
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThreading-gated photochromism in [2]pseudorotaxanes†
Giorgio
Baggi
a,
Lorenzo
Casimiro
 bd,
Massimo
Baroncini
cd,
Serena
Silvi
*bd,
Alberto
Credi
cd and
Stephen J.
Loeb
*a
bd,
Massimo
Baroncini
cd,
Serena
Silvi
*bd,
Alberto
Credi
cd and
Stephen J.
Loeb
*a
aDepartment of Chemistry and Biochemistry, University of Windsor, Windsor, ON N9B 3P4, Canada. E-mail: loeb@uwindsor.ca
bDipartimento di Chimica “G. Ciamician”, Università di Bologna, 40126 Bologna, Italy. E-mail: serena.silvi@unibo.it
cDipartimento di Scienze e Tecnologie Agro-alimentari, Università di Bologna, 40127 Bologna, Italy
dCLAN-Center for Light Activated Nanostructures, Università di Bologna, Consiglio Nazionale delle Ricerche, 40129 Bologna, Italy
First published on 23rd April 2019
Abstract
Rigid, Y-shaped imidazole compounds containing the bis(thienyl)ethene moiety were designed and synthesized. The 4,5-bis(benzothienyl)-2-phenylimidazolium cations were then used as axles for [2]pseudorotaxane formation with 24-membered crown ether wheels. It was demonstrated using 1H NMR spectroscopy, UV-Vis absorption and emission spectroscopies that this host-guest interaction results in significant changes in the photochromic properties of the imidazolium axles. This is a rare example of gated photochromism, which exploits the recognition event of an interpenetrated molecular system to tune the photochromic properties in one of the components.
Introduction
The discovery of new templating motifs for the preparation of interpenetrated and mechanically interlocked molecules (MIMs) has led to the development of a wide variety of pseudorotaxane- and rotaxane-based systems that operate as molecular switches,1 machines and motors.2 In many instances, light has been employed as a highly versatile and “clean” stimulus for both inducing endergonic transformations (i.e. energy supply) and reading the state of the device.3Photochromism – the reversible interconversion of a molecule, with at least one of the reactions being induced by absorption of light, between two forms having different absorption spectra4 – has been thoroughly investigated since the 1980s and become a key component of the supramolecular chemist toolbox. The prominent changes in structure and properties associated with photoisomerization have been exploited to control threading,5 self-assembly processes,6 and non-equilibrium behavior7 in [2]pseudorotaxanes, and molecular movements (shuttling, rotation),8 surface properties,9 catalysis10 and reactivity.11
In molecular devices, photochromism is generally employed to exert light-responsive control over other chemical processes. In contrast, the chemical modulation of the photoreactivity of a photochrome, i.e. “gated photochromism”, is much less common, and has usually been achieved by means of transition metal coordination,12 reversible chemical modification,13 supramolecular self-assembly,14 or formation of inclusion complexes.15 To the best of our knowledge, only one photochromic system gated by a [2]pseudorotaxane formation event has been reported; Hecht et al. described an azobenzene-based axle where photoisomerization can be locked and unlocked by threading or de-threading, respectively, with a crown ether host.16
We have recently developed axles based on an imidazolium recognition site, which, in combination with crown ethers, yields [2]pseudorotaxanes, [2]rotaxanes, [3]rotaxanes, and [1]suitanes.17 The rigidity of these imidazole-based axles also allowed us to incorporate MIMs into metal–organic frameworks (MOFs) and study the dynamics of the interlocked macrocycles in the solid state.18 In order to develop a templating motif endowed with a photoswitchable recognition site, we explored the possibility of including the imidazolium moiety as an integral part of a photochromic unit.
Despite the widespread use of bis(thienyl)ethenes as molecular photoswitches,19 only a few examples of [2]pseudorotaxanes20 and [2]rotaxanes21 containing this moiety as a structural component or signalling unit have been reported. As such, we report herein the design and synthesis of 4,5-bis(benzothienyl)-2-phenylimidazolium axles and show that their association with 24-crown-8 ethers results in significant enhancement of the photochromic properties of the axles. This phenomenon, that could be termed threading-gated photochromism, expands the concept of gated photochromism, by exploiting supramolecular interactions to induce significant changes in the photochromic properties in one of the components involved in an interpenetrated molecular system.
Results and discussion
Design, synthesis, and characterization of axles
