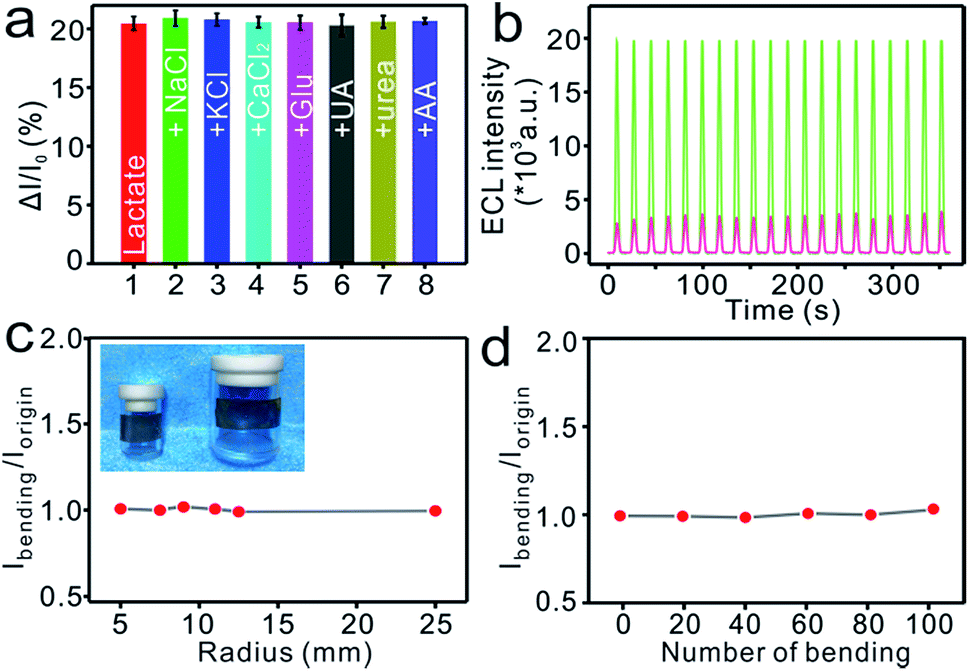 | ||
| Fig. 4 (a) ECL performances of flexible MIP-ECL sensors after they were incubated in separate solutions of 2.5 mM lactate mixed with different interferences each at a concentration of 250 mM. Error bars represent standard deviations, n ≥ 3. (b) Evaluation of the stability of the proposed sensor, accomplished by performing a continuous scanning for 20 cycles apiece after elution (green) and then incubation with 20 mM lactic acid (red). (c, d) ECL intensity changes of the flexible MIP-ECL sensor after it was incubated with a solution of 10 mM lactate undergoing repeated bending with (c) different radii and (d) different numbers of cycles at a fixed radius. | ||
Epidermal deformations are a major cause of concern for wearable devices wherein the devices undergo disfigurations similar to those of the skin. Such deformations of the skin require the flexible devices adhered to the epidermal layer to be able to record signals steadily and accurately during their mechanical deformations. Therefore, the flexibility of MIP-ECL platform was evaluated by flexing it on cylinders with different radii of curvature (Fig. 4c, inset), and the ECL intensity data were recorded before (Iorigin) and after (Ibending) bending for 10 iterations under each curvature. The value of Ibending/Iorigin, which was used to reflect the flexural performance, was nearly 100% with an RSD of 1.6% (Fig. 4c), indicating the high flexibility of the sensor in the face of mechanical bending. Moreover, the conformal nature of the flexible MIP-ECL sensor was verified from the results of repeated bending with an 11 mm radius for 100 iterations. As indicated from Fig. 4d, the resultant minimal deviation with an RSD of 1.2% of the sensor response every 20 bending iterations further demonstrated the structural integrity and ECL stability of the MCP-ECL sensor subjected to mechanical strain.
