
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocopper cross-coupling reaction for C–C bond formation on highly sterically hindered structures†
Miku
Oi
ab,
Ryo
Takita
 *ab,
Junichiro
Kanazawa
a,
Atsuya
Muranaka
b,
Chao
Wang
a and
Masanobu
Uchiyama
*ab
*ab,
Junichiro
Kanazawa
a,
Atsuya
Muranaka
b,
Chao
Wang
a and
Masanobu
Uchiyama
*ab
aGraduate School of Pharmaceutical Sciences, University of Tokyo, Hongo 7-3-1, Bunkyo-ku, Tokyo, Japan. E-mail: takita@mol.f.u-tokyo.ac.jp; uchiyama@mol.f.u-tokyo.ac.jp
bAdvanced Elements Chemistry Research Team, RIKEN Center for Sustainable Resource Science, Elements Chemistry Laboratory, RIKEN, Wako-shi, Saitama 351-0198, Japan
First published on 10th May 2019
Abstract
We describe a powerful, broadly applicable cross-coupling protocol that enables carbon–carbon bond formation at highly sterically hindered carbon centers (both sp2 and sp3) by employing organocopper reagents under palladium catalysis. Experimental studies and theoretical calculations indicated that the key to the unique reactivity of copper is the relatively low activation energy of the compact transmetalation transition state, due to Cu(I)–Pd(II) interaction, which is associated with small values of deformation energy of the reactants. This reaction is applicable to a variety of bulky substrates, including compounds inert to previous cross-coupling chemistry and has high functional group tolerance.
Introduction
Three-dimensionally (3D) bulky carbon frameworks and other bulky substrates have become important scaffolds for a broad range of functional molecules (Scheme 1A). They are useful in many fields due to features such as improved solubility, enhanced stability, and increased control of molecular stacking.1–4 While transition metal-catalyzed cross-coupling reactions are among the most developed of C–C bond-forming reactions,5 bond formation at sterically hindered structures remains a challenging task in cross-coupling chemistry, even with sp2- or sp3-carbon substrates. Recent improvements have focused mostly on the design of (pre)catalysts and customized ligands in order to achieve efficient generation of active palladium species, and on the oxidative addition/reductive elimination step in the catalytic cycle (Scheme 1B).6 These “state-of-the-art” systems, in particular with customized ligands, enable bond formation on sterically hindered substrates.7 However, transmetalation, a fundamental step in cross-coupling reactions,8 can also be targeted to address this challenge. | ||
| Scheme 1 (A) Three-dimensionally (3D) bulky carbon frameworks in a broad range of disciplines. (B) Fundamental steps in cross-coupling reactions and recent developments in the catalytic cycle. (C) Pd-catalyzed organocopper cross-coupling reaction on highly sterically hindered structures (this work). | ||
Herein, we report a powerful and broadly applicable (to both sp2- and sp3-carbons) Pd-catalyzed cross-coupling reaction of organocopper reagents that enables efficient C–C bond formation even on 3D bulky carbon frameworks (Scheme 1C). Notably, this reaction proceeds under mild conditions using readily available reagents and has high functional group tolerance. Experimental and theoretical studies revealed that copper(I)–palladium(II) interaction facilitates the formation of a compact and well-organized transition state in the transmetalation step.
