
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reduction chemistry of mixed-Lewis-base-stabilised chloroborylenes†
Merle
Arrowsmith
 a,
Julia I.
Schweizer
b,
Myron
Heinz
b,
Marcel
Härterich
a,
Ivo
Krummenacher
a,
Max C.
Holthausen
b and
Holger
Braunschweig
*a
a,
Julia I.
Schweizer
b,
Myron
Heinz
b,
Marcel
Härterich
a,
Ivo
Krummenacher
a,
Max C.
Holthausen
b and
Holger
Braunschweig
*a
aInstitut für Anorganische Chemie, The Institute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt am Main, Max-von-Laue-Str. 7, 60438 Frankfurt am Main, Germany
First published on 8th April 2019
Abstract
The one-electron reduction of (CAACMe)BCl3 (CAACMe = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene) yields the dichloroboryl radical [(CAACMe)BCl2]˙. Furthermore, the twofold reduction of (CAACMe)BCl3 in the presence of a range of Lewis bases (L = CAACMe, N-heterocyclic carbene, phosphine) yields a series of doubly base-supported (CAACMe)LBCl chloroborylenes, all of which were structurally characterised. NMR and UV-vis spectroscopic and electrochemical data for (CAACMe)LBCl show that the boron centre becomes more electron-rich and the HOMO–LUMO gap widens as L becomes less π-accepting. A [(CAACMe)BCl2]− boryl anion coordination polymer was isolated as a potential intermediate in these reductions. In most cases the reduction of the chloroborylenes resulted in the formation of the corresponding hydroborylenes or derivatives thereof, as well as ligand C–H activation products.
Introduction
While the field of boron chemistry has long been dominated by the naturally occurring +3 oxidation state of this electron-deficient element, the last decade has seen tremendous advances in the accessibility of lower oxidation state boron species. Among these, borylenes (:BR), which feature a highly reactive boron(I) centre with two empty p orbitals, have attracted a lot of attention owing to their similarities with organic carbenes.1 The parent borylene, :BH, was first generated in the 1930s by exposure of a BCl3/H2 gas stream to an electrical discharge,2 but is so short-lived that it can only be studied spectroscopically. In contrast, boron fluoride (:BF), which calculations have shown to be the most stable diatomic borylene,3 can be generated in over 90% yield from the comproportionation of BF3 with solid boron at 1850 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 and trapped with a variety of small molecules, as for example acetylene (Scheme 1a).5 A variety of methods have been used to generate the significantly less stable boron chloride species, :BCl, including plasma discharge or flash photolysis of BCl3 (Scheme 1b),6 reduction of BCl3 with gaseous copper at −196 °C to generate B2Cl4, which decomposes to :BCl and BCl3 at high temperature (Scheme 1c)5a or comproportionation of Cl2(g) with laser-ablated boron(0) in a solid argon matrix (Scheme 1d).7 The even less stable heavier boron halides, :BBr and :BI, have only been studied spectroscopically.8