Sweat assessment
The ability to use the flexible MIP-ECL platform for sweat sample detection was first evaluated by following a standard addition method, and the corresponding ECL responses were recorded before and after incubation of the platform in artificial sweat samples. As expected, as lactate was added, the ECL signal change ratio (ΔI/I0) increased as well (Fig. S7†), and the calculated recovery ranged from 98.4% to 103.6% with RSDs from 1.4% to 2.2% (Table S1†). To further validate the dependability of the sensor with regards to the lactate excretion profile, authentic sweat was separately collected from four healthy subjects during vigorous exercise. The eluted MIP-ECL sensors were incubated in the collected sweat samples, some of which were subjected to centrifugation pretreatments, and ECL measurements were made to determine the amount of lactate in each of the secreted sweat samples. As exhibited in Fig. 5a, very similar results were obtained for the centrifuged and uncentrifuged samples. This result indicated that the MIP-ECL sensor could effectively resist interferences from the impurities in sweat samples, therefore simplifying the procedure for the subsequent measurements taken on real samples. The ΔI/I0 signals for the four subjects were 31.97%, 36.80%, 35.49%, 33.39%, respectively, and the corresponding concentrations of lactate in the sweat were 16.02 mM, 19.76 mM, 18.75 mM, 17.13 mM, respectively. These results were verified using the standard addition method. Taking the sweat of subject 4#, for example, when the sample was spiked with three concentrations of standard lactate solution, the recovery was found to be 108.4%, 92.4%, 96.7%, respectively, with satisfactory RSDs from 1.2% to 3.3% (Table S2†). These results provided evidence for the ability of the MIP-ECL sensor to specifically recognize the secreted lactate in sweat and hence provide dynamic metabolic information. | ||
| Fig. 5 (a) ΔI/I0 values of flexible MIP-ECL lactate sensors incubated in collected sweat samples subjected to (solid dots) or not subjected to (hollow dots) centrifugation treatment. The short horizontal lines are the average values. The labels 1#, 2#, 3# and 4# denote the respective identification numbers of the subjects. (b) Photograph of the epidermal lactate sensor on a subject. (c, d) Histograms of ΔI/I0 of flexible MIP-ECL platforms designed to sense (c) lactate or (d) urea of the collected sweat samples or epidermal experiments on the neck and wrist regions from subjects 1# and 2#. | ||
Epidermal evaluation
It has been demonstrated that lactate in sweat could be harvested after about 400–1250 seconds of physical activity (depending on individual differences), and less fit individuals excrete greater amounts of lactate.56,60,61 Here, to evaluate the capability of the proposed flexible ECL sensor for on-body sampling and detection, MIP-ECL sensors were discreetly adhered to the sweaty neck (Fig. 5b) and wrist regions of subjects who had continuously exercised for about 900–1200 s. Then in situ absorption of lactate or urea in authentic sweat was performed, and the results are summarized in Fig. 5c and d. As the exercise ensued, the average sweat lactate concentrations on the neck region, derived from the ECL change ratios, were 16.74 mM for subject 1# and 11.35 mM for subject 2#, consistent with the 16.77 mM and 11.40 mM values, respectively, measured for the wrist regions of the two subjects. For urea detection, the average concentrations obtained on the necks (12.59 mM for subject 1# and 9.32 mM for subject 2#) were also consistent with those from the wrists (12.36 mM for subject 1# and 9.38 mM for subject 2#). While different individuals showed different results, the concentrations of lactate (as well as the concentrations of urea) hardly varied at all between different regions of the body from the same person. For comparison, ex situ collected sweat samples were also tested. The resultant data (blue column in Fig. 5c, orange column in Fig. 5d) were observed to be remarkably similar to the data from the on-body sampling experiment (green column in Fig. 5c, cyan column in Fig. 5d). This similarity provided a further strong indication of the high fidelity and favourable stability of the MIP-ECL sensor readings. Moreover, more than 10 cycles of successive elution and incubation were tested, and the results (data not shown) demonstrated the excellent stability and favourable capability of this flexible ECL sensor in successive target detection. Taken together, the flexible MIP-ECL platform can be potentially applied in clinical and other practical settings to offer users valuable information regarding their sweat-based health monitoring and metabolism.Conclusion