Results & discussion
We commenced our search to develop a potent methodology by focusing on the triptycene framework,9 due to its extremely hindered tertiary sp3-carbon at the bridgehead 9-position and unprecedent use in cross-coupling chemistry. The cross-coupling reactions using a boronate ester or zinc(II) complex of 9-triptycene (1a and 1b) failed to afford any coupled product under the representative palladium-catalyzed conditions (runs 1–3, Table 1).7e,g,h On the other hand, we found that the cross-coupling reaction of 9-triptycenylcopper(I) complex 1c10 with 2 smoothly proceeded using 5 mol% of Pd(OAc)2 and tris(o-methoxyphenyl)phosphine (L1) at 80 °C to afford the coupling product 3 in 86% yield (run 4). The direct use of organocopper reagents in Pd-catalyzed cross-coupling reactions is limited,11 and a copper co-catalyst12 as well as relay catalysis using palladium and copper13 have been employed. The absence of a palladium catalyst yielded no desired product (run 5). Under similar reaction conditions, other organometallic reagents (1a, 1b, 1d, and 1e) were also not competent (runs 6–9).|
|
|||
|---|---|---|---|
| Run | Metal | Conditions, time (h) | Yielda (%) |
| a NMR yields determined using dimethylsulfone as an internal standard. Reaction time: 4 h (runs 4 and 5) or 20 h (other runs). b Ref. 7e. c Ref. 7g. d Ref. 7h. e L1: tris(o-methoxyphenyl)phosphine. | |||
| 1 | Bpin (1a) | Pd(OAc)2, SPhos, K3PO4, toluene, 100 °Cb | 0 |
| 2 | ZnCl (1b) | Pd(OAc)2, RuPhos, THF–toluene, 100 °Cc | 0 |
| 3 | ZnCl (1b) | XPhos G3, XPhos, THF–toluene, 100 °Cd | 0 |
| 4 | Cu (1c) | Pd(OAc)2, L1e, THF–toluene, 80 °C | 86 |
| 5 | Cu (1c) | L1 , THF–toluene, 80 °C | 0 |
| 6 | Bpin (1a) | Pd(OAc)2, L1e, K3PO4, THF–toluene, 100 °C | 0 |
| 7 | ZnCl (1b) | Pd(OAc)2, L1e, THF–toluene, 100 °C | Trace |
| 8 | MgCl (1d) | Pd(OAc)2, L1e, THF–toluene, 100 °C | 0 |
| 9 | Li (1e) | Pd(OAc)2, L1e, THF–toluene, 100 °C | 0 |

