DOI:
10.1039/C9SC00678H
(Edge Article)
Chem. Sci., 2019,
10, 4628-4639
Pentanuclear iron catalysts for water oxidation: substituents provide two routes to control onset potentials†
Received
8th February 2019
, Accepted 18th March 2019
First published on 19th March 2019
Abstract
The development of robust and efficient molecular catalysts based on earth-abundant transition metals for water oxidation reactions is a challenging research target. Our group recently demonstrated the high activity and stability of a pentairon-based water oxidation electrocatalyst (M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468). However, the development of strategies to decrease onset potentials for catalysis remains challenging. In this article, we report the construction of a series of pentanuclear iron complexes by introducing electron-donating (methyl) and electron-withdrawing (bromo) substituents on the ligand. Two newly synthesized complexes exhibited five reversible redox processes, similar to what is seen with the parent complex. These complexes can also serve as homogeneous catalysts for water oxidation reactions, and the faradaic efficiencies of the reactions were high. Additionally, the onset potentials of the newly developed complexes were lower than that of the parent complex. Mechanistic insights revealed that there are two methods for decreasing onset potentials: control of the redox potentials of the pentairon complex and control of the reaction mechanism.
Introduction
Water oxidation (2H2O → O2 + 4H+ + 4e−) is considered the main bottleneck in the production of chemical fuels from sunlight and/or electricity;1–7 this is because the reaction requires the transfer of four electrons and the generation of an O–O bond, and is both thermodynamically and kinetically demanding. Therefore, the development of a highly active artificial catalyst for the oxidation of water is of great importance. In this context, since the discovery of the first molecular water oxidation catalyst, “Blue dimer”,8 a significant number of molecular water oxidation catalysts have been reported.9–24 More recently, metal complexes containing earth-abundant transition metal ions such as Mn,25–28 Fe,29–36 Co,37–46 and Cu47–55 ions have also been intensely studied. Nevertheless, the development of efficient metal-complex-based catalysts that consist of earth-abundant transition metals is still very challenging.56,57
In nature, water oxidation is catalysed by the oxygen evolving complex (OEC) in photosystem II.58–60 The OEC is a highly active and robust catalyst for water oxidation that can drive the reaction under mild conditions.61 The active site of the OEC contains a multinuclear metal complex, a Mn4CaO5 cluster, which has several water coordination sites. Due to the multinuclear structure of the Mn4CaO5 cluster, the OEC can smoothly accumulate the oxidative equivalents required for the reaction via the formation of five distinct redox intermediates, the Sn states, where the subscript indicates the number of stored oxidative equivalents (n = 0–4). After the formation of the S4 state, H2O reacts with the S4 state to generate O2 and protons.62
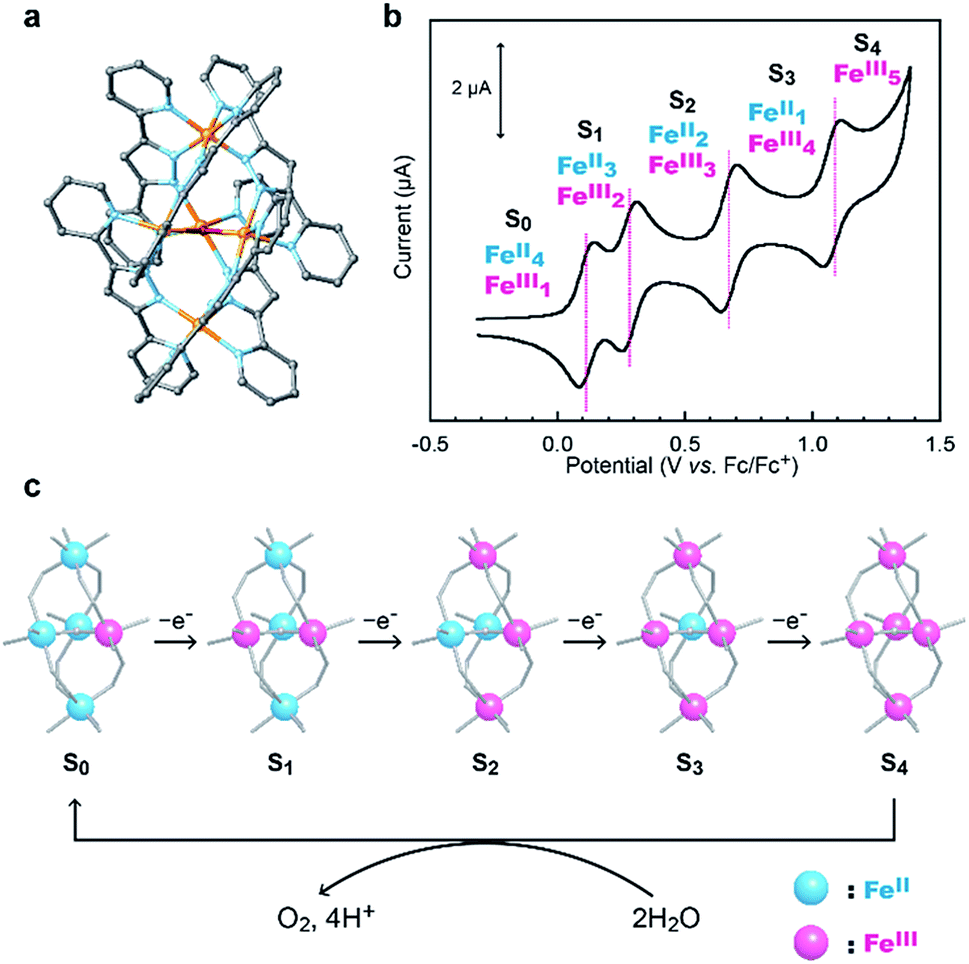
Recently, we demonstrated that a pentanuclear iron complex [FeII4FeIII(μ3-O)(bpp)6]3+, [Fe5-H]3+ (Scheme 1a, Hbpp = bis(pyridyl)pyrazole), can serve as a highly active catalyst for electrocatalytic water oxidation.33[Fe5-H]3+ can also accumulate four oxidative equivalents via the successive oxidation of each of the iron centres in the complex (Scheme 1b). In the catalysis mediated by [Fe5-H]3+, the four-electron-oxidized species, [FeIII5(μ3-O)(bpp)6]7+ ([Fe5-H]7+, S4 state), reacts with H2O to generate O2 (Scheme 1c). The reaction rate and durability of [Fe5-H]3+ are the highest among those of iron-based water oxidation catalysts (Fe-WOCs) reported thus far. However, a relatively large onset potential is required for the catalysis because the S4 state is only generated at high potentials. Therefore, the development of a novel strategy for designing catalysts that can drive the reaction at low onset potentials is essential.
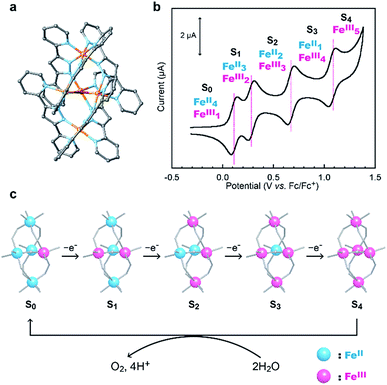 |
| | Scheme 1 (a) Chemical structure of [Fe5-H]3+, (b) a cyclic voltammogram of [Fe5-H]3+ (0.2 mM in MeCN solution containing 0.1 M TBAP under Ar at a scan rate of 10 mV s−1) and (c) sequential oxidation of iron ions in [Fe5-H]3+ and the reaction of the S4 state to generate dioxygen. | |
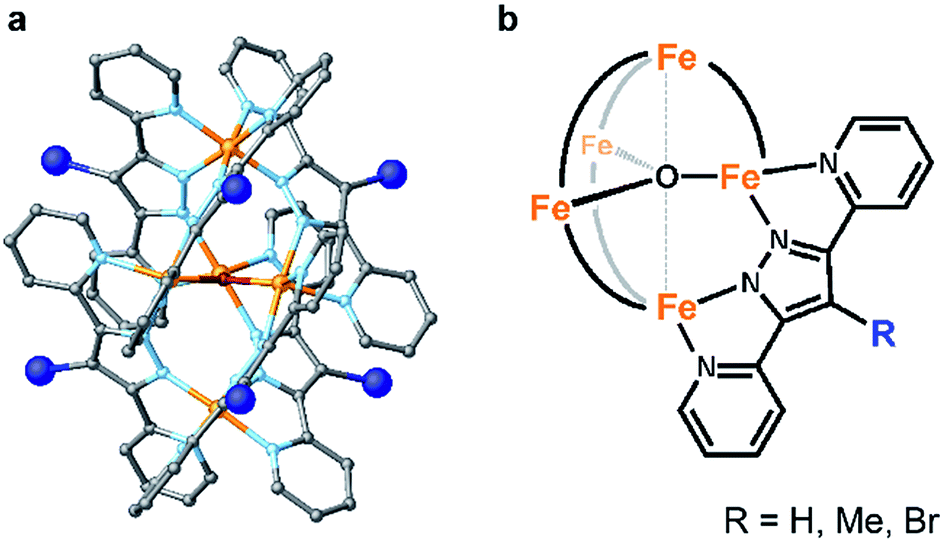
Here, we report two approaches for decreasing the onset potential of pentairon water oxidation systems. Two approaches involving the installation of substituents onto the Hbpp ligand have been demonstrated. Two kinds of ligands, one with electron-donating and the other with electron-withdrawing groups at the 4-position of the Hbpp ligand (Me-Hbpp and Br-Hbpp in Scheme 2), have been employed, and the new pentairon complexes were constructed utilizing these ligands. The newly synthesized complexes catalysed the oxidation of water with high faradaic efficiencies, and the onset potentials of these complexes were lower than that of the parent complex. The mechanistic studies also revealed that two distinct routes exist to decrease the onset potentials for water oxidation in pentanuclear iron systems.
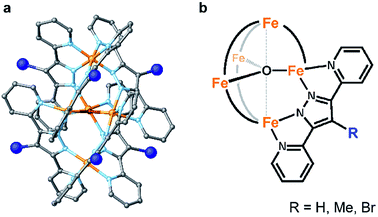 |
| | Scheme 2 Schematic illustrations of the series of pentanuclear iron complexes bearing 4-substituted-3,5-bis(pyridyl)pyrazole (R-Hbpp (R = H, Me, and Br)) ligands investigated in this study. | |
Results
Syntheses and characterization of ligands and pentairon complexes
Me-Hbpp was prepared using the three-step synthetic route shown in Scheme S1 in the ESI.† Initially, 2-acetylpyridine and pyridine-2-carboxylic acid methyl ester were reacted in the presence of sodium ethoxide to yield 1,3-di(2-pyridyl)-1,3-propanedione. The methylation of the product and further treatment with hydrazine monohydrate afforded Me-Hbpp. The total synthetic yield of Me-Hbpp was 26% (see the Experimental section for details). Br-Hbpp was synthesized in a moderate yield (65%) by the bromination of Hbpp.63 Both ligands were characterized by 1H and 13C NMR spectroscopy and elemental analyses. The syntheses of two pentairon complexes with the obtained ligands were performed by reacting the corresponding ligand (6 eq.) with FeSO4·7H2O (5 eq.) in the presence of a base (NaOH, 6 eq.) in methanol at 80 °C (Scheme S2†). The reaction mixture was further treated with a saturated solution of aqueous NaBF4 or NaPF6, and the obtained precipitate was collected by filtration. The precipitate was recrystallized from MeCN/Et2O to afford crystalline products. The electrospray ionization mass spectrometry (ESI-MS) and elemental analysis data of the obtained crystalline samples confirmed the formation of the desired pentairon complexes, [FeII4FeIII(μ3-O)(Me-bpp)6]3+ ([Fe5-Me]3+) and [FeII4FeIII(μ3-O)(Br-bpp)6]3+ ([Fe5-Br]3+). The synthetic yields of the complexes were 62 and 43% for [Fe5-Me]3+ and [Fe5-Br]3+, respectively. The parent complex, [Fe5-H]3+, was synthesized using a reported procedure33 and characterized by ESI-MS and elemental analysis.