In summary, we developed for the first time a flexible ECL platform for in situ sweat analysis. The flexibility resulted from randomly distributed Au NTs on an elastic PDMS membrane. High sensitivity was achieved by closely packing a luminophore-coreactant complex into silica nanospheres and further immobilizing these HLNs on the Au NT network. Coating an elastic MIP membrane on the HLNs/Au NTs provided high selectivity and at the same time yielded excellent flexibility and light emission properties. These features endowed the sensor with favorable mechanical compliance for intimate contact with curvilinear epidermis as well as excellent sensing performance for detection of target molecules. These features allowed on-body sampling and subsequent detection of sweat from different parts of the body with desirable fidelity, reusability and high stability against disturbance. Though continuous monitoring of analytes could not be achieved on the MIP-based ECL sensor since the target molecules must be eluted for successive adsorption and detection, compared with enzyme-based flexible EC sensors, the flexible ECL sensor provided an alternative sensing mode for detection of non-electroactive species. We anticipate that coupling the flexible ECL sensor with a real-time ECL sensing mode and portable detection apparatus would broaden the applications of wearable and flexible devices in non-invasive monitoring of health and physiological metabolism for clinical and biomedical investigations.Experimental
Reagents and apparatus
Tris (4,4′-dicarboxylicacid-2,2′-bipyridyl)ruthenium(II) dichloride (Ru(dcbpy)3Cl2) was obtained from Suna Tech Inc. (Suzhou, China). Branched ethylene imine polymer (PEI, M.W. 10000, 99%), L(+)-lactic acid, methacrylic acid (MAA), ethylene glycol dimethacrylate (EDMA), and azodiisobutyronitrile (AIBN) were acquired from Aladdin Chemistry Co., Ltd. (Shanghai, China). Silver nanowires (Ag NWs) were purchased from Zhejiang Kechuang Advanced Materials Tech Co., Ltd. (Hangzhou, China). PDMS liquid prepolymer and cross-linker were obtained from Momentive Performance Materials (Waterford, NY, USA). Artificial sweat was bought from Kehong Chemical Co., Ltd. (Guangdong, China). Tetraethylorthosilicate (TEOS), cyclohexane, TX-100, n-hexanol, N-hydroxy succinimide (NHS) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimidehydrochloride (EDC) were received from Sigma-Aldrich Co. (St. Louis, MO, U.S.A.). All other chemicals were of analytic grade and used without further purification. Phosphate buffer solution (PBS, 0.1 M) was prepared with 0.1 M K2HPO4, 0.1 M NaH2PO4, and 0.1 M KCl and adjusted to pH 7.4 for standby. Doubly distilled water (18.2 MΩ cm resistivity, Millipore Inc., USA) was used throughout the experiments.
An MPI-A ECL analyzer (Xi'an Remex, China) was employed to generate high-resolution ECL-potential and ECL-wavelength profiles. This set-up contained a classical three-electrode system with a modified working electrode, a saturated Ag/AgCl reference electrode, and a platinum wire auxiliary electrode. A high-resolution transmission electron microscope (HRTEM, JEM-2100, Japan) and scanning electron microscope (SEM, ZEISS SIGMA field-emission, Germany) were used for characterization of morphologies. A fluorescence spectrum was acquired by using a fluorescent spectrophotometer (RF-5301PC, Shimadzu, Japan). An ultraviolet analyzer (ZF-I, Shanghai Guanghao, China) was used along with UV-light irradiation to fabricate MIP.
Preparation of Ru–PEI@SiO2 nanospheres
As shown in detail in Fig. 1a, an amide reaction was carried out to synthesize the Ru–PEI molecular compound. Briefly, a volume of 0.25 mL of 0.01 M [Ru(dcbpy)3]2+ solution was mixed with 0.1 mL of 170 mg mL−1 EDC and 0.15 mL of 8.67 mg mL−1 NHS under stirring for 30 min to activate the carboxyl of [Ru(dcbpy)3]2+. Then 1 mL of a solution of 20 mg mL−1 PEI was added into the above mixture with continuous agitation. The reaction was allowed to proceed at 4 °C for 6 h and the Ru–PEI complex was eventually obtained, and saved for further use.Analogous to the preparation of Ru@SiO2 nanoparticles,42 Ru–PEI@SiO2 nanospheres were obtained by following water-in-oil (W/O) microemulsion procedures. First 1.77 mL of TX-100, 7.5 mL of cyclohexane, and 1.8 mL of n-hexanol were injected into a conical flask. Then, a volume of 0.34 mL of the as-prepared Ru–PEI compound was added in small amounts with stirring for 30 min. Subsequently, a volume of 0.1 mL of TEOS as the precursor was rapidly administered into the above mixed solution and the polymerization was made to proceed by then injecting 0.06 mL of NH3·H2O. After carrying out the reaction for 24 h in the dark, the end products were isolated by adding 10 mL of acetone to break up the emulsion and then subjecting the resulting mixture to centrifugation (10000 rpm, 8 min). Finally, purification was carried out using ethanol, water, and methanol in succession, and the Ru–PEI@SiO2 nanospheres were obtained and dispersed in methanol for storage.