4 and trapped with a variety of small molecules, as for example acetylene (Scheme 1a).5 A variety of methods have been used to generate the significantly less stable boron chloride species, :BCl, including plasma discharge or flash photolysis of BCl3 (Scheme 1b),6 reduction of BCl3 with gaseous copper at −196 °C to generate B2Cl4, which decomposes to :BCl and BCl3 at high temperature (Scheme 1c)5a or comproportionation of Cl2(g) with laser-ablated boron(0) in a solid argon matrix (Scheme 1d).7 The even less stable heavier boron halides, :BBr and :BI, have only been studied spectroscopically.8
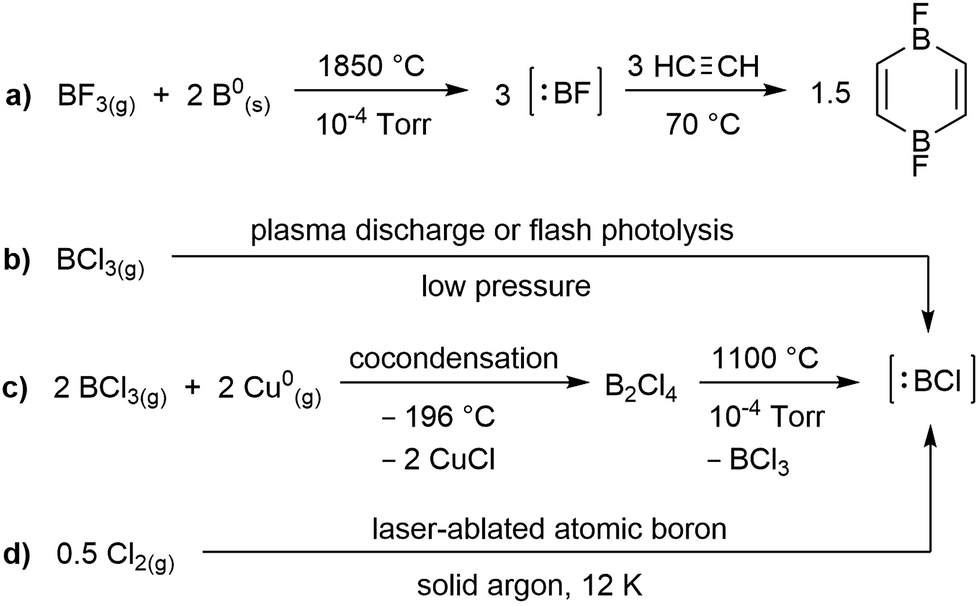 | ||
| Scheme 1 Methods for generating and trapping transient haloborylenes. | ||
Like carbenes, borylenes may be stabilised by adduct formation with electron-rich metal centres. Since the report by our group of the first stable transition metal borylene in 19959 this field has rapidly expanded10 and a number of bridging haloborylene complexes have been structurally characterised,11 while terminal fluoroborylene complexes have only been observed in the matrix at 6 K.12
It was not until 2011 that the first stable metal-free borylene, (CAACCy)2BH (I, CAACCy = 3,3-dimethyl-2-(2,6-diisopropylphenyl)-2-azaspiro[4,5]dec-1-ylidene) was isolated by Bertrand and co-workers.13 Its stability is owed to the two strongly σ-donating/π-acidic cyclic (alkyl)(amino)carbene (CAAC) ligands,14 which compensate for the build-up of electron density at the boron(I) centre by efficient π delocalisation to form two partial B–CCAAC π bonds. In recent years the field of metal-free borylenes has greatly expanded,15 recently culminating in the synthesis of the first bis(borylene)-stabilised borylene, (TMP)B(B(TMP))2 (TMP = 2,2,6,6-tetramethylpiperidinyl).16
In order to vary the electronic properties at sp2-hybridised borylene centres and tune their reactivity, various synthetic routes toward mixed-base-stabilised L1L2BR borylenes have been developed, in which L1 is typically a CAAC ligand and L2 a less π-accepting donor ligand. The simplest approach, which consists in adding a suitably small Lewis base (CO, CNtBu, IMeMe = 1,3,4,5-tetramethylimidazol-2-ylidene) to an isolated dicoordinate borylene, was employed to generate (CAACCy)(CO)BN(SiMe3)2 from linear (CAACCy)BN(SiMe3)217 and the mixed-carbene borylene II-IMeMe (Fig. 1) from the tetrameric precursor [(CAACMe)B(CN)]4,18 as well as a number of mixed-base bis(borylenes) from the diboracumulene precursor [(CAACMe)2B]2.19 While facile, this method remains limited by the dearth of isolable dicoordinate borylenes. A number of L(CO)BR borylenes (L = CAAC, CNR, NHC) have also been obtained by the addition of strongly σ-donating ligands to metal-carbonyl-bound borylenes.20 This is the case of