Crystal structures of the pentairon complexes
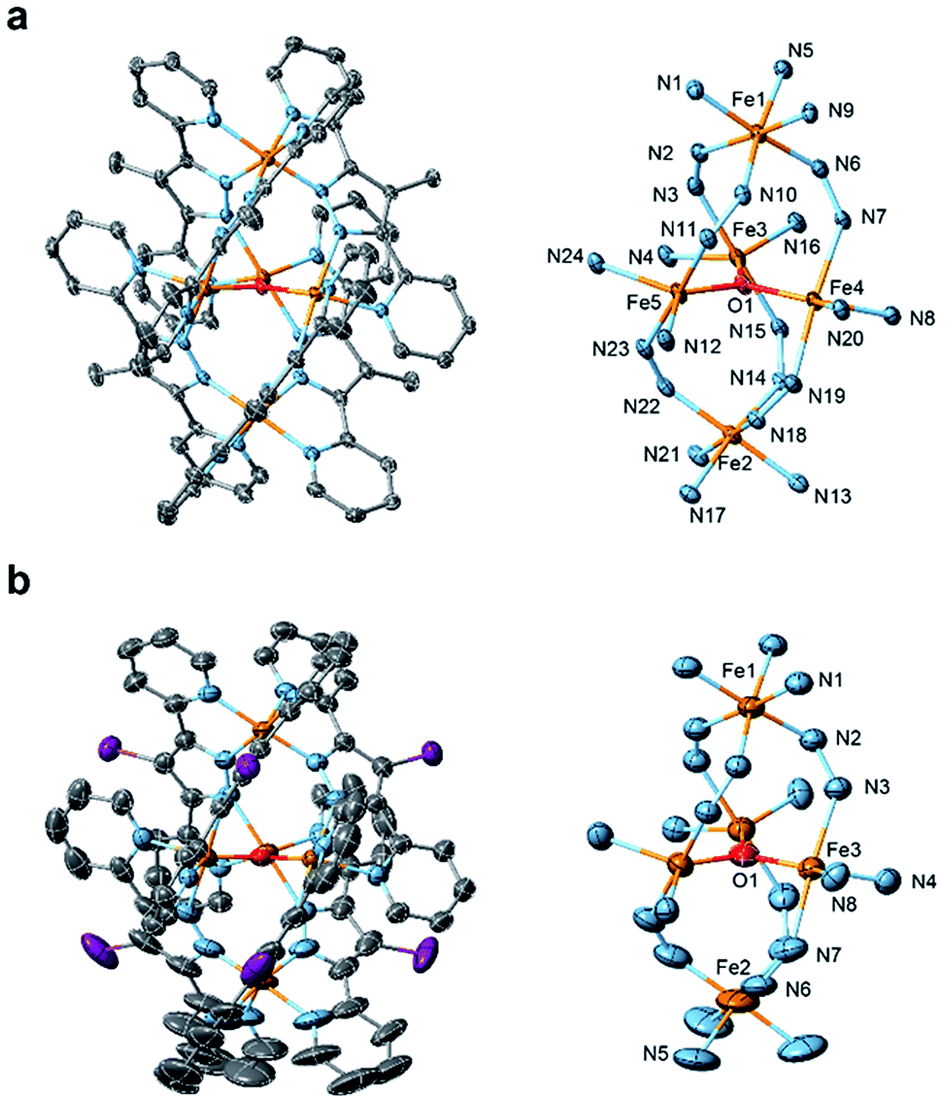
Single crystals of [Fe5-Me]3+ and [Fe5-Br]3+ suitable for single-crystal X-ray diffraction (SCXRD) were obtained by vapor diffusion of diethyl ether (Et2O) into saturated solutions of the respective complexes in acetonitrile (MeCN). Note that a few drops of MeOH were added to the MeCN solution of [Fe5-Me]3+ to prevent the oxidation of the complex. As we previously reported,33 single crystals of [Fe5-H]3+ were obtained by slow evaporation of acetonitrile from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN–H2O (v/v) solution of the complex. [Fe5-Me]3+ was obtained as a PF6 salt, and [Fe5-Br]3+ and [Fe5-H]3+ were obtained as BF4 salts. The ORTEP diagrams of the cationic moieties of the three complexes are shown in Fig. 1 and S1,† and the crystallographic data for the newly synthesized complexes are summarized in Table S1.† All complexes displayed the same structural motif consisting of a central [Fe3(μ3-O)] core connected by two apical Fe ions through six R-bpp− units. [Fe5-Me]3+ crystallized in the P
:1 MeCN–H2O (v/v) solution of the complex. [Fe5-Me]3+ was obtained as a PF6 salt, and [Fe5-Br]3+ and [Fe5-H]3+ were obtained as BF4 salts. The ORTEP diagrams of the cationic moieties of the three complexes are shown in Fig. 1 and S1,† and the crystallographic data for the newly synthesized complexes are summarized in Table S1.† All complexes displayed the same structural motif consisting of a central [Fe3(μ3-O)] core connected by two apical Fe ions through six R-bpp− units. [Fe5-Me]3+ crystallized in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, and the asymmetric unit contains one cationic pentairon complex and three PF6 anions. The asymmetric unit of the R
space group, and the asymmetric unit contains one cationic pentairon complex and three PF6 anions. The asymmetric unit of the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) crystal of [Fe5-Br]3+ is composed of one-third of the cationic pentairon complex and one BF4 anion. The crystal structure of [Fe5-H]3+ belongs to the I
crystal of [Fe5-Br]3+ is composed of one-third of the cationic pentairon complex and one BF4 anion. The crystal structure of [Fe5-H]3+ belongs to the I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) space group, and the asymmetric unit contains half of the cationic pentairon complex and one and half BF4 anions. The bond distances between the iron atoms and the N atoms on R-bpp− ligands are not significantly changed by either bromo or methyl substitution (Table S2†). These results clearly demonstrate that the substituents do not affect the pentanuclear core structure of the complexes (Fig. S2†).
space group, and the asymmetric unit contains half of the cationic pentairon complex and one and half BF4 anions. The bond distances between the iron atoms and the N atoms on R-bpp− ligands are not significantly changed by either bromo or methyl substitution (Table S2†). These results clearly demonstrate that the substituents do not affect the pentanuclear core structure of the complexes (Fig. S2†).
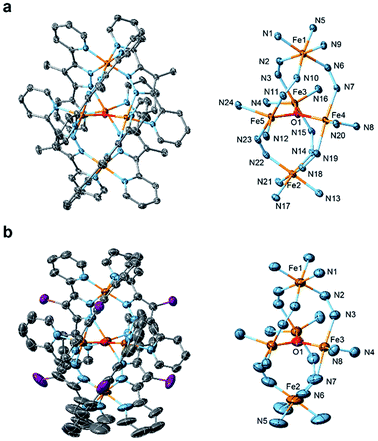 |
| | Fig. 1 ORTEP drawings of the cationic moieties (left) and core structures (right) of (a) [Fe5-Me](PF6)3 and (b) [Fe5-Br](BF4)3. The atoms are represented by the following colours: Fe, orange; O, red; N, blue; C, grey; and Br, purple. Hydrogen atoms and crystal solvent molecules are omitted for clarity. Thermal ellipsoids are shown at 30% probability. | |
UV-Vis absorption spectra
The UV-Vis absorption spectra of [Fe5-Me]3+, [Fe5-Br]3+ and [Fe5-H]3+ in MeCN are shown in Fig. S3.† The complexes exhibited intense bands at approximately 400–410 nm and shoulder peaks at 480–490 nm. These bands are attributed to the charge transfer from the low-spin FeII centre to the ligand (MLCT).33,64 In our previous study, Mössbauer spectroscopic measurements of [Fe5-H]3+ showed that the two iron centres at the apical positions are low-spin FeII ions and that the [Fe3(μ3-O)] core is composed of two high-spin FeII ions and one high-spin FeIII ion.33 The UV-Vis absorption spectra strongly indicate that the oxidation and spin states of the iron centres of [Fe5-Me]3+ and [Fe5-Br]3+ are identical to those of [Fe5-H]3+. Note that the MLCT bands of [Fe5-Me]3+ and [Fe5-Br]3+ are slightly red shifted and blue shifted, respectively, compared to those of [Fe5-H]3+, reflecting the electron-donating or electron-withdrawing nature of the substituents.
Electrochemical properties
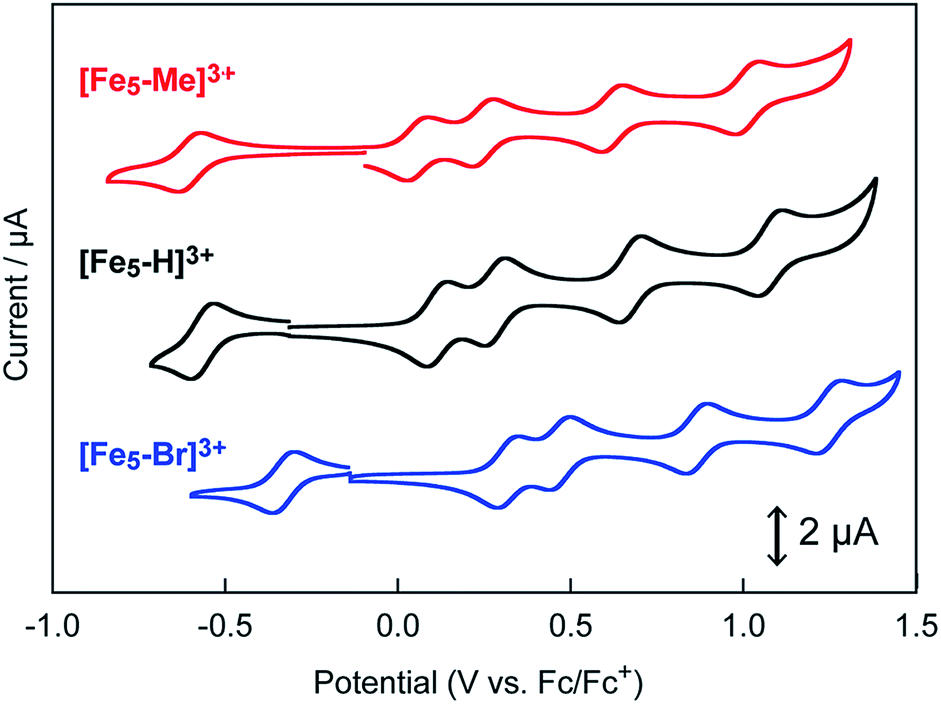
The influence of the electron-donating and electron-withdrawing groups on the redox properties of the pentairon complexes was investigated by cyclic voltammetry. The cyclic voltammograms (CVs) of [Fe5-Me]3+, [Fe5-Br]3+ and [Fe5-H]3+ (0.2 mM) in dry MeCN containing 0.1 M Bu4NClO4 (TBAP) under an Ar atmosphere are shown in Fig. 2, and the electrochemical data are summarized in Table 1. All the complexes displayed one reversible and four successive reversible one-electron oxidation waves assigned to the FeII4FeIII/FeII5, FeII3FeIII2/FeII4FeIII, FeII2FeIII3/FeII3FeIII2, FeIIFeIII4/FeII2FeIII3, and FeIII5/FeIIFeIII4 redox couples, indicating that the electron transfer ability arising from the pentairon structure is preserved even after the introduction of substituents on the ligands. Importantly, all the redox waves of [Fe5-Me]3+ were shifted to a more negative potential relative to those of [Fe5-H]3+, whereas the redox waves of [Fe5-Br]3+ were positively shifted (Fig. 2 and Table 1). These trends are consistent with the electron-donating and electron-withdrawing properties of the methyl and bromo substituents, respectively. This result clearly demonstrates that the redox potentials of the pentairon complexes can be tuned by the introduction of substituents on the ligands. The open-circuit potentials of the complexes, located at −0.10 ([Fe5-Me]3+), −0.18 ([Fe5-Br]3+), and −0.26 V ([Fe5-H]3+), indicate an initial state of FeII4FeIII in solution. These initial oxidation states of the complexes in the solution state determined by the electrochemical measurements are fully consistent with those estimated from the UV-Vis absorption spectra (vide supra).