Fabrication of flexible MIP-ECL sensors
As also described in our recently reported work,14 the overall procedure of fabricating the MIP-ECL sensor is displayed schematically in Fig. 1b. Typically, a solution of isopropanol-dispersed Ag NWs (35–45 nm in diameter, 10–20 μm in length) was spin-coated (400 rpm, 5 s) twice on a dopamine-treated hydrophilic PDMS surface to produce an Ag NW/PDMS film. Then the Ag NW/PDMS film was in situ displaced sacrificially at 90 °C for 2 h to create an Au NT/PDMS film. Relying on conductive carbon adhesives, a thin copper wire was gently adhere to the Au NTs, and the outside of which was insulated with PDMS, producing a flexible Au NT/PDMS electrode (0.5 cm × 0.5 cm).The prefabricated Ru–PEI@SiO2 nanospheres were directly dropped onto the Au NT/PDMS electrode, dried under ambient conditions to achieve Ru–PEI@SiO2-modified Au NTs/PDMS. After immersion in the cross-linking mixture of MAA, EDMA, AIBN and lactate or urea templates for a little while and irradiation under UV radiation of 365 nm for 8 min, the flexible MIP/Ru–PEI@SiO2/Au NTs/PDMS samples were manufactured successfully. The flexible electrode modified with non-molecularly imprinted polymers was fabricated with a uniform flow path, only leaving out the template molecules during cross-linking process.
Signal conditions, processing
ECL emission intensity of the flexible MIP-ECL sensor was measured in a quartz cell containing 0.6 mL of 0.1 M PBS (pH 7.4) with a photomultiplier tube (PMT, a high voltage of 800 V, magnitude 4) and the cyclic voltammetry scanning was carried out (initial potential: 0.4 V; final potential: 1.3 V; sample rate: 40 T s−1; scan rate: 0.1 V s−1). After elution in acetic acid solution for a moment, the ECL signal was measured and denoted as I0. Subsequently, after being immersed in a series of standard solutions for some time, the sensor was evaluated again for another ECL intensity value (I). The ratio of variation (I0–I) to the initial I0, denoted as ΔI/I0, was employed for indirect determination of the concentrations of the imprinted templates. Note that the sensor had to be and indeed was washed thoroughly after the incubation phase to mitigate nonspecific adsorption of interfering species to the electrode surface. The specificity of the lactate-imprinted sensor was tested using other physiological products of authentic human sweat, namely NaCl, KCl, CaCl2, glucose, uric acid, urea, and ascorbic acid, each at 250 mM, a concentration much higher than their physiologically relevant levels.Flexibility test
For mimicking the viscoelasticity of the epidermis, the mechanical integrity of the flexible MIP-ECL sensor was studied by adhering it onto the rigid exteriors of cylinders with different radii (0.5 cm; 0.75 cm; 0.9 cm; 1.1 cm; 1.25 cm; 2.5 cm). The bending and subsequent relaxation of the sensors was repeated 10 times after which the ECL signal (Iorigin) of 10 mM lactate used for incubation was measured, and then Ibending was measured. Then, Ibending/Iorigin was derived to assess the flexibility of the sensor. The stability of the sensor was analogously determined by repeatedly bending it with a fixed radius of 1.25 cm for 100 cycles.Sweat analysis
The ability to apply the flexible MIP-ECL sensor to sweat analysis was confirmed from the results of recovery experiments that were performed. The ECL responses of the freshly eluted sensors were first recorded. Afterward, the sensors were put into various solutions each containing 0.4 mL of artificial sweat plus 0.4 mL of 0.1 M PBS (0.1 M, pH 7.4), and either 5 mM, 10 mM, or 15 mM lactate for separate incubation. After washing, the ECL intensity of each sample was measured again and the corresponding ΔI/I0 values were calculated for ascertaining the lactate levels in artificial sweat.For detection of authentic sweat, four healthy subjects (3 males and 1 female, aged from 25–30) performed a badminton competition. Until the subjects started to sweat profusely, authentic sweat samples were collected from the foreheads and necks of the subjects. Then 0.2 mL of collected sweat sample diluted with 0.4 mL of PBS (0.1 M, pH 7.4) was used for each adsorption to determine the lactate level in the authentic sweat of each subject. Then, a recovery evaluation of the fourth authentic sweat sample as an illustration was carried out for the same inspection process as that in artificial sweat analysis.