The initial design was inspired by the striking similarity between our previously reported Y-shaped 2,4,5-triphenylimidazolium axles17d and the photochromic 4,5-bis(thienyl)imidazoles originally reported by Krayushkin et al. (Scheme 1a and b).22 We thus decorated the 4- and 5-positions of three different 2-phenylimidazolium derivatives with 2-methylbenzo[b]thiophen-3-yl groups to attain photochromic axles capable of association with 24-membered crown ether rings (Scheme 1c). The choice of benzothiophene stems from the increased thermal stability of the ring-closed isomer and the improved fatigue resistance displayed in diarylethenes bearing this type of hetaryl moiety.23 The methoxycarbonyl and methoxy groups in axles [2o-H]BF4, and [3o-H]BF4 were chosen to probe the influence of electron-withdrawing and electron-donating substituents, for comparison with the parent axle [1o-H]BF4 (Scheme 2).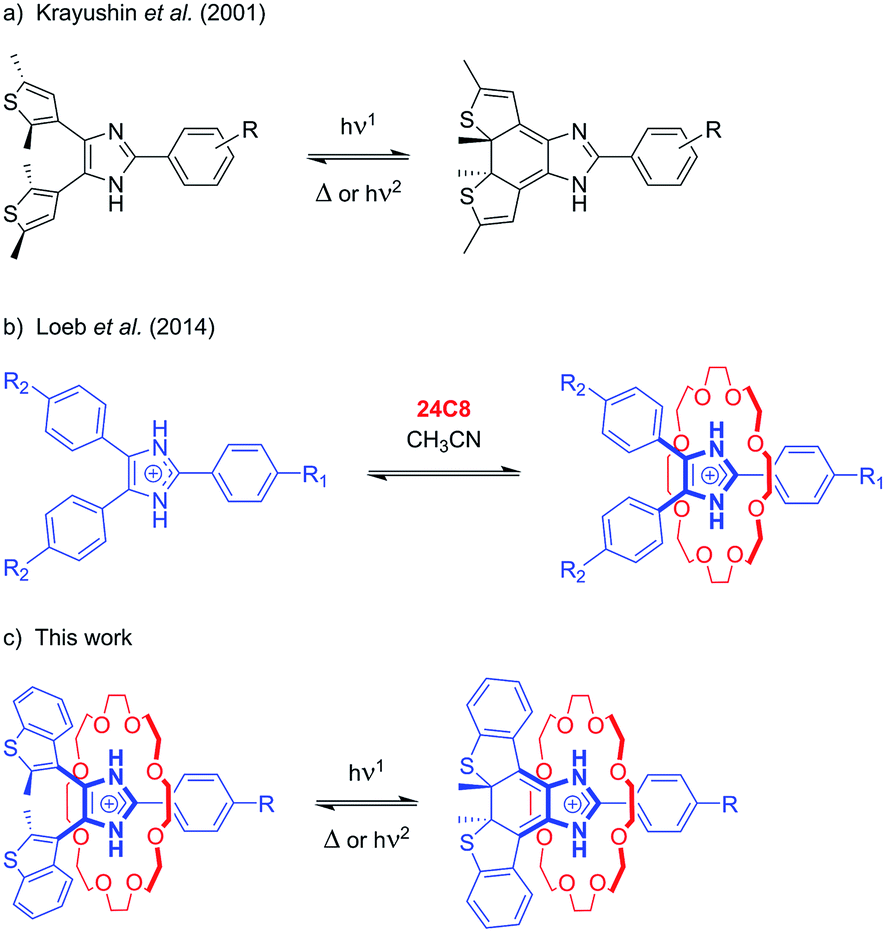 | ||
| Scheme 1 Design of bis(thienyl)imidazolium axles for photochromic [2]pseudorotaxanes. | ||
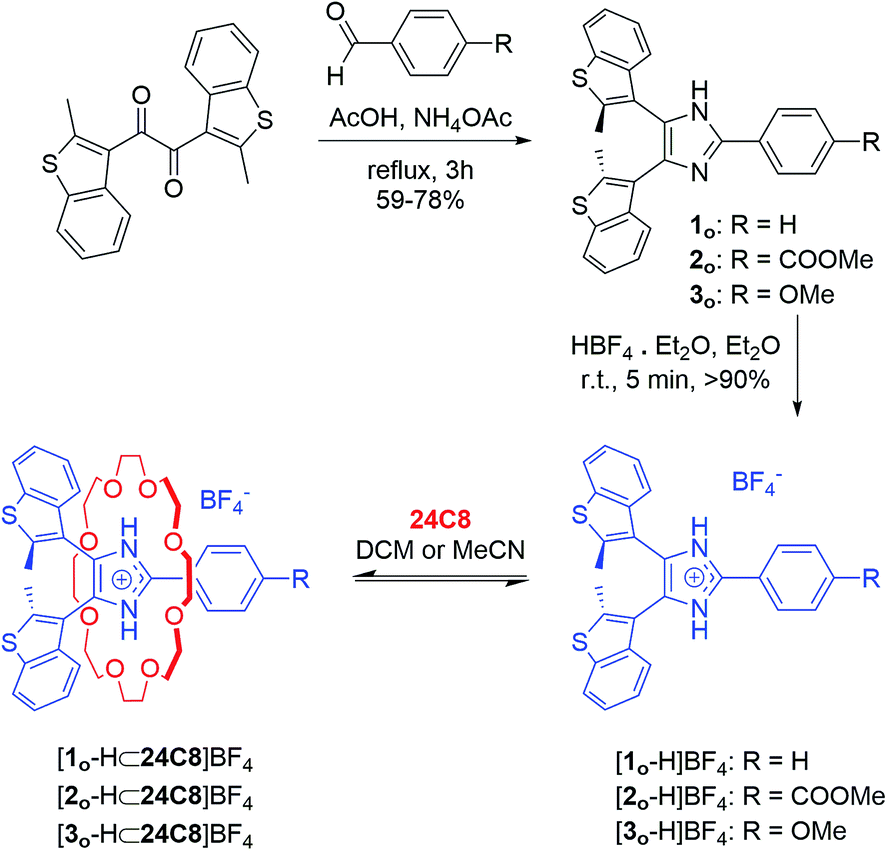 | ||
| Scheme 2 Synthesis of bis(thienyl)imidazolium axles and [2]pseudorotaxane formation. | ||
The synthetic approach followed for the preparation of the open form of the axles [1o-H]BF4, [2o-H]BF4, and [3o-H]BF4 (subscripts o = open, c = closed) is summarized in Scheme 2 and described in detail in the ESI (Scheme S1†). Lithiation of 3-bromo-2-methylbenzo[b]thiophene,24,25 followed by conversion to the corresponding organocopper compound, and subsequent reaction with oxalyl chloride (see ESI†), afforded the 1,2-diketone 7, an important precursor for the synthesis of an array of photochromic bis(thienyl)ethenes with different heterocyclic bridging units.13b,26 Subsequent cyclo-condensation of diketone 7 with the appropriate benzaldehyde and ammonium acetate, and treatment with tetrafluoroboric acid yielded [1o-H]BF4, [2o-H]BF4, and [3o-H]BF4.
The newly synthesized axles were fully characterized by NMR spectroscopy and, in the case of [1o-H]BF4 and [2o-H]BF4, by single-crystal X-ray diffraction. Variable temperature (VT) 1H NMR experiments in CD2Cl2 showed that the interconversion between parallel and anti-parallel conformations of axle [1o-H]BF4 becomes slow enough on the NMR timescale below 273 K (coalescence temperature Tc) to yield separate sets of peaks for each conformer, which become resolved below 233 K (slow-exchange limit), revealing a parallel versus anti-parallel ratio of 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :47. Lineshape analysis of the methyl group resonances in the range 203–298 K allowed determination of the exchange rates k between parallel and anti-parallel conformers at different temperatures; k values were then used to build an Eyring plot and extrapolate the activation parameters: ΔH‡ = 10.6 kcal mol−1; ΔS‡ = −8.54 cal mol−1 K−1; ΔG‡298 = 13.1 kcal mol−1; k298 = 1530 s−1 (ESI, Fig. S18–20†). Axles [1o-H]BF4 and [2o-H]BF4 were isolated as single crystals suitable for X-ray diffraction studies (see ESI†). The crystal structure of [1o-H]BF4 displays a disordered benzothiophene moiety, corresponding to anti-parallel (Fig. 1a) and parallel (Fig. 1b) conformations of the axle within the same crystal lattice. The bis(benzothienyl)imidazolium moiety of [1o-H]+ prefers to adopt a parallel conformation in the crystalline state, as demonstrated by the 94:6 distribution of parallel/anti-parallel conformers in the crystal structure. Conversely, only the anti-parallel conformation of the bis(benzothienyl)imidazolium moiety is displayed in the crystal structure of [2o-H]BF4·2Et2O (ESI, Fig. S8a†). Interestingly, in this crystal structure the two imidazolium NH groups hydrogen bond to two diethyl ether solvent molecules, which adopt a spatial arrangement around the axle resembling two fragments of a crown ether ring (ESI, Fig. S8b†).
:47. Lineshape analysis of the methyl group resonances in the range 203–298 K allowed determination of the exchange rates k between parallel and anti-parallel conformers at different temperatures; k values were then used to build an Eyring plot and extrapolate the activation parameters: ΔH‡ = 10.6 kcal mol−1; ΔS‡ = −8.54 cal mol−1 K−1; ΔG‡298 = 13.1 kcal mol−1; k298 = 1530 s−1 (ESI, Fig. S18–20†). Axles [1o-H]BF4 and [2o-H]BF4 were isolated as single crystals suitable for X-ray diffraction studies (see ESI†). The crystal structure of [1o-H]BF4 displays a disordered benzothiophene moiety, corresponding to anti-parallel (Fig. 1a) and parallel (Fig. 1b) conformations of the axle within the same crystal lattice. The bis(benzothienyl)imidazolium moiety of [1o-H]+ prefers to adopt a parallel conformation in the crystalline state, as demonstrated by the 94:6 distribution of parallel/anti-parallel conformers in the crystal structure. Conversely, only the anti-parallel conformation of the bis(benzothienyl)imidazolium moiety is displayed in the crystal structure of [2o-H]BF4·2Et2O (ESI, Fig. S8a†). Interestingly, in this crystal structure the two imidazolium NH groups hydrogen bond to two diethyl ether solvent molecules, which adopt a spatial arrangement around the axle resembling two fragments of a crown ether ring (ESI, Fig. S8b†).