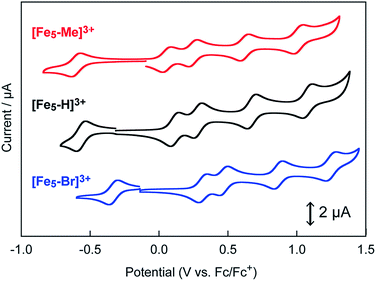 |
| | Fig. 2 Cyclic voltammograms of 0.2 mM solutions of [Fe5-Me]3+ (red line), [Fe5-Br]3+ (blue line) and [Fe5-H]3+ (black line). Measurements were performed in MeCN solutions containing 0.1 M TBAP under Ar at a scan rate of 10 mV s−1. Working electrode, glassy carbon; counter electrode, Pt wire; reference electrode, Ag/Ag+. CV scans were initiated from the respective open-circuit potentials. | |
Table 1 Redox potentials (E1/2, V vs. Fc/Fc+) and the onset potentials for water oxidation (Eonset, V vs. Fc/Fc+) of a series of pentairon complexes in acetonitrile solutions with TBAP (0.1 M). E1/2(−1), E1/2(1), E1/2(2), E1/2(3), and E1/2(4) correspond to the E1/2 values of the FeII4FeIII/FeII5, FeII3FeIII2/FeII4FeIII, FeII2FeIII3/FeII3FeIII2, FeIIFeIII4/FeII2FeIII3, and FeIII5/FeIIFeIII4 redox couples, respectively
| Complex |
E
1/2
|
E
onset
|
|
E
1/2(−1) |
E
1/2(1) |
E
1/2(2) |
E
1/2(3) |
E
1/2(4) |
|
Conditions: [cat] = 0.2 mM, [H2O] = 5 M, working electrode: glassy carbon, scan rate: 10 mV s−1.
|
|
[Fe5-Me]3+
|
−0.60 |
0.05 |
0.24 |
0.62 |
1.01 |
1.09 |
|
[Fe5-Br]3+
|
−0.32 |
0.32 |
0.48 |
0.87 |
1.27 |
1.15 |
|
[Fe5-H]3+
|
−0.55 |
0.13 |
0.30 |
0.68 |
1.08 |
1.18 |
Catalytic activity for water oxidation
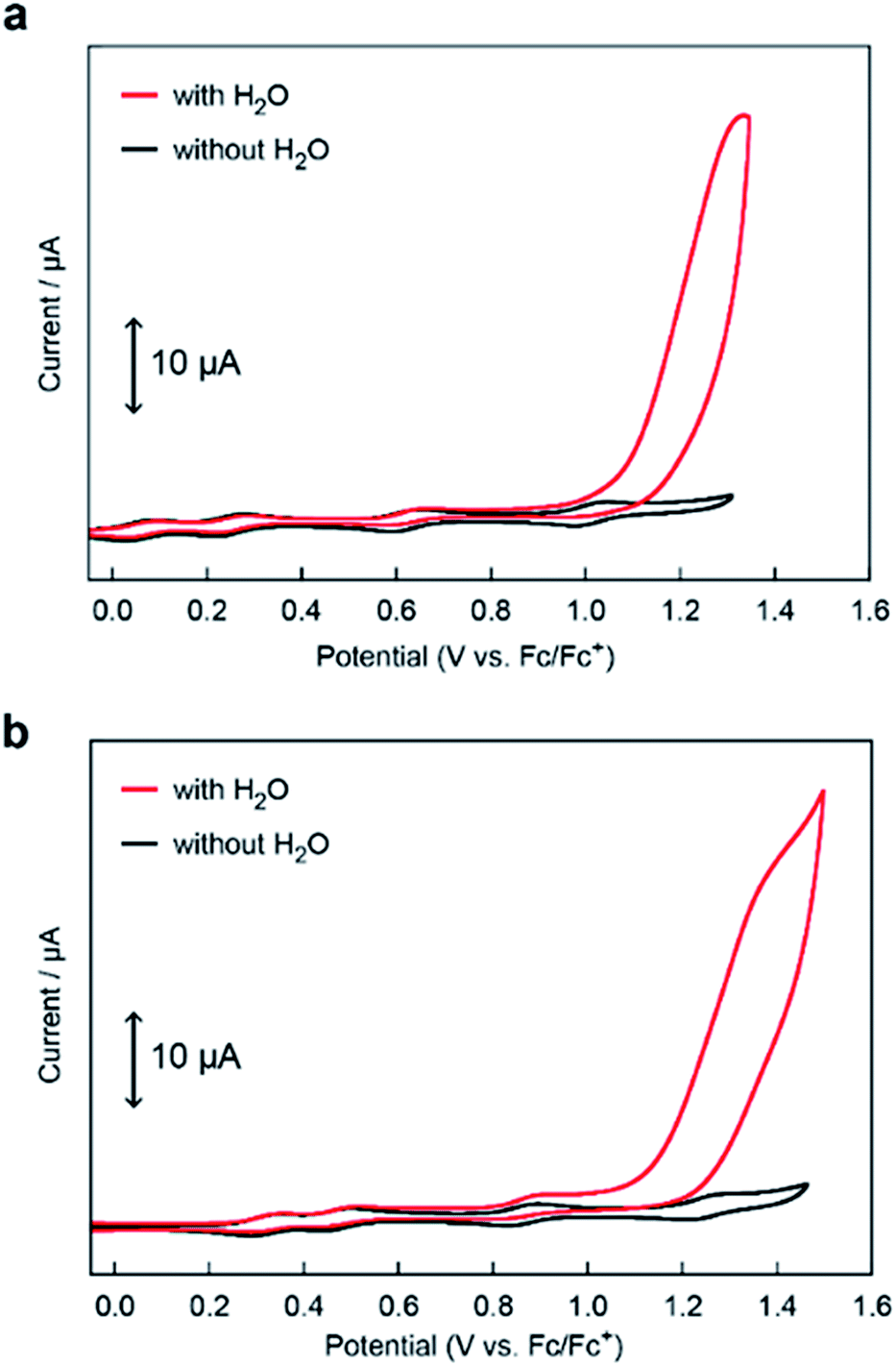
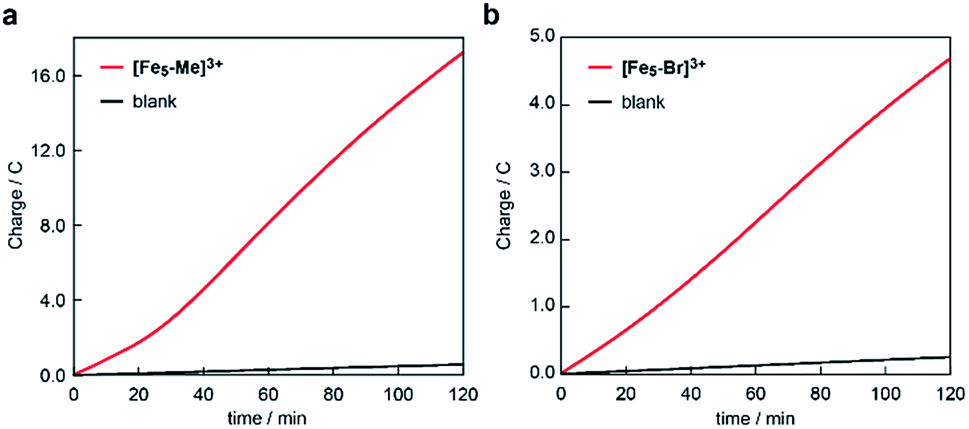
The electrocatalytic activities of [Fe5-Me]3+ and [Fe5-Br]3+ for water oxidation were examined by electrochemical measurements of the 0.2 mM solution of the complexes in the presence of 5 M H2O. Under these conditions, both complexes exhibited a large irreversible current in the >1.0 V region (Fig. 3). As previously reported for [Fe5-H]3+,33 such an increase in the current is indicative of promotion of the electrocatalytic water oxidation reaction. To further verify whether the electrocatalytic current actually corresponds to the activity in the catalytic oxidation of H2O, controlled potential electrolysis (CPE) was carried out at an indium tin oxide (ITO) electrode using a customized two-compartment cell system.33 After 2 h of electrolysis of [Fe5-Me]3+ at 1.42 V (vs. Fc/Fc+), 17.2 C of charge was passed, and gas chromatography (GC) detected 39.5 μmol of O2 as the product (Fig. 4a). In the case of [Fe5-Br]3+, the electrolysis under identical conditions afforded a charge of 4.7 C and generated 9.3 μmol of O2 (Fig. 4b). The faradaic efficiencies of the reaction based on the 4e− process were 92 and 86% for [Fe5-Me]3+ and [Fe5-Br]3+, respectively. Based on the results of CPE experiments, turnover frequencies (TOFs) and turnover numbers (TONs) for water oxidation were roughly estimated. For [Fe5-Me]3+, TOF and TON were 3 × 102 s−1 and 2 × 106, respectively, and TOF and TON values of [Fe5-Br]3+ were estimated to be 20 s−1 and 1 × 105, respectively (for the details of calculation see the ESI (P.S24)†). Although these values were lower than those of [Fe5-H]3+ (1 × 103 s−1 (TOF) and 7.5 × 106 (TON)) estimated by using the same method, they were substantially higher compared to those of the reported iron-complex-based catalysts for water oxidation.29–32,35 In both cases, the electrolyzed solutions were treated with oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV) as a chemical probe65 and the 2e− oxidized product of H2O (H2O2) was not detected (for details of the experimental procedure, see the ESI (P.S22–S23)†). After the CPE experiment, the ITO working electrodes used in the electrolysis were gently rinsed with small amounts of water and MeCN, and then, a second round of electrolysis was performed using the solution without the catalyst. Significantly small currents were observed in the second electrolysis compared to the first electrolysis in both cases (Fig. S4 and S5†), which indicates that the species homogeneously dissolved in the solution are catalytically active. CV measurements of the solution after the CPE experiments also clarified the presence of pentanuclear complexes in the solution phase (Fig. S6†). Additionally, the UV-Vis absorption spectra of the ITO electrodes before and after the CPE experiments remained almost identical (Fig. S7 and S8†), suggesting no formation of heterogeneous deposits during the electrolysis. We also analysed the electrolyte solutions after the electrolysis by dynamic light scattering (DLS) measurements and no formation of heterogeneous nanoparticles was detected (Fig. S9†). These experimental results show that [Fe5-Me]3+ and [Fe5-Br]3+ can serve as homogeneous electrocatalysts for water oxidation.