Epidermal sweat detection
The epidermal MIP-ECL sensor was applied onto the necks and wrists of two subjects (1 male, aged 28; 1 female, aged 30) in order to assess the real-time lactate or urea concentration profile of the secreted sweat. Once sweat appeared during vigorous exercise, the newly eluted sensor was allowed to intimately contact the neck or wrist, i.e. was dipped in the sweat, for 6 min. And then ECL responses were measured in 0.6 mL of PBS (0.1 M, pH 7.4) on the ECL analyzer. The ratio of signal variation to the eluted signal value (ΔI/I0) indirectly provided the corresponding lactate or urea level for each individual. As a comparison, the collected sweat samples of the same regions of the body were investigated as well. Each sample of all the subjects was tested at least 10 times for replicate variability.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21725504, 11674085, 31501568).References
- Y. Khan, A. E. Ostfeld, C. M. Lochner, A. Pierre and A. C. Arias, Adv. Mater., 2016, 28, 4373–4395 CrossRef CAS PubMed.
- L. Li, Z. Lou, D. Chen, K. Jiang, W. Han and G. Shen, Small, 2017, 14, 1702829 CrossRef.
- S. Yao, P. Swetha and Y. Zhu, Adv. Healthc. Mater., 2018, 7, 1700889 CrossRef.
- T. Q. Trung and N. Lee, Adv. Mater., 2016, 28, 4338–4372 CrossRef CAS.
- T. Q. Trung, H. S. Le, T. M. L. Dang, S. Ju, S. Y. Park and N.-E. Lee, Adv. Healthc. Mater., 2018, 7, 1800074 CrossRef.
- C.-L. Choong, M.-B. Shim, B.-S. Lee, S. Jeon, D.-S. Ko, T.-H. Kang, J. Bae, S. H. Lee, K.-E. Byun, J. Im, Y. J. Jeong, C. E. Park, J.-J. Park and U.-I. Chung, Adv. Mater., 2014, 26, 3451–3458 CrossRef CAS.
- Z. Song, W. Li, Y. Bao, W. Wang, Z. Liu, F. Han, D. Han and L. Niu, Adv. Electron. Mater., 2018, 4, 1800252 CrossRef.
- G. S. Jeong, D.-H. Baek, H. C. Jung, J. H. Song, J. H. Moon, S. W. Hong, I. Y. Kim and S.-H. Lee, Nat. Commun., 2012, 3, 977 CrossRef PubMed.
- J. H. Koo, S. Jeong, H. J. Shim, D. Son, J. Kim, D. C. Kim, S. Choi, J.-I. Hong and D.-H. Kim, ACS Nano, 2017, 11, 10032–10041 CrossRef CAS PubMed.
- A. J. Bandodkar, I. Jeerapan and J. Wang, ACS Sens., 2016, 1, 464–482 CrossRef CAS.
- M. Tessarolo, I. Gualandi and B. Fraboni, Adv. Mater. Technol., 2018, 3, 1700310 CrossRef.
- A. J. Bandodkar and J. Wang, Trends Biotechnol., 2014, 32, 363–371 CrossRef CAS.
- J. Li, Z. Peng and E. Wang, J. Am. Chem. Soc., 2018, 140, 10629–10638 CrossRef CAS.
- Y.-L. Liu, Z.-H. Jin, Y.-H. Liu, X.-B. Hu, Y. Qin, J.-Q. Xu, C.-F. Fan and W.-H. Huang, Angew. Chem., Int. Ed., 2016, 55, 4537–4541 CrossRef CAS.
- Y.-L. Liu, Y. Qin, Z.-H. Jin, X.-B. Hu, M.-M. Chen, R. Liu, C. Amatore and W.-H. Huang, Angew. Chem., Int. Ed., 2017, 56, 9454–9458 CrossRef CAS PubMed.
- Y.-W. Wang, Y.-L. Liu, J.-Q. Xu, Y. Qin and W.-H. Huang, Anal. Chem., 2018, 90, 5977–5981 CrossRef CAS PubMed.