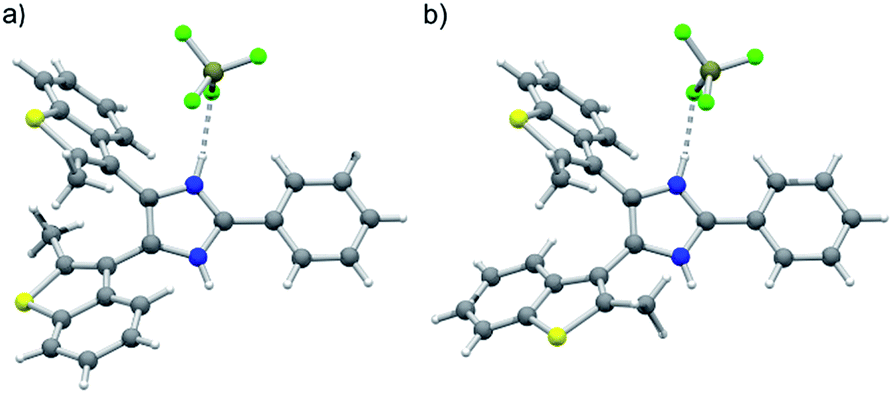 | ||
| Fig. 1 Single-crystal X-ray structure of [1o-H]BF4, displaying both (a) anti-parallel and (b) parallel conformations of the axle within the same crystalline domain. Color key: C dark grey, N blue, S yellow, B olive green, F bright green, H white; covalent bonds = grey, H-bonds = grey dashed. | ||
[2]Pseudorotaxane formation
The formation of [2]pseudorotaxanes was probed by 1H NMR spectroscopy in CD2Cl2 at 298 K. In this non-competitive solvent at a concentration of 20 mM, the association was shown to be complete by mixing equimolar amounts of axle and crown ether. As an example, Fig. 2a–c shows 1H NMR spectra of DB24C8, the axle [1o-H]+, and the [2]pseudorotaxane [1o-H⊂DB24C8]+. Two different species, formed in unequal amounts, are observed in the spectrum of the [2]pseudorotaxane, evidenced by the presence of two NH peaks and two CH3 peaks (Fig. 2b). We propose that threading of the axle to form a [2]pseudorotaxane decreases its conformational freedom, which slows the interconversion between parallel and anti-parallel conformations of the bis(benzothienyl)imidazolium moiety to a rate observable on the NMR timescale.27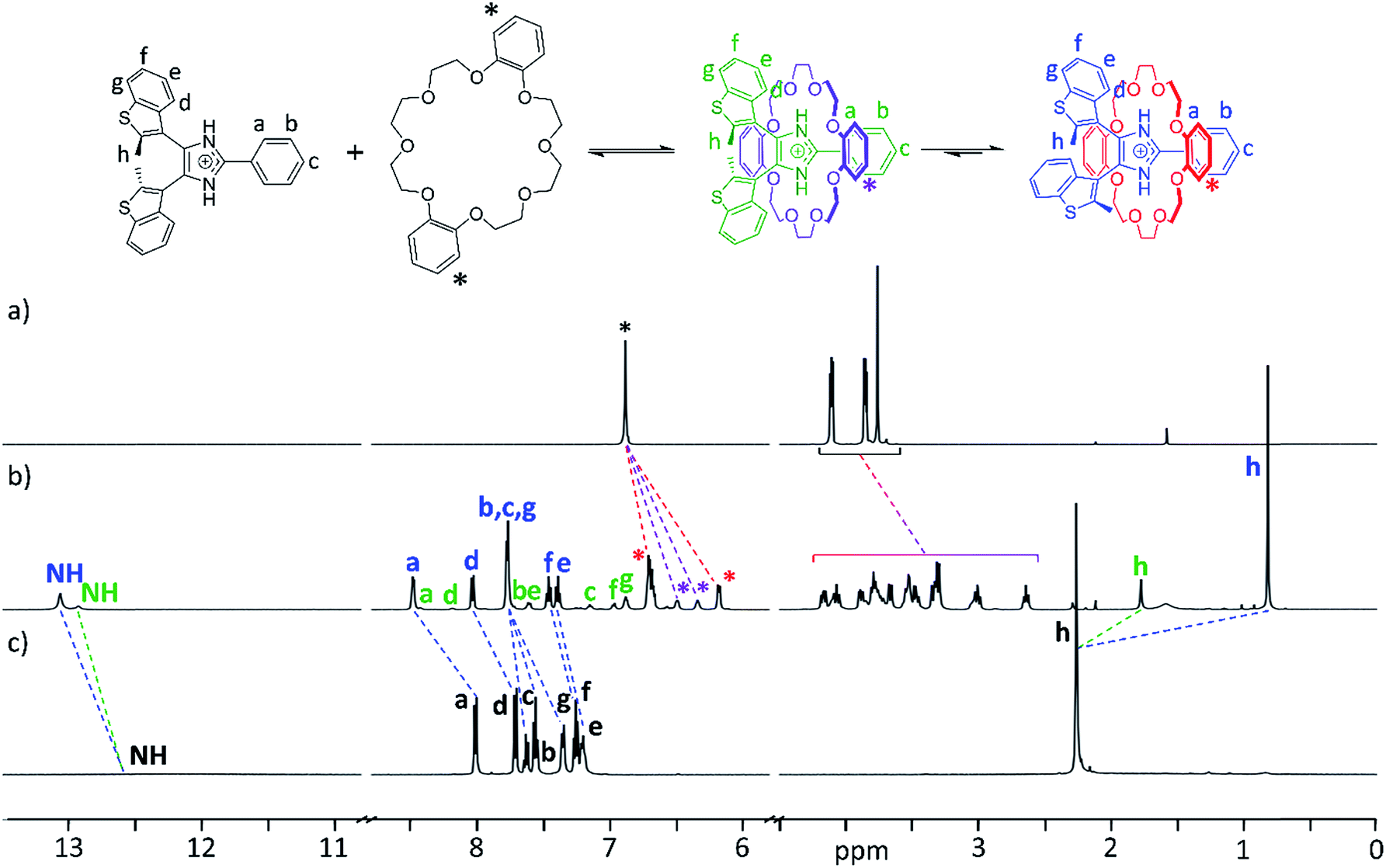 | ||
| Fig. 2 1H NMR spectra of (a) DB24C8, (b) [2]pseudorotaxane [1o-H⊂DB24C8]BF4, and (c) [1o-H]BF4. Corresponding peaks are matched by dashed lines; the axle peaks and the aromatic peaks of the macrocycle are labelled using colours matching the accompanying reaction scheme. | ||
To corroborate our hypothesis of a decreased conformational freedom of the threaded axle and assign each species to the appropriate set of NMR peaks, we recorded 2D 1H–1H COSY, EXSY, and NOESY NMR spectra of the [2]pseudorotaxane [1o-H⊂DB24C8]+ (ESI, Fig. S9–11, S21 and 22†). NOE cross-peak analysis (ESI, Fig. S11†) supported formation of the [2]pseudorotaxane and provided insightful information about the conformations adopted by the axle, allowing complete assignment of the peaks for the two species, and estimation of an 80:20 distribution of parallel versus anti-parallel [2]pseudorotaxane (see ESI†).
The slow interconversion on the NMR timescale between parallel and anti-parallel [2]pseudorotaxanes was studied by exchange spectroscopy (EXSY) experiments in CD2Cl2 at 298 K (ESI, Fig. S21 and 22†). The rate of conversion of anti-parallel to parallel [2]pseudorotaxane was determined to be higher than the reverse process at 298 K (kAP→P = 6.824 s−1; kP→AP = 0.886 s−1), in agreement with the uneven distribution of the two species in favour of the parallel conformer.
The chemical shift variations between naked guest and [2]pseudorotaxane are consistent with an interpenetrated host–guest interaction: the de-shielding of NH, a and d protons for both species is indicative of NH⋯O hydrogen bonding and CH⋯O interactions between axle and crown ether, while shielding of h methyl protons is consistent with π-stacking interactions between the phenyl rings of DB24C8 and the axle. Interestingly, the parallel [2]pseudorotaxane displays a larger shielding for the methyl groups protons (−1.44 ppm) when compared to the anti-parallel version (−0.48 ppm). This is due to the additional ring current arising from the overlap of the methyl group of one benzothienyl unit with the π-cloud of the adjacent benzo unit in the parallel conformation of the [2]pseudorotaxane; only one averaged peak is observed for both methyl groups since the interconversion between the two equivalent parallel conformations is fast in the NMR experiment timescale. Analogous observations can be made for the NMR studies of any other combination of axle and crown ether, although the absence of aromatic rings for 24C8 produces less pronounced shielding and a slight de-shielding, respectively, on the methyl protons of parallel and anti-parallel [2]pseudorotaxanes (ESI, Fig. S12–14†).
The association constants (Ka) between axles [1o-H]+, [2o-H]+, and [3o-H]+, and rings 24C8 and DB24C8 were determined in CH2Cl2 by UV-Vis titrations, and in CD3CN by 1H NMR measurements (single-point method). A 1:1 binding stoichiometry was determined for all combinations of axle and ring as evidenced by integration of complexed and un-complexed peaks in the 1H NMR spectra (ESI, Fig. S15–17†). Due to slow exchange between complexed and un-complexed components on the NMR timescale, association constant (Ka) values were determined using the single-point method from solutions containing equimolar amounts of axle and ring at known concentrations.28 The higher association constants (Ka) in a non-competitive solvent such as CH2Cl2 were determined by fitting the experimental UV-Vis titration data according to a 1:1 binding model.28 The results of these association studies are summarized in Table 1. The estimated association constants are comparable to previously reported ones for 2,4,5-triphenylimidazolium axles. Predictably, the presence of an electron-withdrawing ester group increases the association constant between [2o-H]+ and crown ethers, due to strengthened hydrogen-bonding and ion–dipole interactions between the electron-poor axle and the electron-rich macrocycle. Conversely, the presence of an electron-donating methoxy group on [3o-H]+ decreases the Ka due to weakening of these same interactions17d (ESI, Fig. S34–42†).
| CD3CNa,d | CH2Cl2b,d | |||
|---|---|---|---|---|
| DB24C8 | 24C8 | DB24C8 | 24C8 | |
|
a
K
a values from 1H NMR measurements (single-point) in CD3CN (0.02 M, 298 K).
b
K
a values from UV-Vis titrations in CH2Cl2 (2.6–6.0 × 10−5 M, 298 K) assuming a 1:1 host–guest association model.
c Not available, see ESI for details.
d Errors estimated to be <5%.
|
||||
| [1o-H]+ | 300 M−1 | 380 M−1 | 2.9 × 105 M−1 | 2.3 × 106 M−1 |
| [2o-H]+ | 800 M−1 | 910 M−1 | —c | —c |
| [3o-H]+ | 200 M−1 | 290 M−1 | 2.5 × 105 M−1 | 9.7 × 105 M−1 |
The formation of [2]pseudorotaxanes between bis(benzothienyl)imidazolium axles and crown ethers was further confirmed by the crystal structure of [1o-H⊂DB24C8]BF4·Et2O (Fig. 3). Again, both anti-parallel (Fig. 3a) and parallel (Fig. 3b) conformations of the [2]pseudorotaxane are displayed within the same crystal lattice.