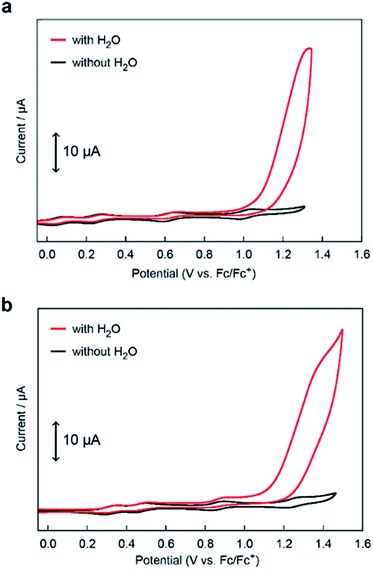 |
| | Fig. 3 CVs of 0.2 mM solutions of (a) [Fe5-Me]3+ and (b) [Fe5-Br]3+ in MeCN containing 0.1 M TBAP in the absence of H2O (black lines) and the presence of 5 M H2O (pH = 5, red lines). The CVs were measured using a GC electrode under an Ar atmosphere at a scan rate of 10 mV s−1. | |
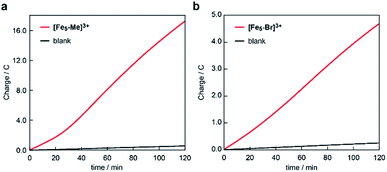 |
| | Fig. 4 Controlled potential electrolysis data of (a) [Fe5-Me]3+ and (b) [Fe5-Br]3+ in a MeCN/H2O mixed solution system containing 0.1 M TBAP using an ITO electrode under an argon atmosphere. Conditions: [cat] = 0.2 mM, [H2O] = 5 M and applied potential: 1.42 V vs. Fc/Fc+, pH = 5. | |
Onset potentials for water oxidation
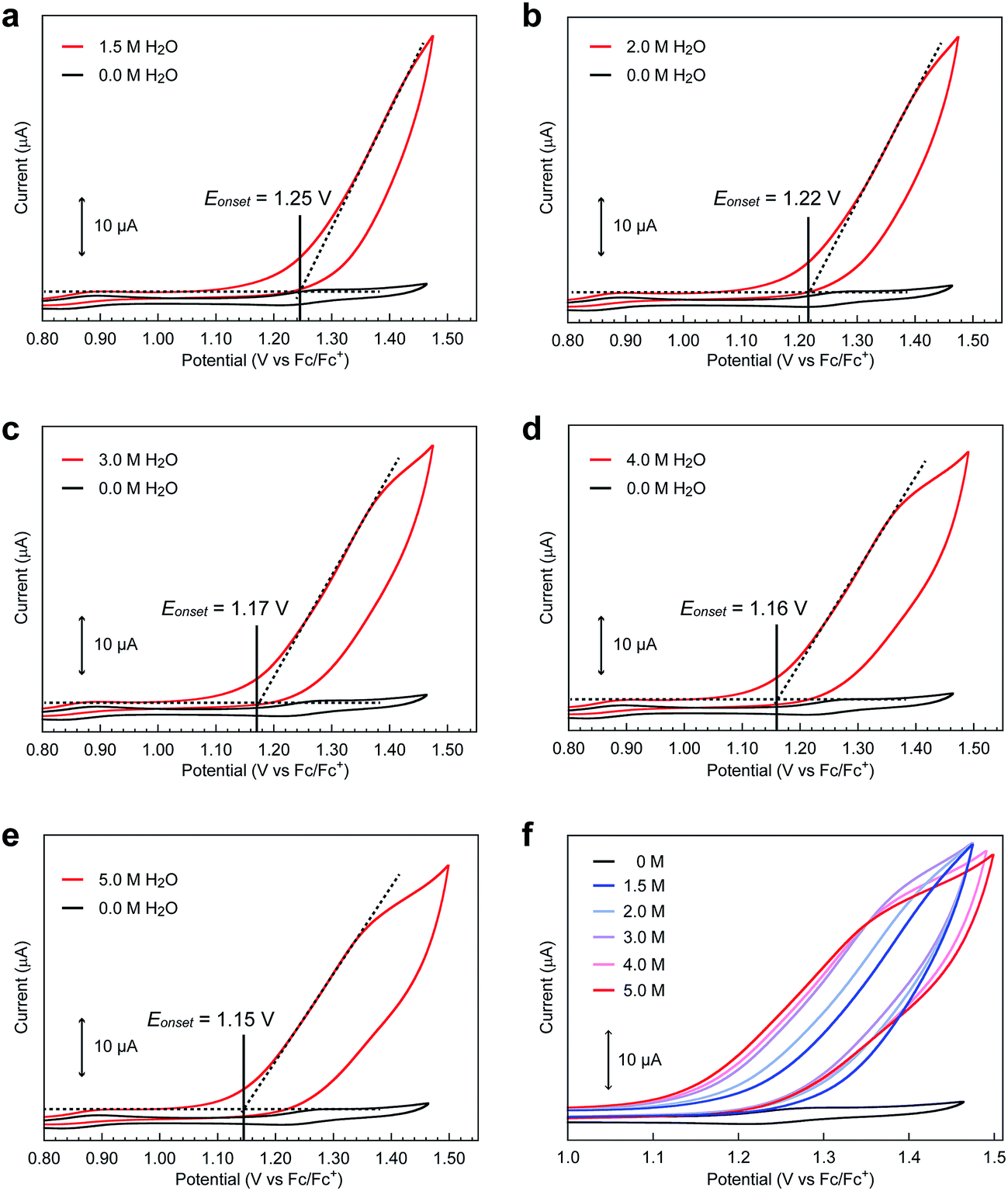
Although both of the novel pentanuclear complexes, [Fe5-Me]3+ and [Fe5-Br]3+, exhibited catalytic activity for water oxidation, the electrochemical responses of these complexes in the presence of H2O were different. In the case of [Fe5-Me]3+, a large irreversible current attributed to the catalytic water oxidation was observed at a potential close to that of the fourth redox couple (FeIII5/FeIIFeIII4, E1/2(4) in Table 1) in the presence of 5 M H2O (Fig. 3a). The onset potential (Eonset, Table 1) for the reaction was estimated from the cross-point of two lines that were obtained by extrapolating the slopes of the catalytic current and non-faradaic current. The Eonset of [Fe5-Me]3+ was determined to be 1.09 V, which corresponds to the overpotential (η) of 0.65 V at pH = 5.0, and was slightly larger than E1/2 of the fourth redox couple (1.01 V). In contrast, the catalytic current for water oxidation for [Fe5-Br]3+ was observed at a more negative potential than that of the fourth redox couple (1.27 V, Fig. 3b); the Eonset of [Fe5-Br]3+ was determined to be 1.15 V (η = 0.71 V at pH = 5.0). These results clearly demonstrate that [Fe5-Me]3+ and [Fe5-Br]3+ catalyse the water oxidation reaction by distinct reaction mechanisms. Notably, the Eonset of these complexes is lower than that of [Fe5-H]3+ (1.18 V, η = 0.74 V at pH = 5.0) under identical experimental conditions.
Discussion
Reaction mechanism of [Fe5-Me]3+
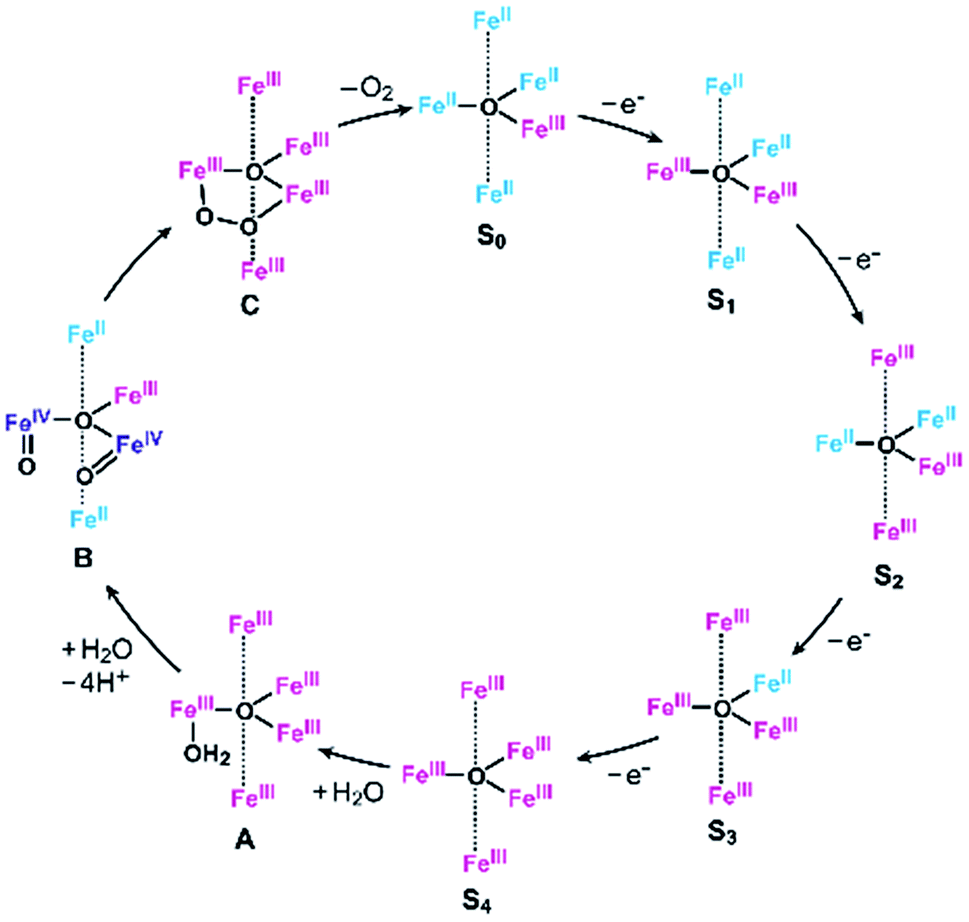
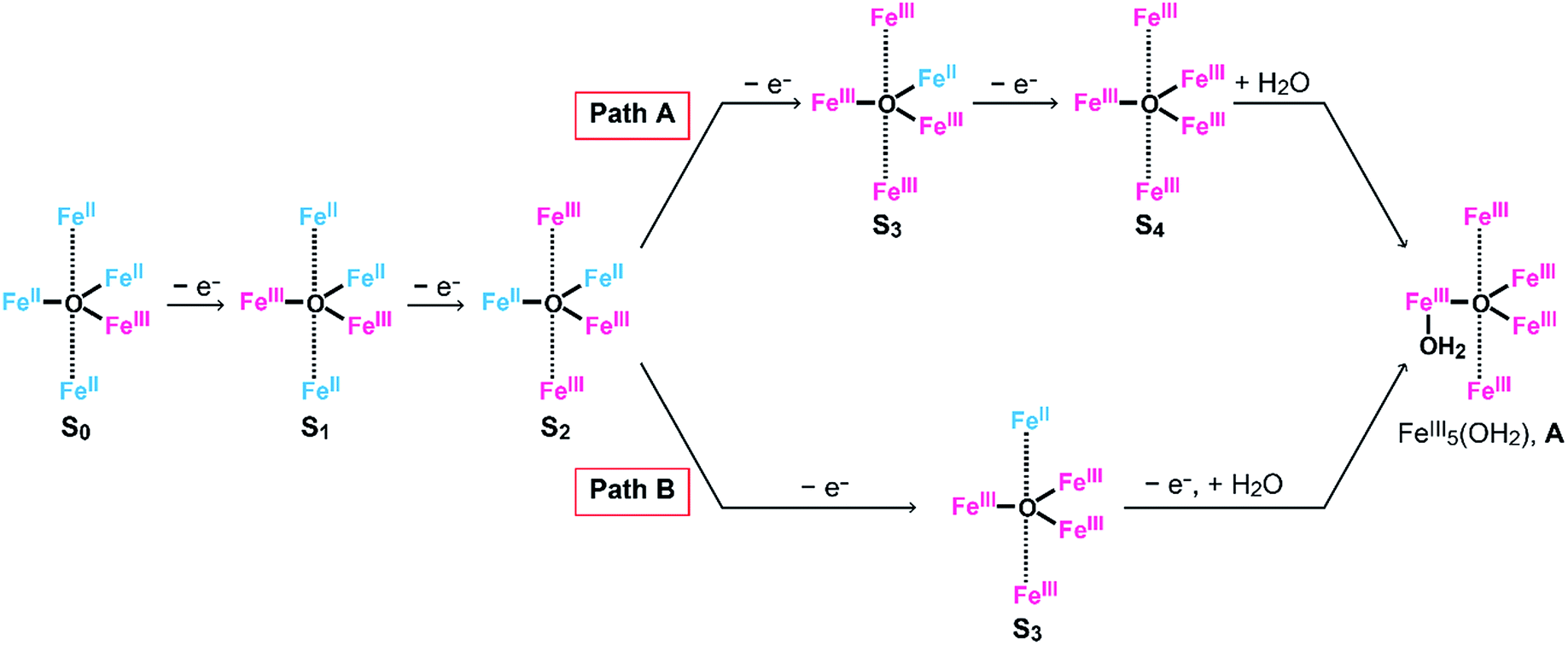
As described above, the Eonset of [Fe5-Me]3+ is located at a slightly more positive potential than the E1/2 of the fourth redox couple (Table 1). In other words, the formation of the four-electron oxidized species (FeIII5, the S4 state) triggers the reaction with a water molecule and the subsequent oxidation of water in this case. A similar trend was also observed in the previously reported electrocatalysis by [Fe5-H]3+; the onset of the catalytic wave is coupled with the formation of the four-electron oxidized species. Therefore, it is suggested that [Fe5-Me]3+ probably promotes electrocatalytic water oxidation through a catalytic cycle similar to that of [Fe5-H]3+, which we previously proposed based on experimental and computational studies33 (Fig. 5, see also the ESI (P.S32)†). In the catalytic cycle, the successive four-step, one-electron oxidation of the resting FeII4FeIII (the S0 state) initially generates the four-electron oxidized species FeIII5 (the S4 state) via the S1, S2 and S3 states. In the S4 state, all the iron atoms in the [Fe3(μ3-O)] core are in the FeIII state. Subsequently, the addition of H2O to this fully oxidized [Fe3(μ3-O)] core of the S4 state generates the water-bound FeIII5(OH2) species (intermediate A). Intermediate A then reacts with an additional H2O molecule to generate O2 and regenerate the initial S0 state. Therefore, the shift of the Eonset of [Fe5-Me]3+ (approximately 90 mV lower than that of [Fe5-H]3+) is attributed to the electron-donating nature of the methyl groups, which reduces the potential to generate the S4 state.