The optimized conditions of the present organocopper cross-coupling reaction were then applied to bond formation using a variety of electrophiles (Scheme 2A). Both electron-withdrawing and -donating groups on aryl halides, containing CO2Me, CF3, CN, acetyl, and MeO groups, were compatible with the reaction conditions, and the desired products were obtained in good yields (3–8). Moreover, sterically demanding ortho-substituted substrates gave 9-arylated triptycene derivatives 9 and 10. X-ray crystallography of 9 confirmed the desired C–C bond formation. A straightforward, one-step synthesis of 9-ferrocenyltriptycene 11 (ref. 4b) was accomplished with the present coupling strategy. Various heteroaromatic moieties were successfully coupled (12–18), and bond formation was also achieved with sp-carbon of an alkynyl substrate 19. The coupling reaction with ethyl bromoacetate proceeded to give the product 20 in 76% yield having a C(sp3)–C(sp3) bond.14 Since existing syntheses of 9-functionalized triptycenes are rather lengthy,9 the present methodology offers substantial synthetic advantages, including high functional group tolerance.
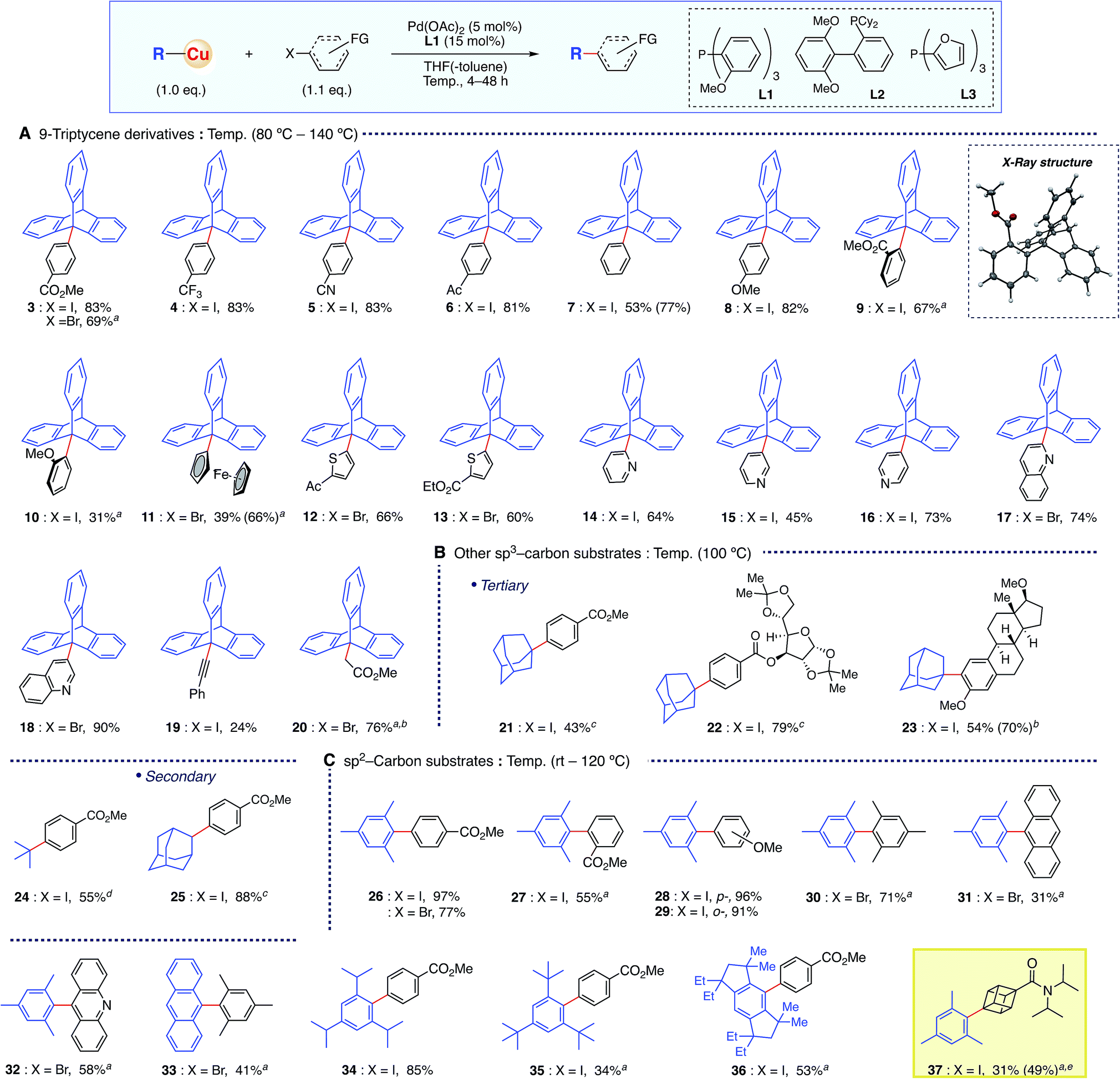 | ||
| Scheme 2 Organocopper cross-coupling reaction of sp3- and sp2-carbon substrates with sterically hindered structures. Isolated yields are shown (1H NMR yields in parentheses). aL2 was used instead of L1. b Ni(acac)2 was used instead of Pd(OAc)2. cL2 was used instead of L1 and 3 eq. of TMEDA were added. dL3 was used instead of L1 at −20 °C and 3 eq. of TMEDA were added. e 10 mol% Pd catalyst and 30 mol% L2 were used. | ||
Other sterically hindered tertiary sp3-carbon compounds were also competent substrates for this reaction (Scheme 2B). The adamantyl group was successfully introduced not only onto a simple aryl group (21), but also onto aryl halide moieties on a sugar skeleton and into a steroid backbone, affording the corresponding products 22 and 23 in good yields. These results indicate that the present methodology would be suitable for late-stage functionalization of biologically relevant compounds and functional molecules with bulky tertiary alkyl moieties. Similarly, the introduction of a tert-butyl group having β-hydrogen was achieved, affording 24 in 63% yield. A secondary 2-adamantyl group was also introduced onto an aromatic ring to give 25 in 88% yield.