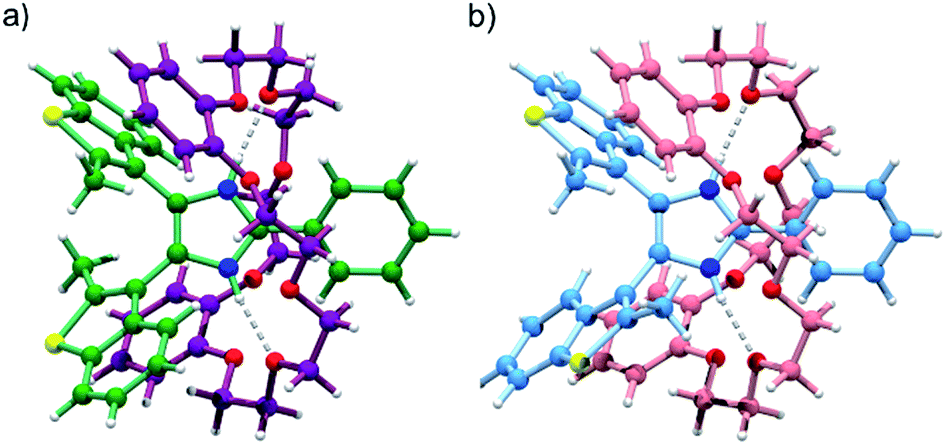 | ||
| Fig. 3 Single-crystal X-ray structure of [1o-H⊂DB24C8]BF4·Et2O, displaying both (a) anti-parallel and (b) parallel conformations of the [2]pseudorotaxane within the same crystalline domain. Color key: C (axle, anti-parallel) green, C (ring, anti-parallel) violet, C (axle, parallel) azure, C (ring, parallel) pink, N blue, O red, S yellow, H white; axle bonds = sea green (anti-parallel) and light blue (parallel), ring bonds = plum (anti-parallel) and light pink (parallel), H-bonds = grey dashed. For the sake of clarity, BF4− and Et2O have been omitted. | ||
However, despite the preference for a parallel conformation in both solution and the solid-state for [1o-H]BF4, and in solution for [1o-H⊂DB24C8]+, this trend is reversed in the crystal structure for [1o-H⊂DB24C8]+ where a 7:93 distribution of parallel/anti-parallel conformers was observed. The imidazolium NH groups of the threaded axle display charge-assisted hydrogen bonding interactions with oxygen atoms of the crown ether ring (N–H⋯O 2.83 Å, 170.5°; and 2.84 Å, 174.0°), while CH–π interactions between methyl groups at the 2-position of the benzothienyl groups and the catechol rings of the macrocycle in the major anti-parallel structure (CH3⋯C6-centroid 3.43 Å, 80.7°; and 3.38 Å, 86.1°), and partial π–π stacking between the benzo portion of one benzothienyl group and one catechol ring of the macrocycle in the minor parallel structure (C6-centroid⋯C6-centroid 4.01 Å, 15.9°), give rise to a C-shaped conformation (“supramolecular hug”) for the DB24C8 wheel.17a,d
Photochromism of the axles
The photochromism of axles [1o-H]+, [2o-H]+, and [3o-H]+ was studied in solution by UV-Vis (CH2Cl2) and 1H NMR (CD2Cl2) spectroscopy. The absorption and emission spectra of the axles (Fig. S33†) and the photophysical data (Table S7†) are summarized in the ESI.† All the compounds show a strong luminescence, with quantum yields of 10–40% and exhibit low fatigue resistance; 1H NMR experiments show that all the axles display less than 30% recovery after only five cycles of alternated UV (313 nm) and visible (580 nm) irradiation (ESI, Chart S1†). Moreover, they appear to degrade after prolonged irradiation, with formation of a photoproduct, which absorbs in the visible region and is still present after the cycloreversion reactions. For this reason, photostationary states were not reached, but all the compounds were irradiated up to the maximum degree of photoconversion (PC), and irradiation stopped before significant photodegradation occurred (see ESI†).The closed isomers [1c-H]+, [2c-H]+ and [3c-H]+ revert very quickly to the corresponding open isomers when irradiated with visible light and undergo relatively fast thermal cycloreversion reactions at room temperature, which prevented full characterization by NMR spectroscopy. The photoisomerized product [1c-H]BF4 could however be detected by irradiating, freezing, and subsequently thawing the sample immediately prior to recording an 1H NMR spectrum (Fig. S23†); the new CH3 and NH peaks of the closed isomer [1c-H]+ appear more shielded (−0.17 and −1.73 ppm, respectively) compared to the corresponding open isomer [1o-H]+. Determination of the quantum yields of the cyclization reactions was not straightforward and was affected by significant error due to the photostability issues and fast thermal cycloreversion reactions. However, percentage conversions could be estimated from the residual luminescence intensity at the emission wavelength of the open isomer (ESI, Table S7†).
Compounds [1o-H]+ and [3o-H]+ can be converted to their closed forms [1c-H]+ and [3c-H]+ upon irradiation at 313 nm; the photoisomerization reactions are accompanied by the appearance of the typical bands in the visible region. [1c-H]+ and [3c-H]+ revert to the open forms in tens of minutes, but, surprisingly, in both cases the absorbance changes linearly with time, i.e. the cycloreversion reactions follow a zero-order kinetic rate law (Fig. 4a and ESI Fig. S44†).
 | ||
| Fig. 4 (a) Absorption spectra of a 3.2 × 10−5 M solution of [1o-H]OTf after irradiation at 313 nm; inset shows the plot of absorption changes at 580 nm in time (white circles) and the fitting of the experimental data with a zero-order kinetic model (solid line). (b) Absorption spectra of a 1:1 mixture of [2o-H]OTf and 24C8 (4.5 × 10−5 M) after irradiation at 340 nm; inset shows the plot of absorption changes at 580 nm in time (white circles) and the fitting of the experimental data with a first order kinetic model (solid line). | ||
This behaviour is quite uncommon and cannot be ascribed to a simple unimolecular process. For example, it is known that, for related diarylethene compounds, a catalytic amount of oxidant can accelerate the thermal cycloreversion and induce zero-order kinetics by triggering a chain reaction.29 To exclude effects related to the presence of impurities or humidity in the solvent, control experiments in different batches of CH2Cl2, including those dried on molecular sieves, were undertaken, but this had no effects on the kinetic traces. We therefore hypothesize that the photoproduct formed upon irradiation of the protonated compounds (vide supra) could react with the latter and activate a chain reaction that ultimately affords the open forms.
Compound [2o-H]+ was irradiated in the UV, but the closed form could not be accumulated, due to the very high thermal cycloreversion reaction rate. Moreover, the initial spectrum of [2o-H]+ could never be completely recovered. It is likely that, in this case, the quantum yield of the photoreaction is lower as corroborated by the larger emission quantum yield, and that photodegradation occurs upon irradiation. The percentage of photoconversion could again be estimated by evaluating the extent of emission quenching at the end of the irradiation experiments, however it was not possible to record the absorption spectrum at this degree of photoconversion, because the thermal reaction was too fast (ESI).
Photochromism of [2]pseudorotaxanes
Following the studies performed on the naked axles, the photochromism of the six [2]pseudorotaxanes [1o-H⊂24C8]+, [2o-H⊂24C8]+, [3o-H⊂24C8]+, [1o-H⊂DB24C8]+, [2o-H⊂DB24C8]+, and [3o-H⊂DB24C8]+ was studied. Mixtures of axles and crown ethers were irradiated with UV light to assess how [2]pseudorotaxane formation might affect photoisomerization of the terarylenes and the thermal stability of the closed isomers.UV-Vis studies were performed on 1:1 mixtures of axles and crown ethers, which were irradiated with UV light followed by thermal cycloreversion at room temperature. The same precautions as for the free axles were taken when irradiating the pseudorotaxane solutions; i.e. irradiation was maintained up to the maximum conversion and stopped before any significant photodegradation occurred. Impressively, compound [2o-H]+ shows significant improvement in photochemical properties upon complexation.
Unlike the free axle, both [2o-H⊂24C8]+ and [2o-H⊂DB24C8]+ could be photoisomerized to the respective closed forms and the thermal cycloreversion reactions were compatible with a first-order mechanism (Fig. 4b and ESI, Fig. S47†). The UV-Vis spectroscopic studies for the [2]pseudorotaxanes are summarized in ESI Table S7.† Unfortunately, some of the data are difficult to interpret because, at the low concentrations utilized, the thermal cycloreversion reactions of the [2]pseudorotaxanes depended on the absolute concentration and/or on the relative ratio of the molecular components. For a thorough discussion of the UV-Vis studies, see the ESI.†
At higher concentrations typical for 1H NMR experiments a greater degree of association between axles and crown ethers occurs. For example, for a 20 mM 1:1 mixture of axle and macrocycle in CH2Cl2 it can be predicted from the Ka values that the [2]pseudorotaxane is the dominant species (>99%) present in solution and any effect on the photochromism attributable to [2]pseudorotaxane formation can be ultimately evidenced and amplified. Furthermore, under these experimental conditions the undesired effects of by-products could be possibly reduced.