- S. Iguchi, H. Kudo, T. Saito, M. Ogawa, H. Saito, K. Otsuka, A. Funakubo and K. Mitsubayashi, Biomed. Microdevices, 2007, 9, 603–609 CrossRef CAS PubMed.
- D. Pankratov, E. González-Arribas, Z. Blum and S. Shleev, Electroanalysis, 2016, 28, 1250–1266 CrossRef CAS.
- J. Kim, G. Valdés-Ramirez, A. J. Bandodkar, W. Jia, A. G. Martinez, J. Ramírez, P. Mercier and J. Wang, Analyst, 2014, 139, 1632–1636 RSC.
- J. Kim, S. Imani, W. R. de Araujo, J. Warchall, G. Valdés-Ramírez, T. R. L. C. Paixão, P. P. Mercier and J. Wang, Biosens. Bioelectron., 2015, 74, 1061–1068 CrossRef CAS PubMed.
- W. Gao, S. Emaminejad, H. Y. Y. Nyein, S. Challa, K. Chen, A. Peck, H. M. Fahad, H. Ota, H. Shiraki, D. Kiriya, D.-H. Lien, G. A. Brooks, R. W. Davis and A. Javey, Nature, 2016, 529, 509–514 CrossRef CAS.
- X. Zhang, Y. Jing, Q. Zhai, Y. Yu, H. Xing, J. Li and E. Wang, Anal. Chem., 2018, 90, 11780–11784 CrossRef CAS PubMed.
- M. Bariya, H. Y. Y. Nyein and A. Javey, Nat. Electron. , 2018, 1, 160–171 CrossRef.
- H. Lee, T. K. Choi, Y. B. Lee, H. R. Cho, R. Ghaffari, L. Wang, H. J. Choi, T. D. Chung, N. Lu, T. Hyeon, S. H. Choi and D.-H. Kim, Nat. Nanotechnol., 2016, 11, 566–572 CrossRef CAS.
- Y. Ling Liu, R. Liu, Y. Qin, Q.-F. Qiu, Z. Chen, S.-B. Cheng and W.-H. Huang, Anal. Chem., 2018, 90, 13081–13087 CrossRef.
- S. Emaminejad, W. Gao, E. Wu, Z. A. Davies, H. Yin Yin Nyein, S. Challa, S. P. Ryan, H. M. Fahad, K. Chen, Z. Shahpar, S. Talebi, C. Milla, A. Javey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4625–4630 CrossRef CAS.
- H. Y. Y. Nyein, W. Gao, Z. Shahpar, S. Emaminejad, S. Challa, K. Chen, H. M. Fahad, L.-C. Tai, H. Ota, R. W. Davis and A. Javey, ACS Nano, 2016, 10, 7216–7224 CrossRef CAS.
- W. Dang, L. Manjakkal, W. T. Navaraj, L. Lorenzelli, V. Vinciguerra and R. Dahiya, Biosens. Bioelectron., 2018, 107, 192–202 CrossRef CAS.
- S. Y. Oh, S. Y. Hong, Y. R. Jeong, J. Yun, H. Park, S. W. Jin, G. Lee, J. H. Oh, H. Lee, S.-S. Lee and J. S. Ha, ACS Appl. Mater. Interfaces, 2018, 10, 13729–13740 CrossRef CAS.
- J. Kim, J. R. Sempionatto, S. Imani, M. C. Hartel, A. Barfidokht, G. Tang, A. S. Campbell, P. P. Mercier and J. Wang, Adv. Sci., 2018, 5, 1800880 CrossRef.
- A. J. Bard, Electrogenerated Chemiluminescence, Marcel Dekker, New York, 2004 Search PubMed.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS.
- Z. Liu, W. Qi and G. Xu, Chem. Soc. Rev., 2015, 44, 3117–3142 RSC.
- L. Li, Y. Chen and J.-J. Zhu, Anal. Chem., 2017, 89, 358–371 CrossRef CAS.
- Y. Zhuo, H.-J. Wang, Y.-M. Lei, P. Zhang, J.-L. Liu, Y.-Q. Chai and R. Yuan, Analyst, 2018, 143, 3230–3248 RSC.
- X.-L. Cai, B. Zheng, Y. Zhou, M. R. Younis, F.-B. Wang, W.-M. Zhang, Y.-G. Zhou and X.-H. Xia, Chem. Sci., 2018, 9, 6080–6084 RSC.