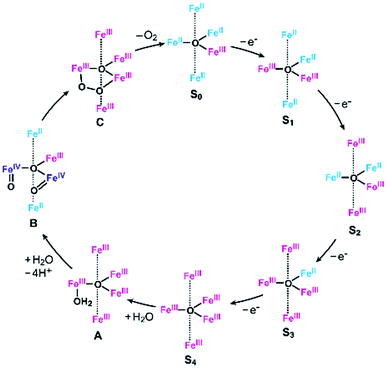 |
| | Fig. 5 Proposed reaction mechanism for the water oxidation catalyzed by [Fe5-Me]3+ and [Fe5-H]3+.33 FeII, blue; FeIII, red; and FeIV, purple. | |
Reactivity of the S3 state of [Fe5-Br]3+
Unlike [Fe5-Me]3+ and [Fe5-H]3+, the catalytic current for [Fe5-Br]3+ was not coupled with the fourth redox wave. Instead, the catalytic current (Eonset = 1.15 V) arises in the potential region between the third (0.87 V) and fourth (1.27 V) redox waves (vide supra). This result indicates that the FeIIFeIII4 state (the S3 state) generated by the third redox process should undergo a chemical reaction (EC process), and the formed species should be further oxidized at 1.15 V. Alternatively, a concerted chemical and electrochemical process should occur at 1.15 V. In any case, the S3 state is considered the key intermediate for water oxidation in the case of [Fe5-Br]3+.
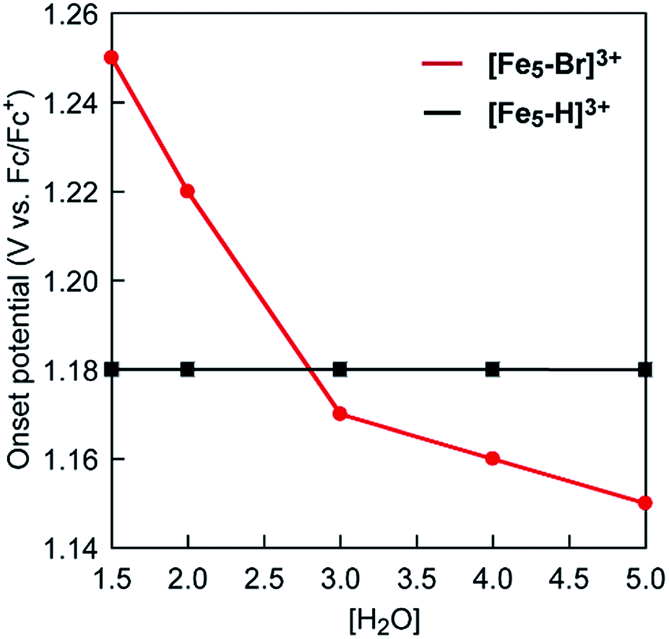
Intrigued by this finding, we set out to investigate the reactivity of the S3 state of [Fe5-Br]3+. Initially, electrochemical measurements were performed at various concentrations of [Fe5-Br]3+ to confirm if the S3 state undergoes unimolecular or bimolecular reactions. As shown in Fig. S12,† the intensity of the catalytic peak current was linearly dependent on the concentration of [Fe5-Br]3+, suggesting that the rate is first order to the catalyst concentration, and therefore a bimolecular path requiring the association of catalysts is ruled out. Second, we hypothesized that the reaction of the S3 state with H2O to form a H2O bound species and subsequent oxidation of the formed species is a possible pathway. To validate this hypothesis, CVs were collected at various scan rates in the presence of 5 M H2O by reversing the scan of potentials at 1.04 V. As shown in Fig. S13a,† the redox potentials (E1/2 values) and the wave shapes of the first three redox couples remained unchanged. Additionally, the reversibility of the third redox couple (FeIIFeIII4/FeII2FeIII3) was investigated by plotting the intensity of the anodic and cathodic peak currents against the square root of the scan rates (Fig. S13b†). The linearity of the obtained plot confirms that the third redox process is fully reversible and that no EC process occurs in this potential range. Therefore, the reaction of the S3 state with H2O does not proceed in this potential region, and this pathway can be excluded. Third, the possibility of the S3 state undergoing a proton-coupled electron transfer (PCET) reaction was considered because catalytic water oxidation reactions often involve such a process.15 However, this process hardly occurs because no dissociative proton exists in the S3 state. Moreover, the CVs of [Fe5-Br]3+ recorded under various pH conditions showed no change in the onset potential for water oxidation (Fig. S14†). Therefore, the S3 state undergoing a PCET process is also unlikely. Finally, an electron transfer reaction coupled with water binding to the S3 state was investigated. CVs of [Fe5-Br]3+ at various concentrations of H2O were acquired. As shown in Fig. 6 and S15,† the onset potentials of the electrocatalytic current gradually shifted to lower potentials as the content of H2O increased. Note that the onset potential of water oxidation was not affected by the concentrations of H2O in the case of [Fe5-H]3+ (Fig. S16†). This result clearly demonstrates that the electron transfer reaction coupled with the binding of H2O to the S3 state is the key step in the [Fe5-Br]3+-catalysed reaction.
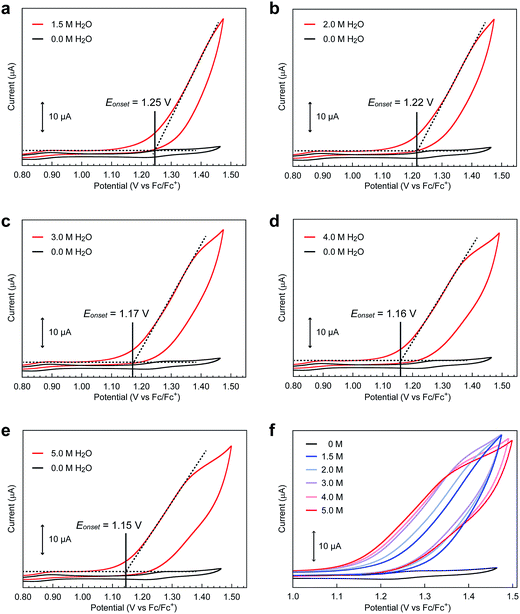 |
| | Fig. 6 Cyclic voltammograms of [Fe5-Br]3+ (0.2 mM) in the presence of (a) 1.5, (b) 2.0, (c) 3.0, (d) 4.0, and (e) 5.0 M H2O at pH = 5.0 (red lines) and in the absence of H2O (black lines). (f) Overlaid CVs of [Fe5-Br]3+ at various concentrations of H2O. CVs were measured in acetonitrile solutions with TBAP (0.1 M) on a GC electrode at a scan rate of 10 mV s−1. The onset potentials were estimated from the cross-points of two lines, which are obtained by extrapolating the slopes of the catalytic current and non-faradaic current. | |
Electronic structures of S3 states
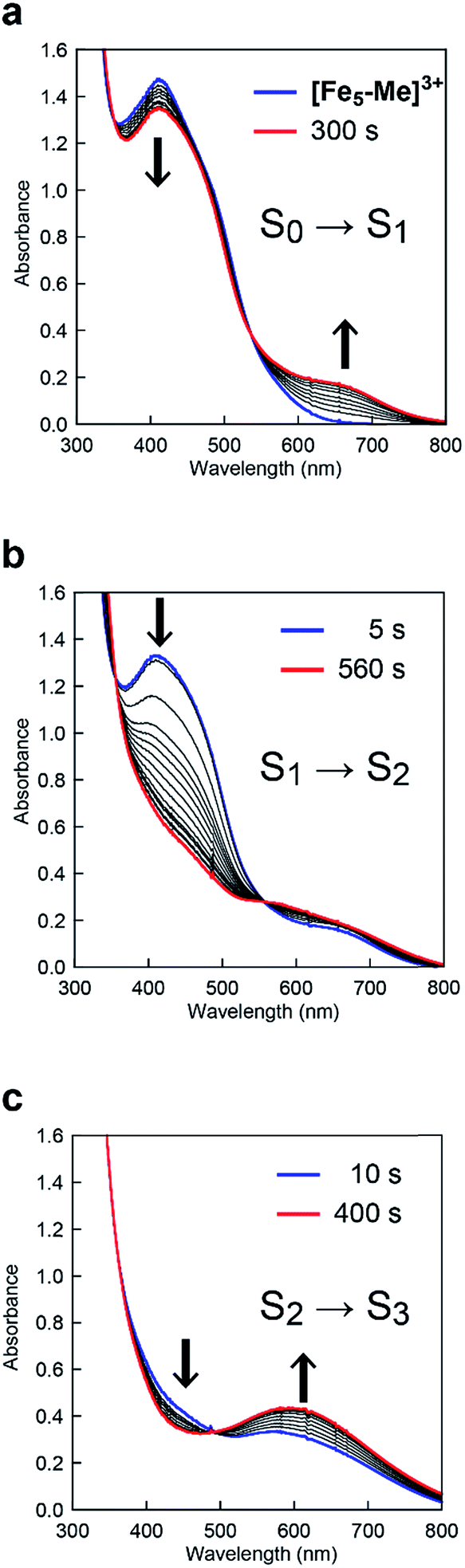
To clarify the origin of the unique reactivity of the S3 state of [Fe5-Br]3+, the electronic structures of the S3 states of a series of pentanuclear iron complexes were investigated. As we previously reported,33 the three-step oxidation of [Fe5-H]3+ affords the S3 state as evidenced by UV-Vis absorption spectroscopy (Fig. S17 and S18†). In the first step, a slight decrease in the MLCT band at 406 nm and the growth of a new broad peak at approximately 640 nm were observed. This newly observed peak is attributed to the formation of the [FeIIFeIII2(μ3-O)] central core.33 In other words, the first step corresponds to the oxidation of the central core, which yields [{FeII(μ-bpp)3}2FeIIFeIII2 (μ3-O)]4+ (the S1 state). In the second step, the intensity of the MLCT band at 406 nm drastically decreased, suggesting that both iron centres at the apical positions are oxidised during the second step. Therefore, in the second step, the oxidation of the complex induces an intramolecular electron transfer process, and a species with two FeIII ions at apical positions and one FeIII and two FeII ions in the central core, [{FeIII(μ-bpp)3}2FeII2FeIII(μ3-O)]5+ (the S2 state), forms. Further oxidation of the complex increased the intensity of the band at approximately 550 nm, which was attributed to the oxidation of the central core affording [{FeIII(μ-bpp)3}2FeIIFeIII2(μ3-O)]6+ (the S3 state). Notably, these redox transformations of the complexes, which involve an intramolecular electron transfer process in the second step, are fully consistent with the previously reported Mössbauer study in the solid state.33,64 A similar trend was also observed for [Fe5-Me]3+, as shown in Fig. 7. Therefore, [Fe5-H]3+ and [Fe5-Me]3+ undergo an electron transfer reaction in an identical manner during their conversions from FeII4FeIII to FeIII5 (Scheme 3, Path A), and the S4 states serve as key intermediates in the catalytic reaction.