The photoisomerization of [2]pseudorotaxanes [1–3c-H⊂(CE)]BF4 (CE = crown ether) was studied by 1H NMR experiments where 0.02 M samples of axle and excess crown ether (3 equivalents) in CD2Cl2 were irradiated for 15 min at 254 nm. These conditions were found to be ideal to achieve maximum photoconversion and minimize any photodecomposition. Interestingly, the free axles would decompose in few minutes if irradiated with such short wavelength under the same experimental conditions but the corresponding [2]pseudorotaxanes would not reach the maximum photoconversion if irradiated with longer wavelengths. The fatigue resistance of [2]pseudorotaxanes was evaluated by alternating UV and visible irradiation and collecting a 1H NMR spectrum after each irradiation event and the thermal cycloreversion rates were probed by recording 1H NMR spectra at increasing time intervals, while keeping the samples in the dark between measurements (ESI).
1H NMR photoisomerization studies of [2]pseudorotaxanes are summarized in Table 2. [2]Pseudorotaxanes formed with axles [1o-H]+ and [3o-H]+ exhibit photoconversion percentages above 50%, whereas slightly lower conversions are obtained in both cases involving [2o-H]+. All the [2]pseudorotaxanes formed with the closed isomers of the axles reverted quickly (within minutes) to the corresponding open isomers when irradiated with visible light. Conversely, when the [2]pseudorotaxanes closed isomers are kept in the dark at room temperature, they exhibit thermal cycloreversion with first-order kinetics and much slower rates, as evidenced by half-lives ranging from few hours to more than two days (Table 2). This observation is in stark contrast to the fast zero-order thermal cycloreversion displayed by the free axles, which prevented any kind of NMR characterization of their closed forms.
| [2]Pseudorotaxanea | % PCb | kc (s−1) | Half-lifed (h) |
|---|---|---|---|
|
a CD2Cl2, 1:3 axle/crown ratio, 0.02 M, 500 MHz, 298 K.
b Photoconversion percentages of [2]pseudorotaxanes reached after irradiation with UV light (254 nm) for 15 min.
c First order cycloreversion rates.
d Half-life for the closed form.
|
|||
| [1o-H⊂(DB24C8)]+ | 52 | 7.5 × 10−6 | 25.8 |
| [1o-H⊂(24C8)]+ | 52 | 3.4 × 10−6 | 56.7 |
| [2o-H⊂(DB24C8)]+ | 43 | 6.0 × 10−5 | 3.2 |
| [2o-H⊂(24C8)]+ | 38 | 1.8 × 10−5 | 10.7 |
| [3o-H⊂(DB24C8)]+ | 51 | 5.8 × 10−6 | 33.3 |
| [3o-H(⊂24C8)]+ | 56 | 3.8 × 10−6 | 50.1 |
The increased half-life of the closed isomers of the axles when threaded into crown ethers is accompanied by an overall enhancement in the photostability of the [2]pseudorotaxanes when compared to the corresponding free axles. As mentioned above, all the 1H NMR irradiation experiments involving [2]pseudorotaxanes have been performed under experimental conditions (254 nm UV light) that would cause fast photodegradation of the free axles. This observation is corroborated by the improvement of the [2]pseudorotaxanes fatigue resistance (Fig. 5 and ESI Chart S3†): while the axles [1o-H]+, [2o-H]+, and [3o-H]+ display 30%, 27%, and 23% recovery of the open isomer, respectively, after only five cycles of alternated UV (313 nm) and visible irradiation, analogous experiments where 254 nm UV light is used yield recoveries twice as high after five irradiation cycles for [2]pseudorotaxanes [1o-H⊂24C8]+ (63%), [2o-H⊂24C8]+ (54%), and [3o-H⊂24C8]+ (59%). When 24C8 is replaced by DB24C8, only a slight fatigue resistance improvement is observed, although, once again, the use of higher energy UV light for [2]pseudorotaxanes must be emphasized.
 | ||
| Fig. 5 Fatigue resistance of free axle [1o-H]BF4 (●, dotted line) [2]pseudorotaxane [1o-H⊂(CE)]BF4 (▲, dashed line: DB24C8; ■, solid line: 24C8) evaluated by 1H NMR experiments (254 nm, CD2Cl2, 0.02 M, 500 MHz, 298 K). | ||
The longer lifetime of the closed isomers of the axles when threaded into crown ethers allowed full characterization of the photoisomerized [2]pseudorotaxanes by 1H NMR spectroscopy. Fig. 6 shows a comparison between the 1H NMR spectrum of a CD2Cl2 solution of the [2]pseudorotaxane [1o-H⊂DB24C8]+ with the same sample after the maximum photoconversion was reached. The newly formed species in the irradiated sample was thermally stable enough in the dark to allow for longer 2D NMR experiments (ESI Fig. S24–26†).
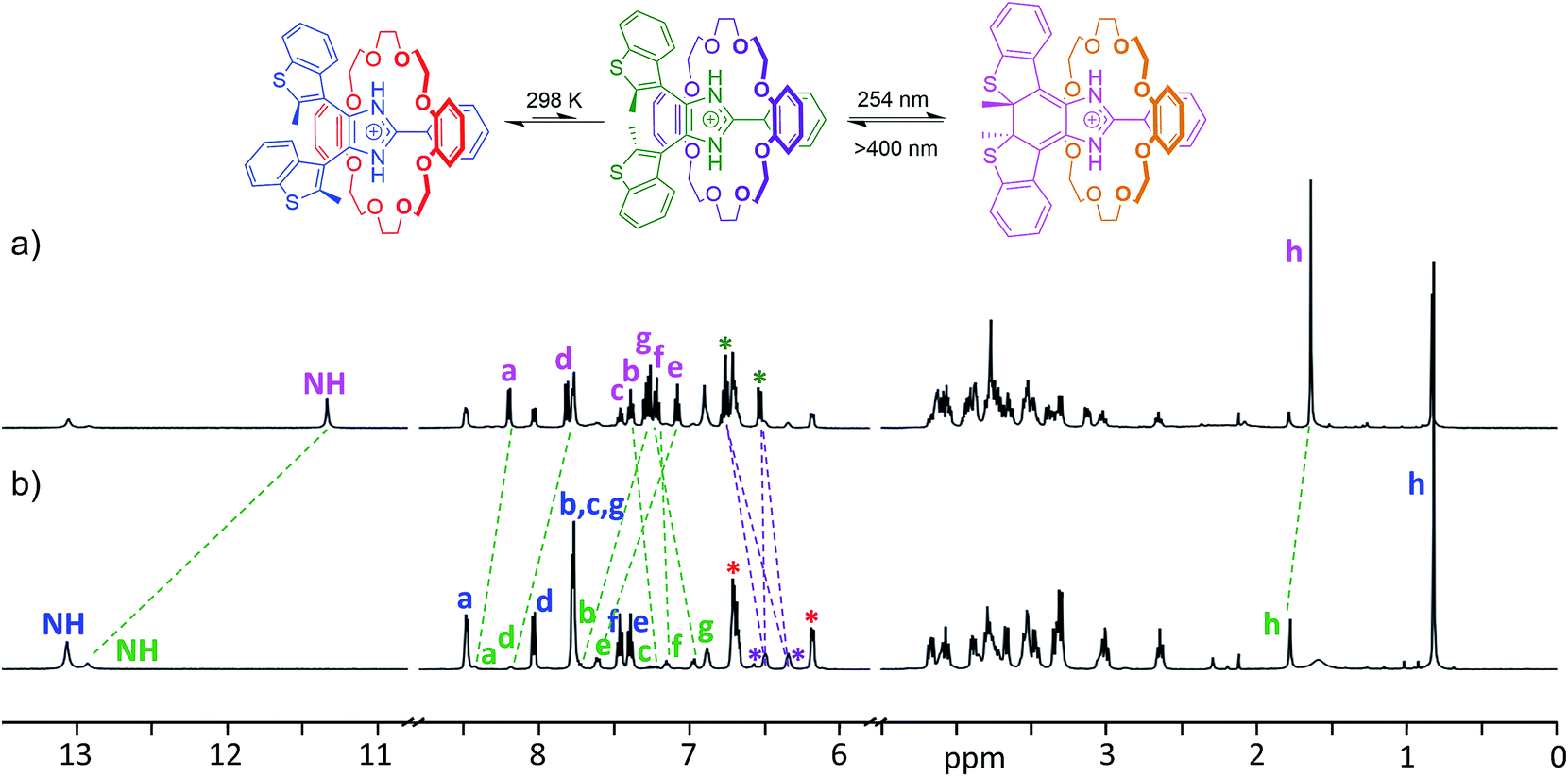 | ||
| Fig. 6 1H NMR spectra of (a) [1c-H⊂DB24C8]BF4 after reaching the photostationary state by irradiation with 254 nm light for 10 min, and (b) [1o-H⊂DB24C8]BF4 [2]pseudorotaxane (CD2Cl2, 500 MHz, 298 K). Corresponding peaks are matched by dashed lines; the axle peaks and the aromatic peaks of the macrocycle are labelled using colours matching the accompanying reaction scheme. | ||
In particular, 1H–1H NOESY of [1c-H⊂DB24C8]+ (ESI Fig. S26†) confirmed that the closed isomer generated by UV irradiation persists in its threaded form, i.e. the observed long-lived species is the closed isomer of the [2]pseudorotaxane. Although only the minor anti-parallel conformer of the [2]pseudorotaxane is photoactive,19b the conversion to the closed isomer at the photostationary state, calculated from the ratio of the integrals of closed and open isomers, reached 52%. Such a degree of photoconversion can only be achieved by way of a dynamic interconversion between parallel and anti-parallel conformers of the [2]pseudorotaxane, which restores the parallel/anti-parallel ratio to equilibrium once the photoactive conformer is depleted because of the photoisomerization reaction. As previously noted for the naked axle, the closed [2]pseudorotaxane isomer [1c-H⊂DB24C8]+ exhibits shielded CH3 and NH peaks (−0.14 and −1.58 ppm, respectively) when compared to the corresponding anti-parallel open isomer. Analogous observations can be made for the NMR studies of any other combination of axle and crown ether (ESI, Fig. S27–32†).