Further examination revealed that this methodology was also applicable to sterically hindered sp2-carbon using conditions essentially identical to those employed for sp3-substrates (Scheme 2C). The reaction between mesitylcopper(I) and 2 proceeded efficiently at room temperature, affording the coupled product 26 in quantitative yield. The same organocopper(I) reagent reacted smoothly under these conditions with various electrophiles, affording the corresponding biaryl products 27–32 in good yields, including hindered ortho-substituted substrates. Furthermore, the present protocol realized the reactions with much bulkier copper reagents, such as “super-mesityl”15 (35) and EMind16 (36) substrates. Importantly, this reaction could also be applied to the cubane skeleton (i.e.37). Although the cubane motif has recently attracted great attention as a bioisostere of benzene in pharmaceuticals,17 there have only been two examples of cross-coupling chemistry, and the yields were not high with respect to the catalyst loading.18 In contrast, the present organocopper cross-coupling reaction enabled catalytic C–C bond formation with an iodocubane derivative for the first time, installing the sterically hindered mesityl group in 49% yield using a 10 mol% Pd catalyst.14
Given that the nature of the metal species should have the greatest influence on the transmetalation step (Scheme 1B), and taking into account the mechanistic implications of experimental findings,19 we performed theoretical calculations for the ligand transfer process from 9-metalated triptycene (boron, zinc, and copper) to an arylpalladium complex as a model of the transmetalation step (Fig. 1) at the ωB97X-D/SDD & 6-31+G* level of theory. DFT calculation for the reaction of 9-triptycenylboronate with trans-Pd(Ph)OH(PMe3)20 indicated that the bulky triptycene group distorts the transition-state structure (TSB1), resulting in a high activation energy (ΔG‡ = +46.4 kcal mol−1, Fig. 1A). Although a lower activation barrier (ΔG‡ = +28.0 kcal mol−1) was observed with 9-triptycenylzinc chloride viaTSZn1, this reaction (IMZn1–TSZn1–IMZn2) is thermodynamically unfavorable; the transmetalated product IMZn2 is quite unstable (+25.5 kcal mol−1, compared with IMZn1), suggesting that efficient transmetalation is improbable (Fig. 1B). These findings are consistent with the experimental results shown in Table 1.
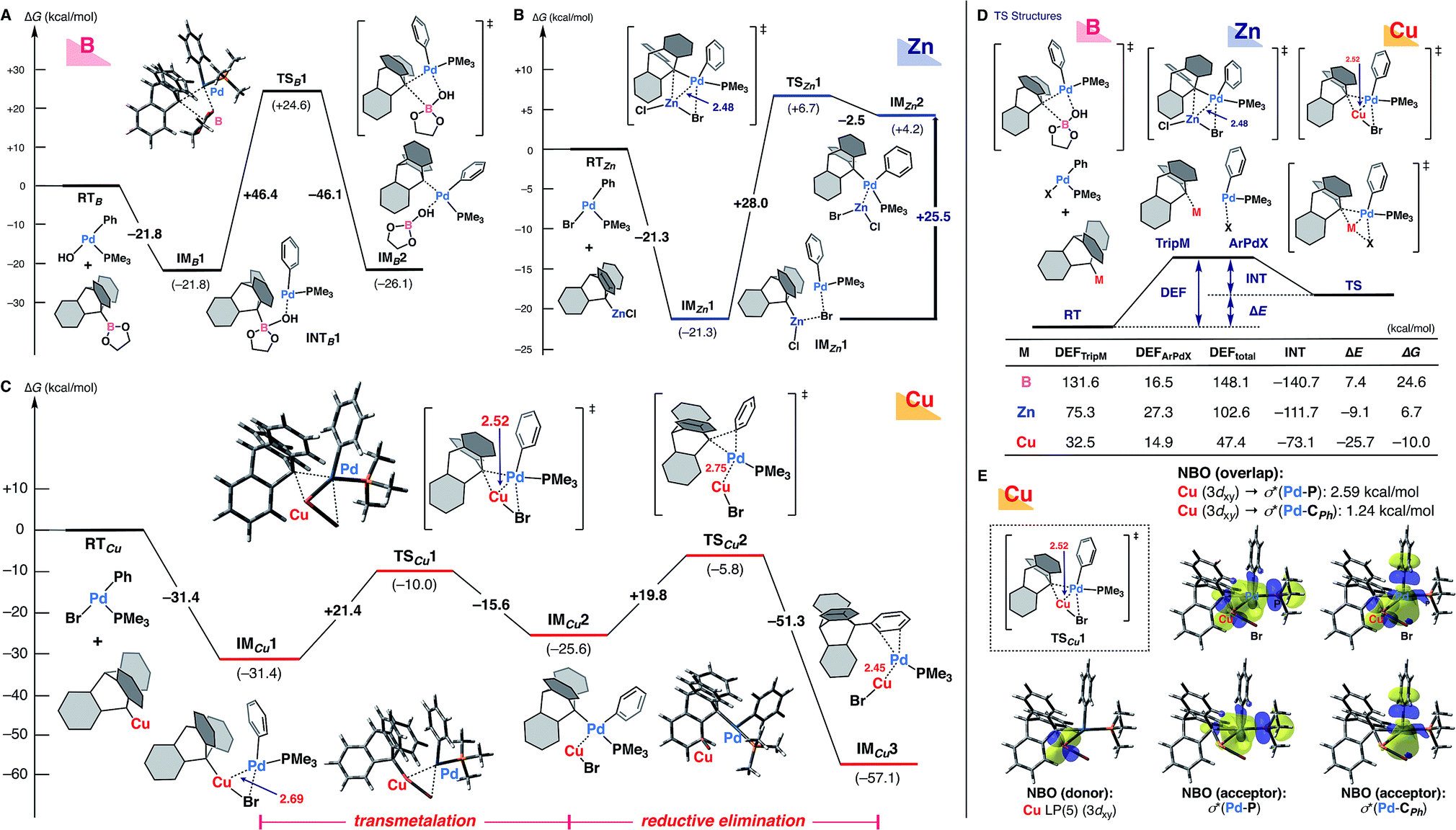 | ||
| Fig. 1 Theoretical calculations for the transmetalation step between an arylpalladium complex and 9-metalated triptycene complexes (A) with a 9-triptycenylboronate reagent, (B) with a 9-triptycenylzinc chloride reagent, and (C) with a 9-triptycenylcopper reagent (the reductive elimination step is also shown). Energy changes and bond lengths at the ωB97X-D/SDD (for Pd, Cu, Zn, and Br) & 6-31+G* (for other atoms) level of theory are shown in kcal mol−1 and Å, respectively. (D) Results of EDA. (E) Results of NBO analysis. | ||