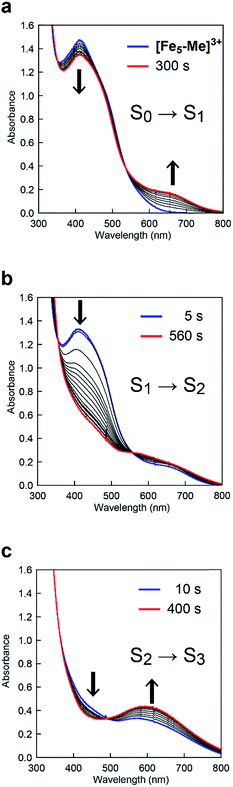 |
| | Fig. 7 UV-Vis absorption spectra of [Fe5-Me]3+ (0.05 mM) at various applied potentials ((a) 0.17, (b) 0.50 and (c) 0.86 V (vs. Fc/Fc+)) in 0.1 M TBAP/MeCN. Solutions were purged with Ar for 15 min prior to measurements. Weak Ar flow was maintained throughout the measurements. | |
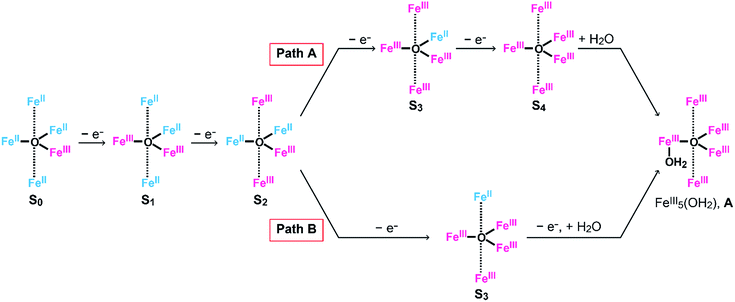 |
| | Scheme 3 Oxidation processes of (Path A) [Fe5-H]n+ and [Fe5-Me]n+ and (Path B) [Fe5-Br]n+. | |
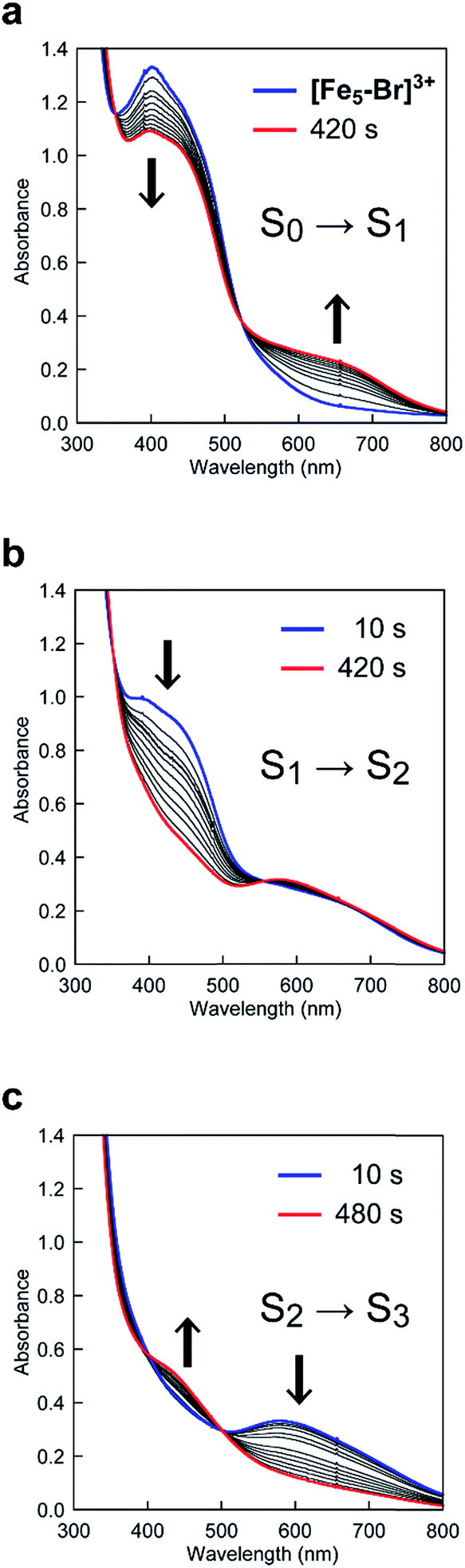
In the case of [Fe5-Br]3+, the spectral changes that occurred upon the first and second oxidations were quite similar to those of [Fe5-H]3+ and [Fe5-Me]3+; a slight decrease in the MLCT band at 402 nm and the emergence of a new band at approximately 660 nm in the first step and a drastic decrease in the MLCT band in the second step (Fig. 8a and b). Therefore, the iron ion at the central core is oxidised in the first step, and an oxidation-induced intramolecular electron transfer affords [{FeIII(μ-Br-bpp)3}2FeII2FeIII(μ3-O)]5+ (the S2 state) in the second step. However, in the third step, the complex exhibited spectral changes that were unlike those of [Fe5-H]3+ and [Fe5-Me]3+. In this step, the intensity of the band at approximately 580 nm decreased (Fig. 8c), whereas the corresponding bands for [Fe5-H]3+ and [Fe5-Me]3+ grew (vide supra). This result indicated that all iron atoms in the [Fe3(μ3-O)] core are oxidized in [Fe5-Br]6+. In other words, an additional intramolecular electron transfer reaction generates a three-electron oxidized species, and the oxidation states of the iron ions can be described as [{FeII(μ-Br-bpp)3}{FeIII(μ-Br-bpp)3}FeIII3(μ3-O)]6+ (the S3 state).
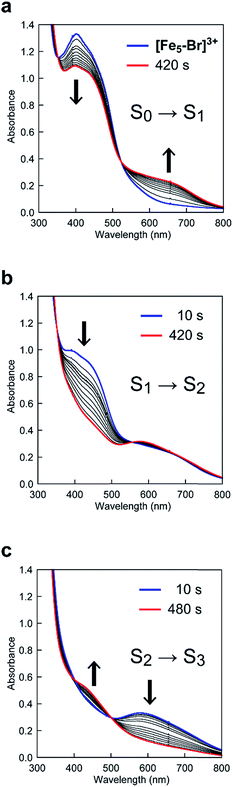 |
| | Fig. 8 UV-Vis absorption spectra of [Fe5-Br]3+ (0.05 mM) at various applied potentials ((a) 0.42, (b) 0.64 and (c) 1.00 V (vs. Fc/Fc+)) in 0.1 M TBAP/MeCN. Solutions were purged with Ar for 15 min prior to measurements. Weak Ar flow was maintained throughout the measurements. | |
In the [Fe5-Br]3+-catalysed reaction, the formed S3 state reacts with water via an electron transfer reaction coupled with the binding of H2O (vide supra). Thus, the oxidation of [Fe5-Br]3+ to afford the key intermediate can be summarized as shown in Scheme 3, Path B. These results clearly demonstrate that the electronic structure of the S3 state of [Fe5-Br]3+ is completely different from that of [Fe5-H]3+ and [Fe5-Me]3+, which may be the origin of the unique reactivity of [Fe5-Br]3+ during catalysis.
Reaction mechanism of [Fe5-Br]3+
Based on the aforementioned experimental evidence, a plausible reaction mechanism for the water oxidation reaction catalysed by [Fe5-Br]3+ was proposed. As depicted in Scheme 3, the first step involves a sequential, stepwise three-electron oxidation of the initial S0 state to produce the S3 state (via the S1 and S2 states), which includes a two-step intramolecular electron transfer process. In the S3 state of the complex, all iron atoms in the [Fe3(μ3-O)] core are in the FeIII state. Subsequently, a concerted process involving water binding to the fully oxidized [Fe3(μ3-O)] core coupled with a one-electron oxidation process gives the water-bound FeIII5(OH2) species, intermediate A (Scheme 3, Path B). Intermediate A then generates intermediates B and C, and the release of O2 from intermediate C regenerates the initial S0 state and produces O2 as a product (Fig. S19†). Thus, the formation of the S4 state is favorably bypassed in the catalytic cycle of [Fe5-Br]3+, whereas the redox potentials to form the S4 states determine the onset potentials for the catalysis in the case of [Fe5-Me]3+ and [Fe5-H]3+. As a result, the onset potential for water oxidation was lower for [Fe5-Br]3+ compared to [Fe5-H]3+ if a sufficient amount of substrate was added to the reaction mixture (Fig. 9). The result also implies that the generation of the fully oxidized [Fe3(μ3-O)] core is essential for initiating catalysis.
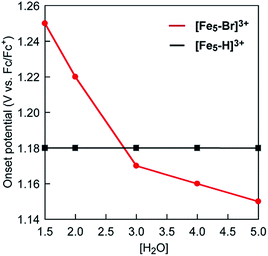 |
| | Fig. 9 A plot of onset potentials as a function of the concentration of H2O for [Fe5-Br]3+ (red line) and [Fe5-H]3+ (black line). | |
Conclusions
In conclusion, we have demonstrated the syntheses, electrochemical behaviour, and catalytic activity of a series of pentanuclear iron complexes. Two types of ligands bearing electron-donating (methyl) or electron-withdrawing (bromo) groups at the 4-position of the Hbpp ligand were successfully synthesized, and the complexation of the ligands (Me-Hbpp and Br-Hbpp) with iron resulted in the formation of pentanuclear complexes, [Fe5-Me]3+ and [Fe5-Br]3+, which have structures similar to that of the parent complex, [Fe5-H]3+.
All three complexes exhibited five reversible redox waves, which can be attributed to the sequential redox processes of iron ions in the absence of H2O. However, the redox potentials of the complexes were significantly changed by the electronic effect of the substituents installed on the ligands. The redox potentials of [Fe5-Me]3+ were shifted to more negative potentials compared to those of [Fe5-H]3+, whereas the redox waves of [Fe5-Br]3+ were observed at more positive potentials than those of [Fe5-H]3+. In the presence of H2O, both [Fe5-Me]3+ and [Fe5-Br]3+ exhibited catalytic currents attributed to water oxidation reactions, similar to [Fe5-H]3+. CPE experiments of the complexes revealed that the faradaic efficiencies for the catalysis were 92 and 86% for [Fe5-Me]3+ and [Fe5-Br]3+, respectively. The onset potentials for water oxidation by [Fe5-Me]3+ and [Fe5-Br]3+ were lower than that of [Fe5-H]3+.