Proposed mechanism
Experimental evidence strongly supports a threading-gated type of photochromism, where the thermal cycloreversion kinetics and the fatigue resistance of axles [1o-H]+, [2o-H]+ and [3o-H]+ can be significantly influenced by threading inside crown ether rings to form [2]pseudorotaxanes.We propose the stepwise cycloreversion mechanism, depicted in Scheme 3a, where, upon UV irradiation, the bis(benzothienyl)imidazolium open isomer (Scheme 3a, left-hand side) undergoes a typical photochemical electrocyclization to yield the corresponding imidazolinium closed isomer (Scheme 3a, middle), which can subsequently undergo a thermal cycloreversion, and/or follow an alternative stepwise ring-opening pathway triggered by the tautomerization of the axle. We can reasonably infer that this newly formed imidazole tautomer (Scheme 3a, right-hand side), accessible by intermolecular proton transfer, exhibits higher stability due to an overall increase in aromaticity. Once formed, the tautomer can quickly revert to the parent imidazolium open isomer through a further proton transfer and heterolytic bond cleavage. In this context, it is worth mentioning a recent report by Hecht et al. describing the acid-catalysed thermal cycloreversion of diarylethenes.30
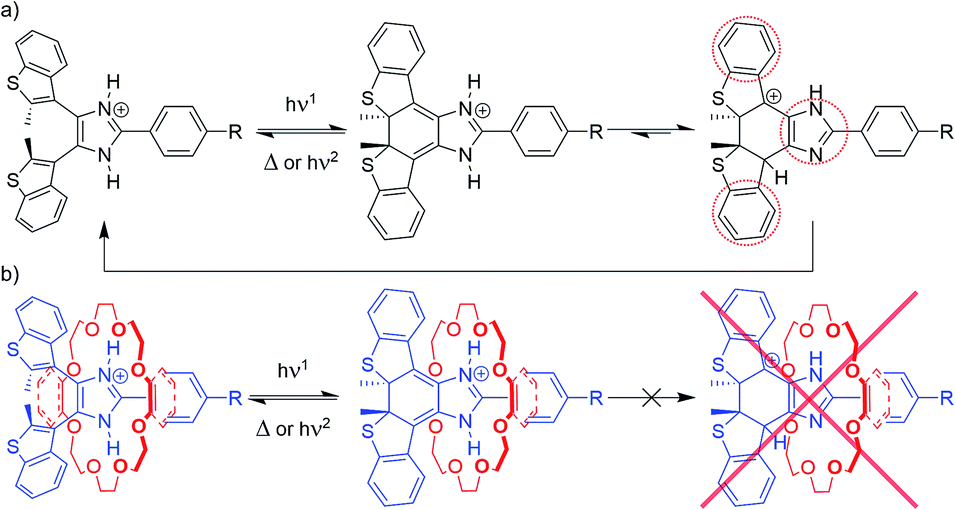 | ||
| Scheme 3 Proposed photoisomerization and cycloreversion pathways for (a) free axles [1–3-H]+, and (b) [2]pseudorotaxanes [1–3-H⊂(CE)]+. | ||
The proposed stepwise thermal cycloreversion pathway presumably exhibits an overall lower kinetic barrier in comparison with the typical electrocyclic thermal cycloreversion (conrotatory, hence forbidden by the Woodward–Hoffmann rules). This supposition agrees with the fast cycloreversion characterizing all the free axles, and such a cycloreversion mechanism might explain the low fatigue resistance of these systems, since the charged intermediates could generate undesirable photoproducts.
This proposed cycloreversion mechanism would also explain why the photochromic properties of the axles involved in this study undergo such significant changes when threaded through crown ethers (Scheme 3b). The strong charge-assisted hydrogen bonds between the NH groups of the axle and the oxygen atoms of the crown ether keep the NH protons in place, in both the open and closed isomers (Scheme 3b, left-hand side and middle), effectively preventing tautomerization from occurring – the tautomerization to the imidazole form of the closed isomer would destroy all the favourable intermolecular interactions which hold the [2]pseudorotaxane together (Scheme 3b, right-hand side) – making this pathway unfavourable. This leaves the closed isomer of the [2]pseudorotaxane with the only option of reverting to the open isomer through the usual electrocyclic thermal cycloreversion, which is kinetically hampered due to its higher activation barrier, which, in turn, explains the slower first-order thermal cycloreversion kinetics and, especially in the cases where 24C8 represents the ring component, the improved fatigue resistance of the [2]pseudorotaxanes when compared to their corresponding free axles.
Further investigations are underway in our laboratories to unambiguously prove the occurrence of this proposed mechanism. Nevertheless, we are confident that this is a plausible representation of the actual mechanism behind this novel example of threading gated-photochromism.
Conclusions
We have reported the design, synthesis, and photochemical characterization of a series of novel photochromic bis(benzothienyl)imidazolium axles, [1o-H]+, [2o-H]+ and [3o-H]+ and shown that their association with the 24 membered crown ethers 24C8 and DB24C8 to form [2]pseudorotaxanes triggers significant changes in their photochromic properties. All the [2]pseudorotaxane closed isomers exhibited longer lifetimes and improved fatigue resistance when compared to the corresponding free axles, and they reverted quickly to the corresponding open isomers only when irradiated with visible light. We have proposed a tautomerization-induced, stepwise thermal cycloreversion mechanism involving a heterolytic bond cleavage to the open isomer to rationalize the poor photochromic properties of the free axles. This proposed mechanism also explains why the thermal cycloreversion kinetics and the fatigue resistance of axles are significantly influenced by threading into crown ethers. In this context, we suggested that the strong supramolecular interactions involved in the [2]pseudorotaxanes prevents the tautomerization of the closed isomer of the axles, leaving the slower electrocyclic thermal cycloreversion as the only possible ring-opening pathway for [2]pseudorotaxanes.This phenomenon, termed threading-gated photochromism, greatly expands the concept of gated photochromism, by exploiting supramolecular interactions to induce significant changes in the photochromic properties in one of the components involved in an interpenetrated molecular system. We believe the scope of this work could be further extended to [2]rotaxanes and other mechanically interlocked systems, where an orthogonal stimulus could be employed to prompt the reciprocal motion (e.g. shuttling or rotation) of interlocked components, and, in response, trigger a change in the photochromic properties of the system, ultimately paving the way towards elegant multi-stimuli photoswitches.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Natural Sciences and Engineering Research Council of Canada (Discovery Grant no. 101694 to SJL) and the European Research Council (Grant no. 692981 to AC). GB and SJL acknowledge M. Revington and J. Auld for their technical assistance with NMR spectroscopy and mass spectrometry experiments respectively.Notes and references
- (a) Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH, Weinheim, 2011 Search PubMed; (b) J. E. M. Lewis, M. Galli and S. M. Goldup, Chem. Commun., 2017, 53, 298–312 RSC; (c) C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, John Wiley & Sons, Inc., Hoboken, NJ, 2016 CrossRef.