III-CO, generated by the addition of CAACMe to [(Me3P)(CO)4Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BDur] (Dur = 2,3,5,6-tetramethylphenyl), and which may itself undergo photolytic CO–L ligand exchange to yield III-L (L = CNtBu, pyridine, IMe = 1,3-dimethylimidazol-2-ylidene).20b A more modular route to L1L2BR borylenes involves the introduction of both donors at the borane stage, prior to reduction. Thus compounds IV-L were obtained via multi-step syntheses from a simple (CAAC)BH3 precursor undergoing (a) replacement of one hydride by a triflate group, (b) displacement of triflate by L, (c) replacement of a second hydride by triflate, and finally, (d) reduction to IV-L.21 A much simpler approach, involving the twofold reduction of a (CAAC)BRBr2 precursor in the presence of a second donor was used for the synthesis of the first phosphine-supported borylene, compound II-PEt3.18
BDur] (Dur = 2,3,5,6-tetramethylphenyl), and which may itself undergo photolytic CO–L ligand exchange to yield III-L (L = CNtBu, pyridine, IMe = 1,3-dimethylimidazol-2-ylidene).20b A more modular route to L1L2BR borylenes involves the introduction of both donors at the borane stage, prior to reduction. Thus compounds IV-L were obtained via multi-step syntheses from a simple (CAAC)BH3 precursor undergoing (a) replacement of one hydride by a triflate group, (b) displacement of triflate by L, (c) replacement of a second hydride by triflate, and finally, (d) reduction to IV-L.21 A much simpler approach, involving the twofold reduction of a (CAAC)BRBr2 precursor in the presence of a second donor was used for the synthesis of the first phosphine-supported borylene, compound II-PEt3.18
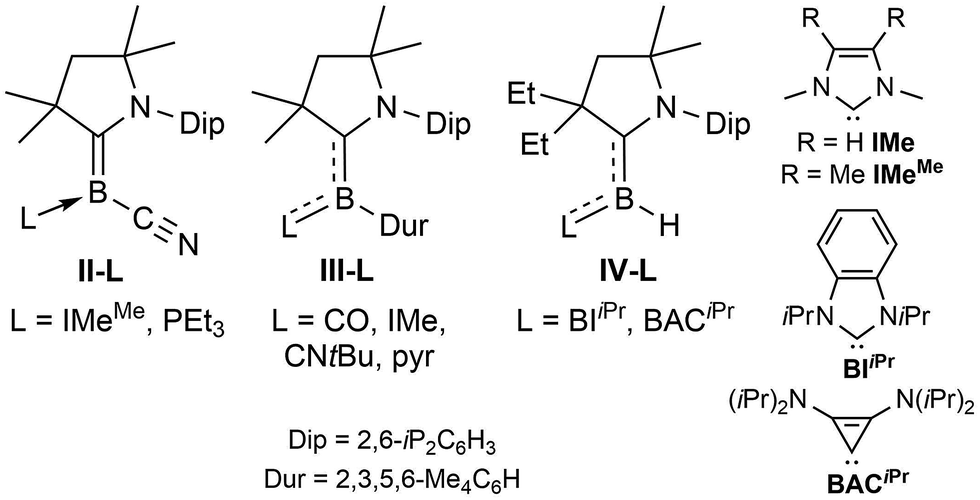 | ||
| Fig. 1 Selected examples of mixed-base-stabilised (CAAC)LBR borylenes. | ||
Despite all these advances, isolable metal-free haloborylenes, which are of particular interest because of their potentially reactive B–X bond, remain rather elusive. Only one bromoborylene, compound V, supported by a complex, chelating carborane-bridged bis(silylene) ligand, has been reported thus far and has already shown promising reduction and ligand exchange reactivity, providing access to the first borylene cation, VI (Scheme 2).22
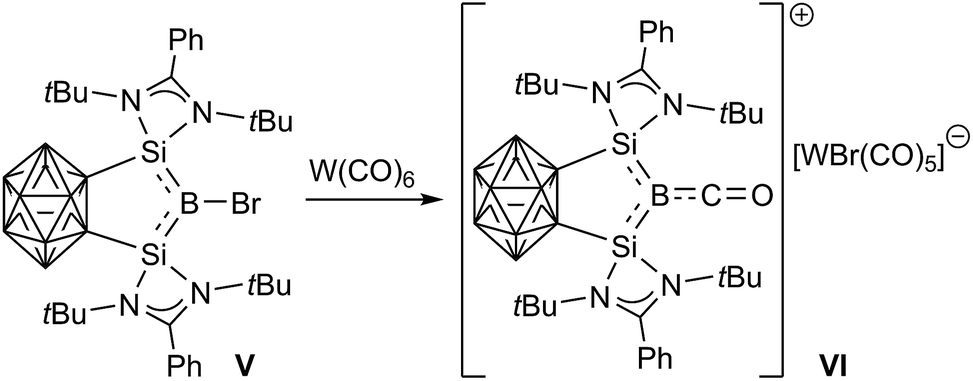 | ||
| Scheme 2 Halide abstraction at bromoborylene V. | ||
In this contribution we present a facile route towards chloroborylenes of the form (CAAC)LBCl (L = CAAC, NHC, phosphine) and examine the steric and electronic effects of L on the structural and spectroscopic properties of these borylenes. We also present the first stable dichloroboryl radical and a dichloroboryl anion, both likely intermediates in the formation of these borylenes. Finally, we show that further reduction of these species leads to hydrogen abstraction from the solvent, ligand metalation and C–H activation products.