To clarify the origin of the lower onset potentials of the complexes, their catalytic mechanisms were investigated. In the case of [Fe5-Me]3+, the formation of the S4 state triggers the catalytic reaction, which is similar to the pathway seen with the parent [Fe5-H]3+ complex. Therefore, the decrease in the onset overpotential for [Fe5-Me]3+ is attributed to the electron-donating nature of the methyl substituents, which allows the generation of the S4 state in the more negative potential region. In contrast, in the catalysis mediated by [Fe5-Br]3+, the three-electron oxidized species (the S3 state) served as a key intermediate due to its unique electronic structure, and the state undergoes a water binding reaction coupled with an electron transfer to initiate the catalytic reaction. In other words, the generation of the S4 state was bypassed in this system, which enables the catalytic reaction to occur at a lower onset potential. Our results reveal that not only the simple tuning of the redox potentials by the introduction of an electron-donating group but also the control over the reaction mechanism by the introduction of an electron-withdrawing group can be a valuable strategy for controlling onset potentials.
Experimental section
Materials
Pyridine-2-carboxylic acid methyl ester, 2-acetyl pyridine, 3,5-bis(2-pyridyl)pyrazole, methyl iodide, oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV), 1,8-diazabicyclo[5.4.0]undec-7-ene and tetraethylammonium perchlorate were purchased from Tokyo Chemical Industry Co., Ltd. NaBF4 and NaOH were purchased from Wako Pure Chemical Industries, Ltd. FeSO4·7H2O was purchased from Kanto Chemical Co., Inc. Potassium bis(trimethylsilyl)amide and tris(4-bromophenyl)ammoniumyl hexachloroantimonate were purchased from Aldrich and were used as received. An ITO-coated glass working electrode (0.7 mm thick, 10 Ω sq−1) was purchased from Furuuchi Chemical Co. Ltd. [FeII4FeIII(μ3-O)(bpp)6](BF4)3·7H2O ([Fe5-H](BF4)3·7H2O) was synthesized using the reported method.33 All solvents and reagents were of the highest quality available and were used as received except for tetrabutylammonium perchlorate (TBAP). Tetrabutylammonium perchlorate was recrystallized from absolute ethanol.
General methods
NMR spectra were recorded on a JEOL JNM-LA 400 spectrometer. UV-Vis spectra were recorded on a SHIMADZU UV-2550UV-Vis spectrophotometer or a UV-Vis Agilent Cary8454 spectrophotometer with a conventional quartz cuvette (path length, l = 1 cm). Spectroelectrochemical studies were performed using a BAS Inc. spectroelectrochemical quartz cell (l = 1 mm) containing Pt gauze (working electrode), a Pt wire (auxiliary electrode) and Ag/Ag+ (reference electrode) in conjunction with a CH Instruments potentiostat. Elemental analyses were performed on a J-SCIENCE LAB MICRO CORDER JM10 elemental analyser. ESI-TOF mass spectra were recorded on a JEOL JMS-T100LP mass spectrometer. Gas chromatography analysis of O2 was performed using a Shimadzu GC-2014 gas chromatograph equipped with a thermal conductivity detector and fitted with a molecular sieve (5 Å) column, and the system was calibrated with air. Dynamic light scattering (DLS) data were measured using a Photal OTSUKA ELECTRONICS ELSZ-1000 zeta-potential and particle size analyser, equipped with a 785 nm red laser source (detection limit: 0.6 nm particle diameter).
X-ray crystallography
Data collection for [Fe5-Me]3+ and [Fe5-Br]3+ was performed at 123 K on a ROD, Synergy Custom system (Rigaku Oxford Diffraction) equipped with confocal monochromated Mo-Kα radiation, and data were processed using CrysAlisPro 1.171.39.43c (Rigaku Oxford Diffraction). The structures were solved by direct methods using SIR-92 (ref. 66) and refined by the full-matrix least squares techniques on F2 (SHELXL-97).67 All nonhydrogen atoms were refined anisotropically and refined with a riding model with Uiso constrained to be 1.2 times Ueq of the carrier atom. The diffused electron densities resulting from the residual solvent molecules were removed from the data set using the SQUEEZE routine of PLATON68 and refined further using the generated data. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre: deposition numbers CCDC 1872481 and 1872482 for [Fe5-Me](PF6)3 and [Fe5-Br](BF4)3, respectively.
Electrochemical studies
Electrochemical experiments were performed on a BAS ALS Model 650 DKMP electrochemical analyser at room temperature under Ar. Cyclic voltammetry experiments were performed using a one-compartment cell with a standard three-electrode configuration, which consisted of a glassy carbon disk (diameter 3 mm, from BAS Inc.), a Ag/Ag+ couple, and a platinum wire as the working, reference and auxiliary electrodes, respectively. Between scans, the working electrode was polished with 0.05 μm alumina paste (from BAS Inc.) and washed with purified H2O. All the redox potentials of the samples presented in this paper were calibrated against the redox potential of the ferrocene/ferrocenium couple (Fc/Fc+).
Controlled potential electrolysis
Controlled potential electrolysis experiments were performed in a custom-designed gas-tight two-compartment cell separated by an anion-exchange membrane.33 In the first compartment, the ITO working electrode (1.0 cm × 1.5 cm) and Ag/Ag+ reference electrode were immersed in an electrolyte solution (0.1 M Bu4NClO4 in acetonitrile/water (10:1) mixed solvent) containing the catalyst (0.2 mM). In the second compartment, the platinum auxiliary electrode was immersed in the electrolyte solution. The amount of evolved oxygen in the headspace of the reaction cell was quantified by gas chromatography. Subsequently, the potential production of liquid products (e.g., H2O2) in the reaction was analysed by treating the electrolyzed solution with oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV) as a chemical probe.65
Syntheses
1,3-Bis(2-pyridyl)-propane-1,3-dione (I).
To a solution of pyridine-2-carboxylic acid methyl ester (2 g, 16.5 mmol) in anhydrous toluene (40 mL) under an argon atmosphere was added freshly prepared sodium ethoxide solution (9.9 mL, 2 M, 19.8 mmol). After heating the reaction mixture to 55 °C, a solution of 2-acetyl pyridine (2.26 g, 16.5 mmol) in anhydrous toluene (10 mL) was added. After stirring the resulting mixture for 2 h at 55 °C, a dark yellow precipitate appeared, and the reaction was stirred overnight at room temperature. The solvent was then evaporated, and the crude product was poured onto ice and neutralized to pH 7 with acetic acid (50%). The resulting solid was collected by filtration and dried under vacuum to give compound I (1.92 g, 52%). The product was used for the next step without further purification.
1,3-Bis(2-pyridyl)-propane-2-methyl-1,3-dione (II).
Under an argon atmosphere, compound I (0.5 g, 2.21 mmol) was dissolved in 30 mL of anhydrous toluene. Potassium bis(trimethylsilyl)amide (6.63 mL, 0.5 M, 3.32 mmol) was then added to the solution. The resulting suspension was heated to 80 °C, and the colour of the reaction mixture changed from orange to green. Then, methyl iodide (1.57 g, 11.05 mmol) was added. After heating the resulting mixture at 80 °C for 5 h, the reaction was stirred overnight at 50 °C. After cooling to room temperature, the reaction was quenched by the addition of 10% NaHCO3 (10 mL) followed by brine (10 mL) and extracted with CH2Cl2. The organic phases were dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated under reduced pressure. The crude oily product was purified by silica gel column chromatography using 30% EtOAc/n-hexane as the eluent to give compound II as a light yellow solid (0.43 g, 82%). 1H NMR (400 MHz, CDCl3): δ = 8.48 (m, 2H), 8.07 (dt, J = 7.7 Hz, J = 1.2 Hz, 2H), 7.81 (td, J = 7.7 Hz, J = 1.7 Hz, 2H), 7.40 (m, 2H), 5.72 ppm (q, J = 7.06 Hz, 1H), 1.54, J = 7.02 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ = 199.3, 152.4, 148.9, 137.2, 127.1, 122.5, 50.2, 13.2 ppm; elemental analyses calcd (%) for C14H12N2O2: C 69.99, H 5.03, N 11.66; found C 69.98, H 4.92, N 11.60.
4-Methyl-3,5-bis(2-pyridyl)pyrazole (Me-Hbpp).
Compound II (0.33 g, 1.4 mmol) was dissolved in anhydrous ethanol (25 mL), and the solution was degassed with Ar for 30 min. To this solution was added hydrazine monohydrate (0.28 g, 5.6 mmol), and the reaction mixture was refluxed at 95 °C under Ar for 3 h. After concentrating the resulting solution by rotary evaporation, the solution was kept in a refrigerator overnight. A precipitate formed, and it was collected by filtration and washed with a small amount of cold ethanol to give Me-Hbpp as a white solid (0.19 g, 61%). 1H NMR (400 MHz, CDCl3): δ = 12.04 (s, 1H), 8.69 (m, 2H), 7.80 (d, J = 7.8 Hz, 2H), 7.78 (m, 2H), 7.25 (m, 2H), 2.74 ppm (s, 3H); 13C NMR (101 MHz, CDCl3): δ = 151.5, 149.5, 136.9, 122.5, 121.8, 113.8, 11.1 ppm; elemental analysis calcd (%) for C14H12N4: C 71.17, H 5.12, N 23.71; found C 70.74, H 5.13, N 23.43.
4-Bromo-3,5-bis(2-pyridyl)-pyrazole (Br-Hbpp).
This compound was synthesized using the reported procedure.63 3,5-Bis(2-pyridy)pyrazole (0.4 g, 1.8 mmol) was dissolved in CH2Cl2 (60 mL) at 0 °C. A solution of bromine (0.4 mL) in aqueous Na2CO3 (1 N, 25 mL) was then added dropwise, and the reaction was allowed to stir for 30 min. The reaction mixture was neutralized to pH 7 with aqueous 1 M NaOH. The aqueous phase was extracted with CH2Cl2 (80 mL). The organic phase was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated under reduced pressure to yield the crude product, which was purified by column chromatography on silica gel using 5% MeOH/CH2Cl2 to afford Br-Hbpp as a pale-yellow solid (0.35 g, 65%). 1H NMR (400 MHz, CD2Cl2): δ = 12.01 (br, 1H), 8.70 (m, 2H), 8.21 (d, J = 7.9 Hz, 2H), 7.84 (td, J = 7.9 Hz, J = 1.8 Hz), 7.34 (m, 2H); 13C NMR (101 MHz, CD2Cl2): δ = 149.4, 149.3, 144.9, 136.8, 123.4, 121.8, 91.1 ppm; elemental analysis calcd (%) for C13H9N4Br: C 51.59, H 3.01, N 18.60; found C 51.59, H 3.13, N 18.28.
[FeII4FeIII(μ3-O)(Me-bpp)6](PF6)3 ([Fe5-Me](PF6)3).
Me-Hbpp (0.040 g, 0.17 mmol) was dissolved in degassed methanol (10 mL), NaOHaq (1 M, 0.17 mL, 0.17 mmol) was added, and the mixture was stirred to dissolve the Me-Hbpp. Subsequently, FeSO4·7H2O (0.038 g, 0.14 mmol) was added to the stirred solution, and the resulting dark red solution was refluxed at 80 °C for 1 h under Ar. After cooling the reaction mixture to room temperature, the mixture was filtered to remove the undissolved residue. An aqueous solution of NaPF6 (excess) was added to the filtrate, and a small amount of water was added to the solution. The solution was kept in a refrigerator for 30 min to generate a red brown precipitate. The precipitate was collected by filtration, washed with water and dried under vacuum. The obtained precipitate was dissolved in a mixture of acetonitrile and MeOH and subjected to vapour diffusion in diethyl ether to afford dark red crystals of [FeII4FeIII(μ3-O)(Me-bpp)6](PF6)3·5H2O. The crystals were collected by filtration and dried under vacuum. Yield 0.038 g (62%). Elemental analysis calcd (%) for Fe5C84H76N24P3F18O6: C 45.21, H 3.43, N 15.06; found C 45.22, H 3.16, N 15.08. ESI-TOF MS (positive ion, acetonitrile): m/z: 569.08 [FeII4FeIII(μ3-O)(Me-bpp)6]3+.