- C. Ma, W. Wu, L. Li, S. Wu, J. Zhang, Z. Chen and J.-J. Zhu, Chem. Sci., 2018, 9, 6167–6175 RSC.
- X.-L. Huo, N. Zhang, H. Yang, J.-J. Xu and H.-Y. Chen, Anal. Chem., 2018, 90, 13723–13728 CrossRef CAS PubMed.
- W. Gao, K. Muzyka, X. Ma, B. Loua and G. Xu, Chem. Sci., 2018, 9, 3911–3916 RSC.
- W. Wen, X. Yan, C. Zhu, D. Du and Y. Lin, Anal. Chem., 2017, 89, 138–156 CrossRef CAS.
- Y. Zhou, S. Chen, X. Luo, Y. Chai and R. Yuan, Anal. Chem., 2018, 90, 10024–10030 CrossRef CAS.
- M.-M. Chen, Y. Wang, S.-B. Cheng, W. Wen, X. Zhang, S. Wang and W.-H. Huang, Anal. Chem., 2018, 90, 5075–5081 CrossRef CAS PubMed.
- W. Zhang, H. Xiong, M. Chen, X. Zhang and S. Wang, Biosens. Bioelectron., 2017, 96, 55–61 CrossRef CAS PubMed.
- G. Liang, M. Yi, H. Hu, K. Ding, L. Wang, H. Zeng, J. Tang, L. Liao, C. Nan, Y. He and C. Ye, Adv. Electron. Mater., 2017, 3, 1700401 CrossRef.
- J. D. Pegan, A. Y. Ho, M. Bachman and M. Khine, Lab Chip, 2013, 13, 4205–4209 RSC.
- T. Wang, D. Wang, J. W. Padelford, J. Jiang and G. Wang, J. Am. Chem. Soc., 2016, 138, 6380–6383 CrossRef CAS.
- S. Carrara, F. Arcudi, M. Prato and L. De Cola, Angew. Chem., Int. Ed., 2017, 56, 4757–4761 CrossRef CAS PubMed.
- Y. Zhuo, N. Liao, Y.-Q. Chai, G.-F. Gui, M. Zhao, J. Han, Y. Xiang and R. Yuan, Anal. Chem., 2014, 86, 1053–1060 CrossRef CAS.
- Y. Gao, X. Liu, W. Qi, W. Gao, Y. Li and G. Xu, Analyst, 2015, 140, 3996–4000 RSC.
- L. Zhou, J. Huang, B. Yu and T. You, Sci. Rep., 2016, 6, 22234 CrossRef CAS PubMed.
- Y. Feng, F. Sun, N. Wang, J. Lei and H. Ju, Anal. Chem., 2017, 89, 7659–7666 CrossRef CAS PubMed.
- R. Schirhagl, Anal. Chem., 2013, 86, 250–261 CrossRef PubMed.
- Y. Zhang, D. Song, L. M. Lanni and K. D. Shimizu, Macromolecules, 2010, 43, 6284–6294 CrossRef CAS.
- H. Yan and K. H. Row, Int. J. Mol. Sci., 2006, 7, 155–178 CrossRef CAS.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- W. Jia, A. J. Bandodkar, G. Valdés-Ramírez, J. R. Windmiller, Z. Yang, J. Ramírez, G. Chan and J. Wang, Anal. Chem., 2013, 85, 6553–6560 CrossRef CAS PubMed.
- J. F. Nichols, S. L. Phares and M. J. Buono, Int. J. Sports Med., 1997, 18, 458–463 CrossRef CAS PubMed.
- J. M. Green, R. C. Pritchett, D. C. Tucker, T. R. Crews and J. R. McLester, J. Sport. Sci., 2004, 22, 321–327 CrossRef CAS PubMed.
- C. J. Harvey, R. F. LeBouf and A. B. Stefaniak, Toxicol. In Vitro, 2010, 24, 1790–1796 CrossRef CAS PubMed.
- W. Jia, G. V. Ramirez, A. J. Bandodkar, J. R. Windmiller and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 7233–7236 CrossRef CAS PubMed.
- A. J. Bandodkar, W. Jia and J. Wang, Electroanalysis, 2015, 27, 562–572l CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01937e |
| This journal is © The Royal Society of Chemistry 2019 |