Results and discussion
The reduction of (CAACMe)BCl3 (1) with one molar equivalent of Li sand in THF at room temperature yielded a pale green solution, which displayed no 11B NMR resonance (Scheme 3). Solvent removal and crystallisation of the hexane extract at −25 °C provided a small crop of orange crystals, which X-ray crystallographic analysis showed to be the [(CAACMe)BCl2]˙ radical, compound 2 (Fig. 2a).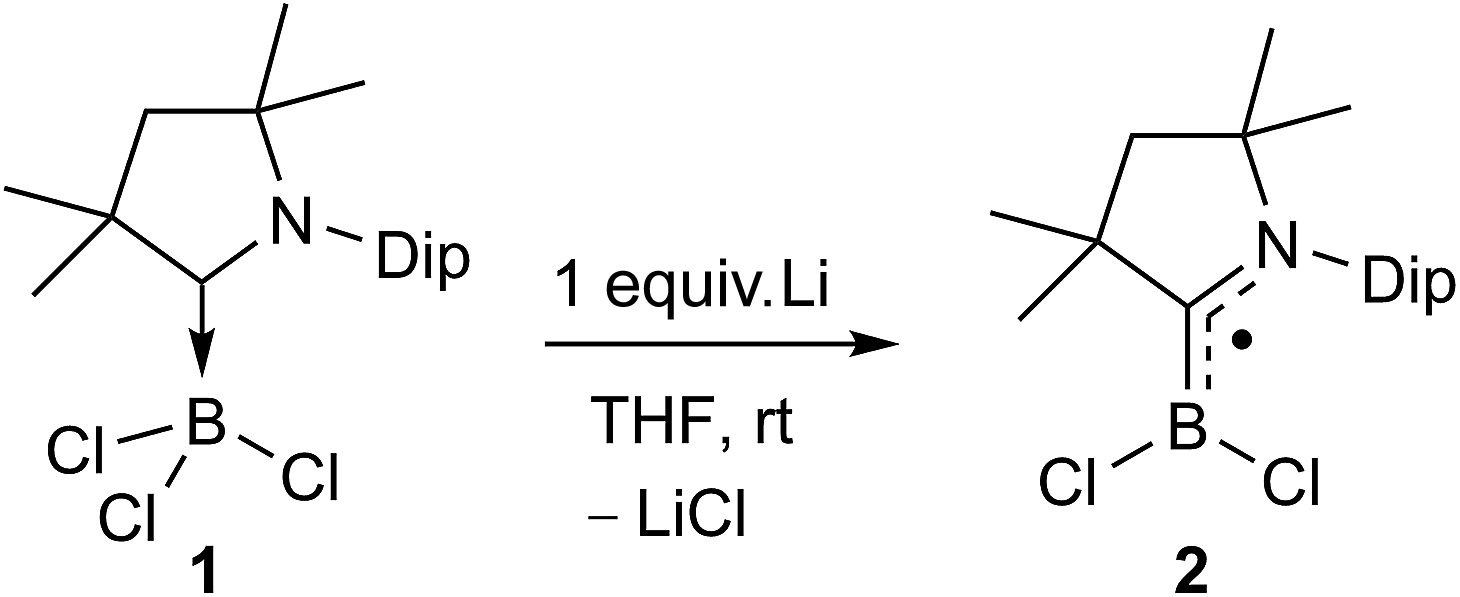 | ||
| Scheme 3 One-electron reduction of 1. | ||
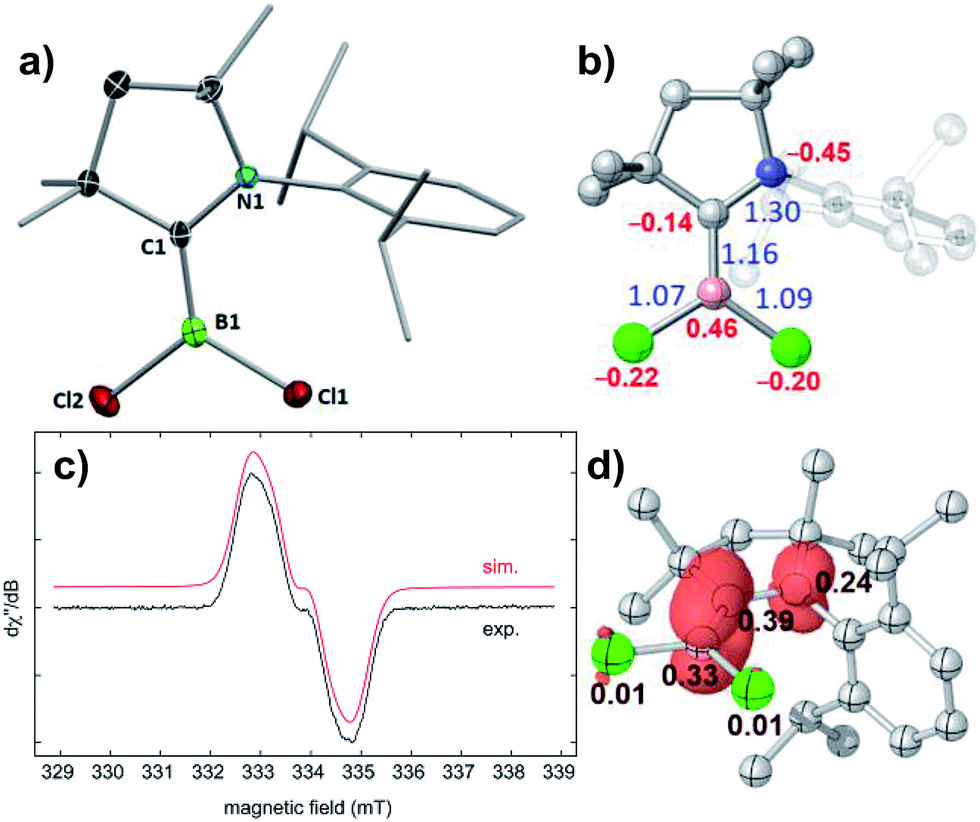 | ||
| Fig. 2 (a) Crystallographically-derived molecular structure of 2. Thermal ellipsoids at 50% probability level. Ellipsoids on the CAACMe ligand periphery and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): B1–C1 1.498(3), B1–Cl1 1.780(2), B1–Cl2 1.790(2), Σ∠B1 359.96(13). (b) Optimised structure of 2, Wiberg bond indices in blue, NPA charges in red. (c) Experimental (black) and simulated (red) EPR spectrum of [(CAACMe)BCl2]˙, a(14N) = 17.3 MHz (6.2 G) and a(11B) = 6.70 MHz (2.4 G). (d) Plot of calculated spin density of 2 (isovalue 0.005a0−3) with Mulliken atomic spin densities. | ||
The geometry around the boron atom is trigonal planar (Σ∠B1 359.96(13)°), while the B1–C1 distance (1.498(3) Å) is indicative of a double bond and significantly shorter than in precursor 1 (1.644(2) Å, see Fig. S41 in the ESI† for the solid-state structure of 