- (a) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS; (b) C. O. Dietrich-Buchecker, M. C. Jimenez-Molero, V. Sartor and J.-P. Sauvage, Pure Appl. Chem., 2003, 75, 1383–1393 CAS; (c) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2006, 46, 72–191 CrossRef PubMed; (d) Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, ed. V. Balzani, A. Credi and M. Venturi, Wiley-VCH, Weinheim, 2008 Search PubMed; (e) From Non-Covalent Assemblies to Molecular Machines, ed. J.-P. Sauvage and P. Gaspard, Wiley-VCH, Weinheim, 2010 Search PubMed; (f) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC; (g) A. Credi, S. Silvi and M. Venturi, Top. Curr. Chem., 2014, 354, 1–34 CrossRef CAS PubMed; (h) E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC; (i) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (j) E. R. Kay and D. A. Leigh, Angew. Chem., Int. Ed., 2015, 54, 10080–10088 CrossRef CAS PubMed; (k) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed; (l) S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- (a) S. Saha and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 77–92 RSC; (b) V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC; (c) A. Credi, M. Venturi and V. Balzani, ChemPhysChem, 2010, 11, 3398–3403 CrossRef CAS PubMed; (d) T. Muraoka and K. J. Kinbara, J. Photochem. Photobiol., C, 2012, 13, 136–147 CrossRef CAS; (e) T. Avellini, M. Baroncini, G. Ragazzon, S. Silvi, M. Venturi and A. Credi, Isr. J. Chem., 2014, 54, 553–567 CrossRef CAS; (f) M. Kathan and S. Hecht, Chem. Soc. Rev., 2017, 46, 5536–5550 RSC.
- H. Bouas-Laurent and H. Dürr, Pure Appl. Chem., 2001, 73, 639–665 CAS.
- (a) V. Balzani, A. Credi, F. Marchioni and J. F. Stoddart, Chem. Commun., 2001, 37, 1860–1861 RSC; (b) Y. Tokunaga, K. Akasaka, K. Hisada, Y. Shimomura and S. Kakuchi, Chem. Commun., 2003, 39, 2250–2251 RSC; (c) M. Baroncini, S. Silvi, M. Venturi and A. Credi, Chem.–Eur. J., 2010, 16, 11580–11587 CrossRef CAS PubMed; (d) L. Zhu, D. Zhang, D. Qu, Q. Wang, X. Ma and H. Tian, Chem. Commun., 2010, 46, 2587–2589 RSC; (e) M. Baroncini, S. Silvi, M. Venturi and A. Credi, Angew. Chem., Int. Ed., 2012, 51, 4223–4226 CrossRef CAS PubMed; (f) M. Baroncini, C. Gao, V. Carboni, A. Credi, E. Previtera, M. Semeraro, M. Venturi and S. Silvi, Chem.–Eur. J., 2014, 20, 10737–10744 CrossRef CAS PubMed; (g) D. Hernández-Melo and J. Tiburcio, Chem. Commun., 2015, 51, 17564–17567 RSC.
- (a) I. A. Banerjee, L. Yu and H. Matsui, J. Am. Chem. Soc., 2003, 125, 9542–9543 CrossRef CAS PubMed; (b) S. Dong, L. Gao, J. Li, D. Xu, Q. Zhou, Polym. Chem., 2013, 4, 3968–3973 Search PubMed; (c) F. B. Schwarz, T. Heinrich, J. O. Kaufmann, A. Lippitz, R. Puttreddy, K. Rissanen, W. E. S. Unger and C. A. Schalley, Chem.–Eur. J., 2016, 22, 14383–14389 CrossRef CAS PubMed.
- (a) G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70–75 CrossRef CAS PubMed; (b) L. Casimiro, J. Groppi, M. Baroncini, M. La Rosa, A. Credi and S. Silvi, Photochem. Photobiol. Sci., 2018, 17, 734–740 RSC.
- (a) H. Murakami, A. Kawabuchi, K. Kotoo, M. Kunitake and N. Nakashima, J. Am. Chem. Soc., 1997, 119, 7605–7606 CrossRef CAS; (b) C. A. Stanier, S. J. Alderman, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2002, 41, 1769–1772 CrossRef; (c) F. G. Gatti, S. León, J. K. Y. Wong, G. Bottari, A. Altieri, M. A. F. Morales, S. J. Teat, C. Frochot, D. A. Leigh, A. M. Brouwer and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10–14 CrossRef CAS PubMed; (d) A. Altieri, G. Bottari, F. Dehez, D. A. Leigh, J. K. Y. Wong and F. Zerbetto, Angew. Chem., Int. Ed., 2003, 42, 2296–2300 CrossRef CAS PubMed; (e) E. M. Pérez, D. T. F. Dryden, D. A. Leigh, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2004, 126, 12210–12211 CrossRef PubMed; (f) V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS PubMed; (g) P. Raiteri, G. Bussi, C. S. Cucinotta, A. Credi, J. F. Stoddart and M. Parrinello, Angew. Chem., Int. Ed., 2008, 47, 3536–3539 CrossRef CAS PubMed; (h) H. Li, A. C. Fahrenbach, A. Coskun, Z. Zhu, G. Barin, Y. L. Zhao, Y. Y. Botros, J.-P. Sauvage and J. F. Stoddart, Angew. Chem., Int. Ed., 2011, 50, 6782–6788 CrossRef CAS PubMed; (i) T. Avellini, H. Li, A. Coskun, G. Barin, A. Trabolsi, A. N. Basuray, S. K. Dey, A. Credi, S. Silvi, J. F. Stoddart and M. Venturi, Angew. Chem., Int. Ed., 2012, 51, 1611–1615 CrossRef CAS PubMed; (j) D. A. Leigh, V. Marcos, T. Nalbantoglu, I. J Vitorica-Yrezabal, F. T. Yasar and X. Zhu, J. Am. Chem. Soc., 2017, 139, 7104–7109 CrossRef CAS PubMed.
- (a) J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef PubMed; (b) C. Jia, H. Li, J. Jiang, J. Wang, H. Chen, D. Cao, J. F. Stoddart and X. Guo, Adv. Mater., 2013, 25, 6752–6759 CrossRef CAS PubMed; (c) F. B. Schwarz, T. Heinrich, A. Lippitz, W. E. S. Unger and C. A. Schalley, Chem. Commun., 2016, 52, 14458–14461 RSC.
- A. Martinez-Cuezva, A. Saura-Sanmartin, T. Nicolas-Garcia, C. Navarro, R.-A. Orenes, M. Alajarin and J. Berna, Chem. Sci., 2017, 8, 3775–3780 RSC.
- A. G. Cheetham, M. G. Hutchings, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2006, 45, 1596–1599 CrossRef CAS PubMed.
- (a) H. S. Tang, N. Zhu and V. W. W. Yam, Organometallics, 2007, 2, 22–25 CrossRef; (b) S. Wang, X. Li, W. Zhao, X. Chen, J. Zhang, H. Ågren, Q. Zou, L. Zhu and W. J. Chen, J. Mater. Chem. C, 2017, 5, 282–289 RSC.
- (a) V. Lemieux and N. R. Branda, Org. Lett., 2005, 7, 2969–2972 CrossRef CAS PubMed; (b) J. Kühni and P. Belser, Org. Lett., 2007, 9, 1915–1918 CrossRef PubMed.
- (a) M. S. Vollmer, T. D. Clark, C. Steinem and M. R. Ghadiri, Angew. Chem., Int. Ed., 1999, 38, 1598–1601 CrossRef CAS; (b) M. Yamamura, Y. Okazaki and T. Nabeshima, Chem. Commun., 2012, 48, 5724–5726 RSC.
- (a) P. Bortolus and S. J. Monti, J. Phys. Chem., 1987, 91, 5046–5050 CrossRef CAS; (b) A. M. Sanchez and R. H. de Rossi, J. Org. Chem., 1996, 61, 3446–3449 CrossRef CAS; (c) M. Takeshita, N. Kato, S. Kawauchi, T. Imase, J. Watanabe and M. J. Irie, J. Org. Chem., 1998, 63, 9306–9313 CrossRef CAS; (d) E. Arunkumar, C. C. Forbes and B. D. Smith, Eur. J. Org. Chem., 2005, 19, 4051–4059 CrossRef; (e) M. A. Petersen, B. Rasmussen, N. N. Andersen, S. P. A. Sauer, M. B. Nielsen, S. R. Beeren and M. Pittelkow, Chem.–Eur. J., 2017, 23, 17010–17016 CrossRef CAS PubMed.