[FeII4FeIII(μ3-O)(Br-bpp)6](BF4)3 ([Fe5-Br](BF4)3).
A solution of FeSO4·7H2O (0.08 g, 0.28 mmol) in methanol (3 mL) was added to a stirred solution of Br-Hbpp (0.10 g, 0.33 mmol) and NaOHaq (1 M, 0.33 mL, 0.33 mmol) in MeOH (10 mL). The resulting dark red solution was refluxed under air at 80 °C for 12 h. The reaction mixture was then filtered to remove the undissolved residue. The obtained filtrate was precipitated with a saturated aqueous solution of NaBF4 to give a brown precipitate, which was collected by filtration, washed with water, and dried under vacuum. The obtained precipitate was dissolved in acetonitrile and subjected to vapour diffusion in diethyl ether to afford dark red crystals of [FeII4FeIII(μ3-O)(Br-bpp)6](BF4)3·4H2O. The crystals were collected by filtration and dried under vacuum. Yield 0.06 g (43%). Elemental analysis calcd (%) for Fe5C78H56Br6N24B3F12O5: C 38.58, H 2.32, N 13.84; found C 38.79, H 2.45, N 13.77. ESI-TOF MS (positive ion, acetonitrile): m/z: 698.54 [FeII4FeIII(μ3-O)(Br-bpp)6]3+.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (B) (No. 16H04125, to S. M.), a Grant-in-Aid for Young Scientists (A) (No. 15H05480, to M. K.) and a Grant-in-Aid for Challenging Research (Exploratory) (No. 17K19185, to M. K.) from the Japan Society for the Promotion of Science. This work was also supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis” (No. 17H06444, to S. M.), a Grant-in-Aid for Scientific Research on Innovative Areas “Coordination Asymmetry” (No. 17H05391, to M. K.), and JST ACT-C Grant Number JPMJCR12YH, Japan. M. K. is grateful to the Tokuyama Science Foundation.
Notes and references
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163 CrossRef CAS
 .
.
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001 CrossRef CAS .
- D. Gust, T. Moore and A. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS .
- J. J. Concepcion, R. L. House, J. M. Papanikolas and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15560 CrossRef CAS PubMed .
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC .
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974 CrossRef CAS PubMed .
- S. Fukuzumi, Y.-M. Lee and W. Nam, ChemPhotoChem, 2017, 2, 121 CrossRef .
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS .
- H. Tseng, R. Zong, J. T. Muckerman and R. Thummel, Inorg. Chem., 2008, 47, 11763 CrossRef CAS PubMed .
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462 CrossRef CAS PubMed .
- S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 182 CrossRef CAS .
- N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210 CrossRef CAS PubMed .
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, G. W. Brudvig, R. H. Crabtree and N. Haven, J. Am. Chem. Soc., 2009, 131, 8730 CrossRef CAS PubMed .
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418 CrossRef CAS PubMed .
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786 CrossRef CAS PubMed .
- M. Okamura, M. Yoshida, R. Kuga, K. Sakai, M. Kondo and S. Masaoka, Dalton Trans., 2012, 41, 13081 RSC .
- M. Yoshida, M. Kondo, S. Torii, K. Sakai and S. Masaoka, Angew. Chem., Int. Ed., 2015, 54, 7981 CrossRef CAS PubMed .
- Y. Xie, D. W. Shaffer, A. Lewandowska-Andralojc, D. J. Szalda and J. J. Concepcion, Angew. Chem., Int. Ed., 2016, 55, 8067 CrossRef CAS PubMed .
- M. Yoshida, M. Kondo, M. Okamura, M. Kanaike, S. Haesuwannakij, H. Sakurai and S. Masaoka, Faraday Discuss., 2017, 198, 181 RSC .
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, J. Benet-Buchholz, X. Sala and A. Llobet, ACS Catal., 2017, 7, 6525 CrossRef CAS .
- G. Menendez Rodriguez, G. Gatto, C. Zuccaccia and A. Macchioni, ChemSusChem, 2017, 10, 4503 CrossRef CAS PubMed .
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 1 Search PubMed .
- R. Matheu, M. Z. Ertem, M. Pipelier, J. Lebreton, D. Dubreuil, J. Benet-Buchholz, X. Sala, A. Tessier and A. Llobet, ACS Catal., 2018, 8, 2039 CrossRef CAS .
- Q. Daniel, L. Duan, B. J. J. Timmer, H. Chen, X. Luo, R. Ambre, Y. Wang, B. Zhang, P. Zhang, L. Wang, F. Li, J. Sun, M. Ahlquist and L. Sun, ACS Catal., 2018, 8, 4375 CrossRef CAS .
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524 CrossRef CAS PubMed .
- Y. Shimazaki, T. Nagano, H. Takesue, B. H. Ye, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2004, 43, 98 CrossRef PubMed .
- Z. Han, K. T. Horak, H. B. Lee and T. Agapie, J. Am. Chem. Soc., 2017, 139, 9108 CrossRef CAS PubMed .
- C. Chen, Y. Li, G. Zhao, R. Yao and C. Zhang, ChemSusChem, 2017, 10, 4403 CrossRef CAS PubMed .
- W. C. Ellis, N. D. Mcdaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990 CrossRef CAS PubMed .
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gàmez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807 CrossRef CAS PubMed .
- M. K. Coggins, M. T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531 CrossRef CAS PubMed .
- L. D. Wickramasinghe, R. Zhou, R. Zong, P. Vo, K. J. Gagnon and R. P. Thummel, J. Am. Chem. Soc., 2015, 137, 13260 CrossRef CAS PubMed .
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465 CrossRef CAS PubMed .
- A. Annunziata, R. Esposito, G. Gatto, M. E. Cucciolito, A. Tuzi, A. Macchioni and F. Ruffo, Eur. J. Inorg. Chem., 2018, 2018, 3304 CrossRef CAS .
- K. G. Kottrup, S. D'Agostini, P. H. Van Langevelde, M. A. Siegler and D. G. H. Hetterscheid, ACS Catal., 2018, 8, 1052 CrossRef CAS PubMed .
- Z. Codolà, I. Garcia-Bosch, F. Acuña-Parés, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem.–Eur. J., 2013, 19, 8042 CrossRef PubMed .
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249 RSC .
- D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178 CrossRef CAS PubMed .
- M. L. Rigsby, S. Mandal, W. Nam, L. C. Spencer, A. Llobet and S. S. Stahl, Chem. Sci., 2012, 3, 3058 RSC .
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325 RSC .
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579 CrossRef CAS PubMed .
- H. Y. Wang, E. Mijangos, S. Ott and A. Thapper, Angew. Chem., Int. Ed., 2014, 53, 14499 CrossRef CAS PubMed .
- A. M. Ullman, C. N. Brodsky, N. Li, S. L. Zheng and D. G. Nocera, J. Am. Chem. Soc., 2016, 138, 4229 CrossRef CAS PubMed .
- T. Nakazono and K. Sakai, Dalton Trans., 2016, 45, 12649 RSC .
- F. Song, R. Moré, M. Schilling, G. Smolentsev, N. Azzaroli, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2017, 139, 14198 CrossRef CAS PubMed .
- H. Y. Du, S. C. Chen, X. J. Su, L. Jiao and M. T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557 CrossRef CAS PubMed .
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498 CrossRef CAS PubMed .
- M. T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048 CrossRef CAS PubMed .
- T. Zhang, C. Wang, S. Liu, J. L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273 CrossRef CAS PubMed .
- M. K. Coggins, M. T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226 CrossRef CAS PubMed .
- C. Lu, J. Du, X. J. Su, M. T. Zhang, X. Xu, T. J. Meyer and Z. Chen, ACS Catal., 2016, 6, 77 CrossRef CAS .
- V. K. K. Praneeth, M. Kondo, P. M. Woi, M. Okamura and S. Masaoka, ChemPlusChem, 2016, 81, 1123 CrossRef CAS .
- J. Shen, M. Wang, J. Gao, H. Han, H. Liu and L. Sun, ChemSusChem, 2017, 10, 4581 CrossRef CAS PubMed .
- S. Nestke, E. Ronge and I. Siewert, Dalton Trans., 2018, 47, 10737 RSC .
- X. Jiang, J. Li, B. Yang, X.-Z. Wei, B.-W. Dong, Y. Kao, M.-Y. Huang, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2018, 57, 7850 CrossRef CAS PubMed .
- M. D. Kärkäs and B. Åkermark, Dalton Trans., 2016, 45, 14421 RSC .
- M. Kondo and S. Masaoka, Chem. Lett., 2016, 45, 1220 CrossRef CAS .
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55 CrossRef CAS PubMed .
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J. R. Shen, Nature, 2015, 517, 99 CrossRef CAS PubMed .
- I. D. Young, M. Ibrahim, R. Chatterjee, S. Gul, F. D. Fuller, S. Koroidov, A. S. Brewster, R. Tran, R. Alonso-Mori, T. Kroll, T. Michels-Clark, H. Laksmono, R. G. Sierra, C. A. Stan, R. Hussein, M. Zhang, L. Douthit, M. Kubin, C. De Lichtenberg, L. Vo Pham, H. Nilsson, M. H. Cheah, D. Shevela, C. Saracini, M. A. Bean, I. Seuffert, D. Sokaras, T. C. Weng, E. Pastor, C. Weninger, T. Fransson, L. Lassalle, P. Bräuer, P. Aller, P. T. Docker, B. Andi, A. M. Orville, J. M. Glownia, S. Nelson, M. Sikorski, D. Zhu, M. S. Hunter, T. J. Lane, A. Aquila, J. E. Koglin, J. Robinson, M. Liang, S. Boutet, A. Y. Lyubimov, M. Uervirojnangkoorn, N. W. Moriarty, D. Liebschner, P. V. Afonine, D. G. Waterman, G. Evans, P. Wernet, H. Dobbek, W. I. Weis, A. T. Brunger, P. H. Zwart, P. D. Adams, A. Zouni, J. Messinger, U. Bergmann, N. K. Sauter, J. Kern, V. K. Yachandra and J. Yano, Nature, 2016, 540, 453 CrossRef CAS PubMed .
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861 CrossRef CAS PubMed .
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935 CrossRef CAS PubMed .
- W. Zhang, J. Liu, K. Jin and L. Sun, J. Heterocycl. Chem., 2006, 43, 1669 CrossRef CAS .
- E. Gouré, B. Gerey, M. Clémancey, J. Pécaut, F. Molton, J. M. Latour, G. Blondin and M. N. Collomb, Inorg. Chem., 2016, 55, 9178 CrossRef PubMed .
- F. Kuttassery, S. Mathew, S. Sagawa, S. N. Remello, A. Thomas, D. Yamamoto, S. Onuki, Y. Nabetani, H. Tachibana and H. Inoue, ChemSusChem, 2017, 10, 1909 CrossRef CAS PubMed .
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef .
-
G. M. Sheldrick, SHELX-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed .
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, characterization, crystal structures and experimental details. CCDC 1872481 and 1872482. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00678h |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  abcd,
Masaya
Okamura
a,
Takuya
Akai
ab,
Hitoshi
Izu
ab and
Shigeyuki
Masaoka
abcd,
Masaya
Okamura
a,
Takuya
Akai
ab,
Hitoshi
Izu
ab and
Shigeyuki
Masaoka