1). This has been observed previously for other CAAC-supported boryl radicals and is indicative of the delocalisation of the unpaired electron over the B–C–N π framework.23 The EPR spectrum of 2 shows a very broad singlet centred at giso = 2.003 (Fig. 2c), unlike its relative [(CAACMe)B(Dur)Cl]˙, which displays a triplet from the hyperfine interaction with the 14N nucleus (a(14N) = 19 MHz).23b A simulation of the EPR spectrum of 2 provided the following hyperfine coupling parameters: a(14N) = 17.3 MHz and a(11B) = 6.70 MHz, the latter being significantly higher than that in the simulated EPR spectrum of [(CAACMe)B(Dur)Cl]˙ (ca. 2.7 MHz),23b which contributes to the strong signal broadening and loss of resolution. The spin density distribution in 2 was further analysed by density functional theory (DFT) calculations.‡ The unpaired electron is delocalised over the B–C–N π system with atomic spin densities of 0.39, 0.33, and 0.24 on C1, B1, and N1, respectively (Fig. 2d). The lower hyperfine coupling with 11B, as well as the lower spin density at boron for [(CAACMe)B(Dur)Cl]˙ (0.277)23bversus2 are due to the delocalisation of spin density to the duryl group in the former, whereas no such delocalisation to the chloride ligands is observed in 2. The B–C π-bonding character of the SOMO of 2 (Fig. S43†) results in a partial double bond, which is reflected in a B–C Wiberg bond index of 1.16 (Fig. 2b).