- M. Lohse, K. Nowosinski, N. L. Traulsen, A. J. Achazi, L. K. S. von Krbek, B. Paulus, C. A. Schalley and S. Hecht, Chem. Commun., 2015, 51, 9777–9780 RSC.
- (a) N. Noujeim, K. Zhu, V. N. Vukotic and S. J. Loeb, Org. Lett., 2012, 14, 2484–2487 CrossRef CAS PubMed; (b) K. Zhu, V. N. Vukotic and S. J. Loeb, Angew. Chem., Int. Ed., 2012, 51, 2168–2172 CrossRef CAS PubMed; (c) K. Zhu, V. N. Vukotic, N. Noujeim and S. J. Loeb, Chem. Sci., 2012, 3, 3265–3271 RSC; (d) N. Farahani, K. Zhu, N. Noujeim and S. J. Loeb, Org. Biomol. Chem., 2014, 12, 4824–4827 RSC; (e) K. Zhu, V. N. Vukotic and S. J. Loeb, Chem.–Asian J., 2016, 11, 3258–3266 CrossRef CAS PubMed; (f) G. Baggi and S. J. Loeb, Angew. Chem., Int. Ed., 2016, 55, 12533–12537 CrossRef CAS PubMed; (g) N. Farahani, K. Zhu and S. J. Loeb, ChemPhysChem, 2016, 17, 1875–1880 CrossRef CAS PubMed; (h) V. N. Vukotic, K. Zhu, G. Baggi and S. J. Loeb, Angew. Chem., Int. Ed., 2017, 56, 6136–6141 CrossRef CAS PubMed; (i) K. Zhu, G. Baggi, V. N. Vukotic and S. J. Loeb, Chem. Sci., 2017, 8, 3898–3904 RSC; (j) G. Baggi and S. J. Loeb, Chem.–Eur. J., 2017, 23, 14163–14166 CrossRef CAS PubMed; (k) G. Gholami, K. Zhu, G. Baggi, E. Schott, X. Zarate and S. J. Loeb, Chem. Sci., 2017, 8, 7718–7723 RSC; (l) K. Zhu, G. Baggi and S. J. Loeb, Nat. Chem., 2018, 10, 625–630 CrossRef CAS PubMed.
- (a) V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456–460 CrossRef CAS PubMed; (b) K. Zhu, V. N. Vukotic, C. A. O'Keefe, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2014, 136, 7403–7409 CrossRef CAS PubMed; (c) K. Zhu and S. J. Loeb, Top. Curr. Chem., 2014, 354, 213–251 CrossRef CAS PubMed; (d) K. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514–519 CrossRef CAS PubMed; (e) V. N. Vukotic, C. A. O'Keefe, K. Zhu, K. J. Harris, C. To, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2015, 137, 9643–9651 CrossRef CAS PubMed; (f) N. Farahani, K. Zhu, C. A. O'Keefe, R. W. Schurko and S. J. Loeb, ChemPlusChem, 2016, 81, 836–841 CrossRef CAS; (g) G. Gholami, K. Zhu, J. S. Ward, P. E. Kruger and S. J. Loeb, Eur. J. Inorg. Chem., 2016, 27, 4524–4529 CrossRef; (h) G. Gholami, G. Baggi, K. Zhu and S. J. Loeb, Dalton Trans., 2017, 46, 2462–2470 RSC.
- (a) H. Tian and S. Wang, Chem. Commun., 2007, 8, 781–792 RSC; (b) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- (a) J. Liu, Y. Xu, X. Li and H. Tian, Dyes Pigm., 2008, 76, 294–298 CrossRef; (b) H. B. Cheng, H. Y. Zhang and Y. J. Liu, J. Am. Chem. Soc., 2013, 135, 10190–10193 CrossRef CAS PubMed.
- (a) H. Zhang, X.-X. Kou, Q. Zhang, D.-H. Qu and H. Tian, Org. Biomol. Chem., 2011, 9, 4051–4056 RSC; (b) F. Hu, J. Huang, M. Cao, Z. Chen, Y.-W. Yang, S. H. Liu and J. Yin, Org. Biomol. Chem., 2014, 12, 7712–7720 RSC; (c) Z. Li, X. Han, H. Chen, D. Wu, F. Hu, S. H. Liu and J. Yin, Org. Biomol. Chem., 2015, 13, 7313–7322 RSC.
- (a) M. M. Krayushkin, S. N. Ivanov, A. Y. Martynkin, B. V. Lichitsky, A. A. Dudinov and B. M. Uzhinov, Russ. Chem. Bull., 2001, 50, 116–121 CrossRef CAS; (b) M. M. Krayushkin, S. N. Ivanov, A. Y. Martynkin, B. V. Lichitsky, A. A. Dudinov, L. G. Vorontsova, Z. A. Starikova and B. M. Uzhinov, Russ. Chem. Bull., 2002, 51, 1731–1736 CrossRef CAS.
- M. Hanazawa, R. Sumiya, H. Yukio and I. Masahiro, Chem. Commun., 1992, 28, 206–207 RSC.
- S. Kawai, T. Nakashima, Y. Kutsunugi, H. Nakagawa, H. Nakano and T. Kawai, J. Mater. Chem., 2009, 19, 3606–3611 RSC.
- A. El Yahyaoui, G. Félix, A. Heynderickx, C. Moustrou and A. Samat, Tetrahedron, 2007, 63, 9482–9487 CrossRef CAS.
- Usually, access to bis(thienyl) 1,2-diketones is achieved by direct Friedel–Crafts acylation of thiophene derivatives with oxalyl chloride, or by a lengthy sequence of stepwise acylation and oxidation reactions; both synthetic pathways suffer from low overall yields and formation of multiple by-products. The synthetic pathway to the 1,2-diketone 7 adopted here exhibits higher overall yields (the first two steps are quantitative), and, potentially, a broader scope. The key step, adapted from a known procedure, takes advantage of the milder nature of organocopper compounds: in this specific case, the highly-reactive and unstable 3-lithium-2-methylbenzo[b]thiophene, which yielded only by-products when reacted directly with oxalyl chloride, was converted to the analogous organocopper intermediate, which, when submitted to similar conditions, yielded the desired diketone in good yield. For more details see: (a) B. M. Neilson and C. W. Bielawski, J. Am. Chem. Soc., 2012, 134, 12693–12699 CrossRef CAS PubMed; (b) J. T. Price and P. J. Ragogna, Chem.–Eur. J., 2013, 19, 8473–8477 CrossRef CAS PubMed; (c) J.-F. Chen, D.-P. Gong, J. Wen, H. Ma and D.-K. Cao, Chem. Sci., 2016, 7, 451–456 RSC; (d) L. I. Belen'kii, V. Z. Shirinyan, G. P. Gromova, A. V. Kolotaev, Y. A. Strelenko, S. N. Tandura, A. N. Shumskii and M. M. Krayushkin, Chem. Heterocycl. Compd., 2003, 39, 1570–1579 CrossRef; (e) S. N. Ivanov, B. V. Lichitskii, A. A. Dudinov, A. Y. Martynkin and M. M. Krayushkin, Chem. Heterocycl. Compd., 2001, 37, 85–90 CrossRef CAS; (f) F. A. Arroyave, C. A. Richard and J. R. Reynolds, Org. Lett., 2012, 14, 6138–6141 CrossRef CAS PubMed; (g) T. Asai, A. Takata, Y. Ushiogi, Y. Iinuma, A. Nagaki and J. Yoshida, Chem. Lett., 2011, 40, 393–395 CrossRef CAS.
- S. J. Loeb, J. Tiburcio, S. J. Vella and J. A. Wisner, Org. Biomol. Chem., 2006, 4, 667–680 RSC.
- (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; (b) D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- (a) T. Nakashima, Y. Kajiki, S. Fukumoto, M. Taguchi, S. Nagao, S. Hirota and T. Kawai, J. Am. Chem. Soc., 2012, 134, 19877–19883 CrossRef CAS PubMed; (b) J. P. Calupitan, T. Nakashima, Y. Hashimoto and T. Kawai, Chem.–Eur. J., 2016, 22, 10002–10008 CrossRef CAS PubMed.
- J. Gurke, M. Quick, N. P. Ernsting and S. Hecht, Chem. Commun., 2017, 53, 2150–2153 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details describing the synthesis and characterization of all new compounds including solution NMR assignments, VT and 2D NMR experiments, crystal structures details, kinetic and thermodynamic studies, UV-Vis and photochromism studies. CCDC 1877446–1877448. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00913b |
| This journal is © The Royal Society of Chemistry 2019 |