The situation with the copper reagent is completely different; the initial coordination of 9-triptycenylcopper and trans-Pd(Ph)Br(PMe3) (RTCu) affords IMCu1 with a large stabilization energy (−31.4 kcal mol−1) (Fig. 1C). Furthermore, the distance between the copper and palladium atoms is shorter (2.69 Å) than the sum of their van der Waals radii (3.03 Å), supporting the existence of strong metal–metal interaction.21 This Cu–Pd interaction results in the formation of a compact transition-state structure (TSCu1, Cu–Pd: 2.52 Å) that facilitates delivery of the bulky triptycenyl group from the copper to the palladium center with a reasonable activation energy (ΔG‡ = +21.4 kcal mol−1). The resultant IMCu2 having both phenyl and triptycenyl ligands on palladium in a cis-fashion then undergoes reductive elimination. The reductive elimination also reasonably proceeds (ΔG‡ = +19.8 kcal mol−1) to achieve the C–C bond formation on the triptycene framework (IMCu3).
Thus, the unique observed reactivity of the copper complex, in contrast to the boron or zinc complex, is supported by the theoretical calculations. Although the possibility of a similar metal–metal interaction in transmetalation has been suggested previously,22–24 our DFT calculations directly compare the transmetalation transition-state structures of an arylpalladium complex with these organometallic reagents. Energy decomposition analysis (EDA)25 of these transition-state structures clearly indicates that small values of the deformation energy (DEF) are mainly responsible for the low activation barrier in the case of the copper reagent (Fig. 1D). In particular, the 9-triptycenylcopper unit involves a much lower DEFTripM than those in the cases of boron and zinc, probably due to the favorable Cu(I)–Pd(II) interaction, as well as the abundance of vacant sites around the copper center. Interestingly, the Zn(II) center also shows some interaction with Pd(II), but the DEFs of both the 9-triptycenyl zinc reagent and the arylpalladium complex are much larger. In the less-distorted TSCu1, smooth delivery of the triptycenyl ligand is facilitated by the well-organized triangular arrangement of copper–palladium–carbon (Cu(I)–Pd(II)–C). In addition, natural bond orbital (NBO)26 analysis showed that electron donation from Cu(I) to Pd(II) is predominant in IMCu1 and TSCu1 (Fig. 1E), while the reverse donation from the Pd(II) center to Lewis acidic Zn(II) was observed in TSZn1.14,23d,e These results reflect the characteristic reactivity of organocopper reagents arising from the high-energy d orbitals and vacant coordination sites of copper. Thus, transfer of the bulky triptycenyl ligand is promoted not only by the adjacency of the two metal centers, but also by the favorable electronic interaction between them.