Cyclic voltammetry performed on 2 showed a single irreversible reduction wave at Epc = −2.35 V (against the ferrocene (Fc/Fc+) couple) and no oxidation event, unlike [(CAACMe)B(Dur)Cl]˙, for which a first reversible reduction wave (Epc = −2.03 V) and an irreversible oxidation wave (E1/2 = −0.53 V) were observed.23b This implies that a 2+ cation is unlikely to be chemically accessible, whereas further chemical reduction of 2 should be achievable with a suitable reducing agent.
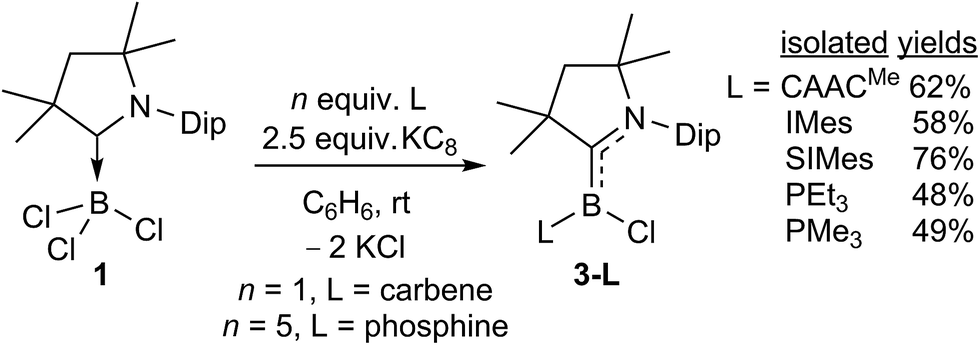
The room-temperature reduction of 1 in the presence of 1 equivalent of CAACMe with 2.5 molar equiv. of KC8 in benzene yielded a strikingly purple-blue reaction mixture, the colour of which intensified over a period of five hours (Scheme 4). Removal of volatiles, extraction with hexanes and subsequent solvent removal yielded a crude purple solid displaying a broad 11B NMR resonance at 18.7 ppm and a single set of 1H NMR CAACMe resonances, indicating a symmetrical compound. Crystallisation from pentane at −25 °C over a period of one week yielded large purple crystals suitable for X-ray crystallographic analysis, which provided the structure of a doubly CAACMe-stabilised chloroborylene, 3-CAACMe, presenting a trigonal planar boron centre (Σ∠B1 359.97(15)°, Fig. 3). The molecule is C2 symmetric and the two B1–C1 and C1–N1 bonds (1.530(2) and 1.389(2) Å, respectively) are intermediate between typical lengths of sp2–sp2 single (B–C 1.56; C–N 1.47 Å)24 and double bonds (BC 1.44; CN 1.35 Å),24,25 suggesting delocalisation of the borylene lone pair over the entire N–C–B–C–N π framework. The compound is thereby reminiscent of Bertrand's parent borylene I, the B–C bonds of which are slightly shorter (1.5175(15) and 1.5165(15) Å),13 presumably because of the smaller size and lower electronegativity of the hydride compared to that of the chloride ligand.
 | ||
| Scheme 4 Synthesis of doubly base-stabilised chloroborylenes. | ||
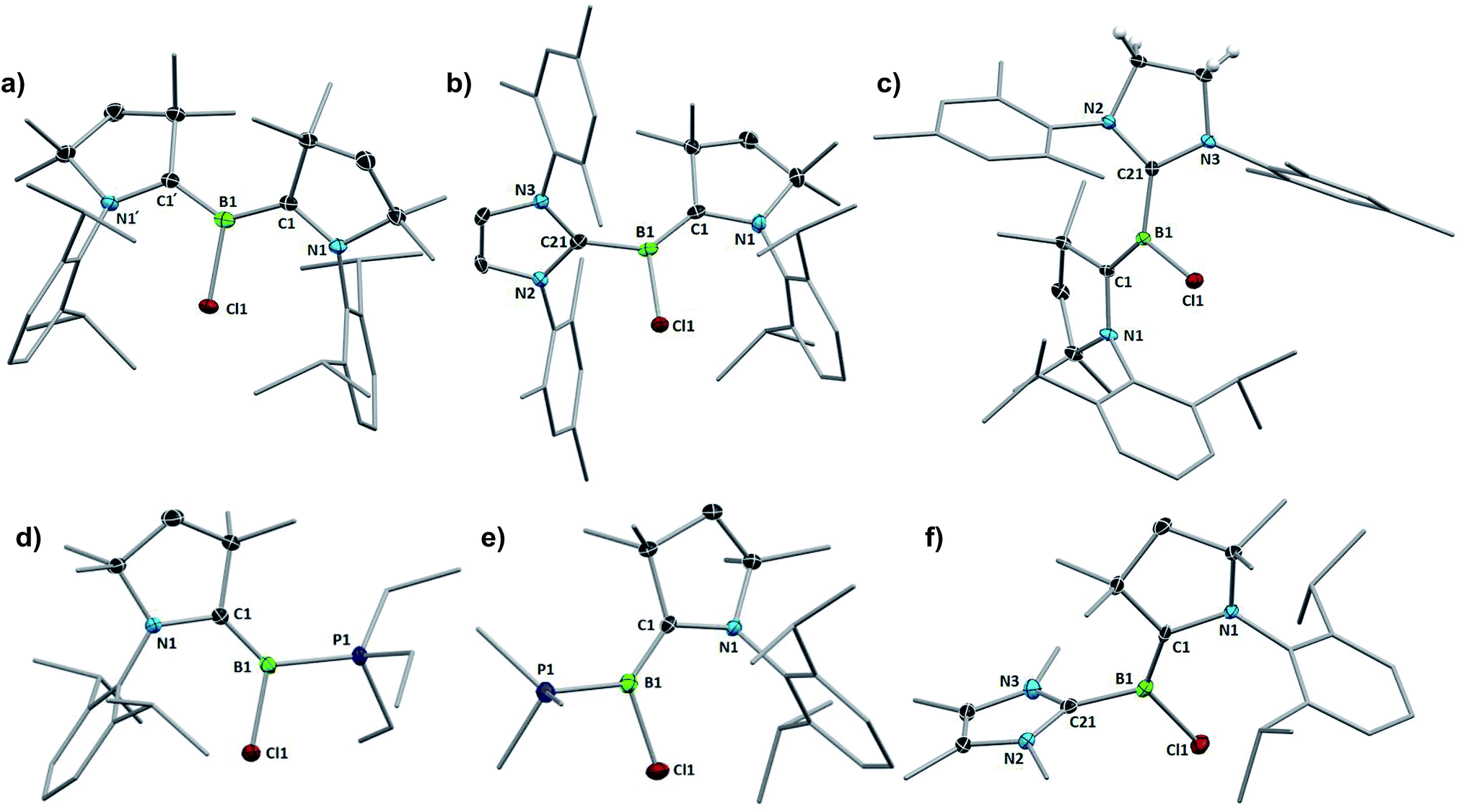 | ||
| Fig. 3 Crystallographically-derived molecular structures of 3-L, L = (a) CAACMe, (b) IMes, (c) SIMes, (d) PEt3, (e) PMe3, and (f) IMeMe. Thermal ellipsoids drawn at 50% probability level. Ellipsoids on the CAACMe ligand periphery and hydrogen atoms omitted for clarity. | ||
The analogous reductions of 1 in the presence of one molar equivalent of IMes (1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene) or SIMes (1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene) with 2.5 molar equivalents of KC8 in benzene similarly yielded deep-pink reaction mixtures from which the corresponding mixed-base chloroborylenes 3-L (L = IMes, SIMes) were extracted in moderate to good yields (58–76%). For smaller L ligands, such as IMeMe, PEt3 or PMe3 using a 1:1 1-to-L ratio for the reduction resulted in mixtures of the desired chloroborylene 3-L and (CAACMe)2BH (δ11B 12.5 ppm, vide infra compound 5-CAACMe). For PEt3 or PMe3 a fivefold excess of the phosphine could be used to obtain the mixed CAAC-phosphine-stabilised chloroborylenes in moderate isolated yields (ca. 50%), with the excess phosphine simply being removed in vacuo prior to extraction of the product. Unlike the bis(CAAC)- or CAAC–NHC-stabilised chloroborylenes, these compounds crystallise as pale yellow solids. Finally, 3-IMeMe was obtained as a bright red compound by quantitative NHC/phosphine ligand exchange with 3-PMe3 at room temperature (Scheme 5). Ligand exchange with the more sterically demanding carbenes IMes, SIMes and CAACMe was also attempted but remained limited to 2–5% conversion.§