Conclusions
In conclusion, the experimental results and DFT calculations confirm that this organocopper cross-coupling reaction is a potent C–C bond-forming methodology with unprecedented applicability to 3D bulky molecules. The unique ability of copper to facilitate efficient transmetalation via a compact transition state arising from an efficient metal–metal interaction is the key to the success of this reaction. Thus, this reaction is quite efficient with high functional group tolerance, and applicable to both sp2- and sp3-substrates. Further investigations on the substrate scope and detailed reaction mechanism are the subjects of ongoing research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from JSPS KAKENHI (S) (17H06173), the Asahi Glass Foundation, the Kobayashi International Scholarship Foundation, the NAGASE Science & Technology Development Foundation, and the Sumitomo Foundation (to M. U.), as well as a JSPS Grant-in-Aid for Young Scientists (A) (25713001) and grants from the Tokyo Biochemical Research Foundation, Takeda Science Foundation, FUGAKU Trust for Medicinal Research, and Uehara Memorial Foundation (to R. T.). M. O. is grateful for the Graduate Program for Leaders in Life Innovation (GPLLI) of the University of Tokyo and a JSPS Research Fellowship for Young Scientists. The computations were performed using HOKUSAI GreatWave at the RIKEN and Research Center for Computational Science at Okazaki, Japan.Notes and references
-
(a)
G. L. Patrick, An Introduction to Drug Synthesis; Oxford University Press, 2015 Search PubMed
; (b) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed
- For a recent representative example, see: Y. Wada, S. Kubo and H. Kaji, Adv. Mater., 2018, 30, 1705641 CrossRef PubMed
- For representative reviews, see:
(a) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277 CrossRef CAS PubMed
-
(a) T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150 CrossRef CAS PubMed
-
(a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed
-
(a) P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497 CrossRef CAS
- For the representative examples of the Pd-catalyzed reactions of sterically hindered substrates with bulky phosphine or NHC ligands, see:
(a) N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440 CrossRef CAS PubMed
- For a book and reviews, see:
(a)
J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed
- For representative utilization of triptycene derivatives, see:
(a) T. M. Swager, Acc. Chem. Res., 2008, 41, 1181 CrossRef CAS PubMed
- 1c was prepared by the addition of CuI to 9-lithiated triptycene in THF–toluene solution. Much of copper reagents in Table 2 were prepared from the corresponding lithiated substrates. 1- and 2-adamantyl reagents were prepared from the commercially available zinc bromide solutions. For details, see the ESI.†.
-
(a) N. Jabri, A. Alexakis and J. F. Normant, Tetrahedron Lett., 1981, 22, 959 CrossRef CAS
-
(a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef
- For a review, see:
(a) D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705 RSC
- For details, see the ESI.†.
-
(a) S. Ito, H. Miyake and M. Yoshifuji, ARKIVOC, 2012, 6 Search PubMed
- M. Kobayashi, T. Matsuo, T. Fukunaga, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, J. Am. Chem. Soc., 2010, 132, 15162 CrossRef CAS PubMed
-
(a) K. F. Biegasiewicz, J. R. Griffiths, G. P. Savage, J. Tsanaktsidis and R. Priefer, Chem. Rev., 2015, 115, 6719 CrossRef CAS PubMed
-
(a) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132 CrossRef CAS PubMed
- Control experiments gave some mechanistic insights, including the necessity of Pd catalyst (run 5, Table 1), no effects on the reaction outcome in the presence of radical scavengers, and the product formation in the reaction of the prepared 1c and Pd(Ar)I(PPh3)2 complex at 80 °C (Ar = p-MeO2C-C6H4–). For details, see the ESI.†.
-
(a) A. A. Thomas and S. E. Denmark, Science, 2016, 352, 329 CrossRef CAS PubMed
-
(a) P. Pyykkö, Chem. Rev., 1997, 97, 597 CrossRef
-
(a) R. J. Oeschger and P. Chen, J. Am. Chem. Soc., 2017, 139, 1069 CrossRef CAS PubMed
-
(a) D. G. Musaev and L. S. Liebeskind, Organometallics, 2009, 28, 4639 CrossRef CAS PubMed
- During the preparation of the manuscript, it was reported that the ligand transfer between Rh(I) and Au(I) species should proceed via an oxidative addition/reductive elimination pathway, see: M. N. Peñas-Defrutos, C. Bartolomé, M. García-Melchor and P. Espinet, Angew. Chem., Int. Ed., 2019, 58, 3501 CrossRef PubMed
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236 CrossRef CAS
-
(a)
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0., Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1566502. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00891h |
| This journal is © The Royal Society of Chemistry 2019 |