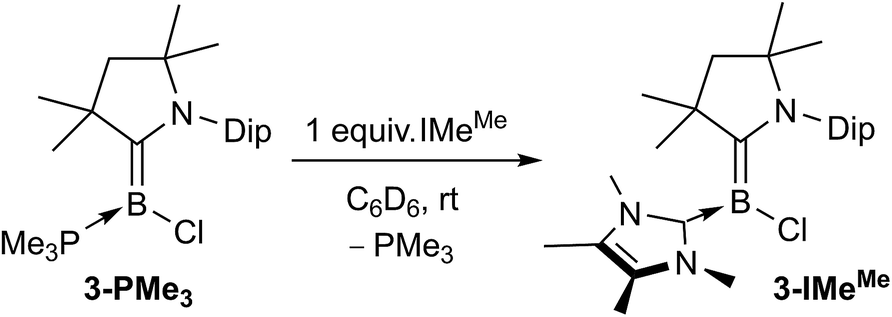 | ||
| Scheme 5 Synthesis of 3-IMeMe. | ||
From the attempted synthesis of the dimethylsulfide-stabilised borylene (CAACMe)(SMe2)BCl using 20 equivalents SMe2, recrystallisation of the toluene–hexane extract at −25 °C yielded a few isolated crystals of a linear coordination polymer of the dichloroboryl anion 4, [{(CAACMe)BCl2}2K2(SMe2)]n (Fig. 4). The boron atoms in this structure are trigonal planar with respect to their attached chloride and CAACMe ligands (Σ(∠B1) 359.95(14)°) and present a relatively short BCCAAC double bond (1.427(3) Å) compared to other known [(CAAC)BXY]− boryl anions (XY = (CN)2, H(CN), HCl, H2:B–CCAAC 1.432(6) to 1.473(2) Å).26 Each potassium cation lies in the plane of the sp2-boron atom, coordinating to both chloride ligands and bridging between three adjacent boryl anion units, once via a Cl1–K1–Cl1′ bridge and once via a Cl2–K1–Cl2′′ bridge. The adjacent (N1,C1,B1,Cl1,Cl2,K1) planes form an angle of 75° between each other. The two potassium cations bridging the Cl and Cl′ atoms, K1 and K1′, are further bridged by one SMe2 ligand and their coordination sphere is completed by π-interactions with the adjacent Dip residue. Unfortunately, due to its virtual insolubility in hydrocarbon solvents and extreme air- and moisture-sensitivity, no NMR spectroscopic or elemental analysis data could be obtained for 4. It is likely, however, that similar [{(CAACMe)BCl2}KL]n coordination oligomers or polymers could be intermediates in the synthesis of the 3-L chloroborylenes shown in Scheme 4.
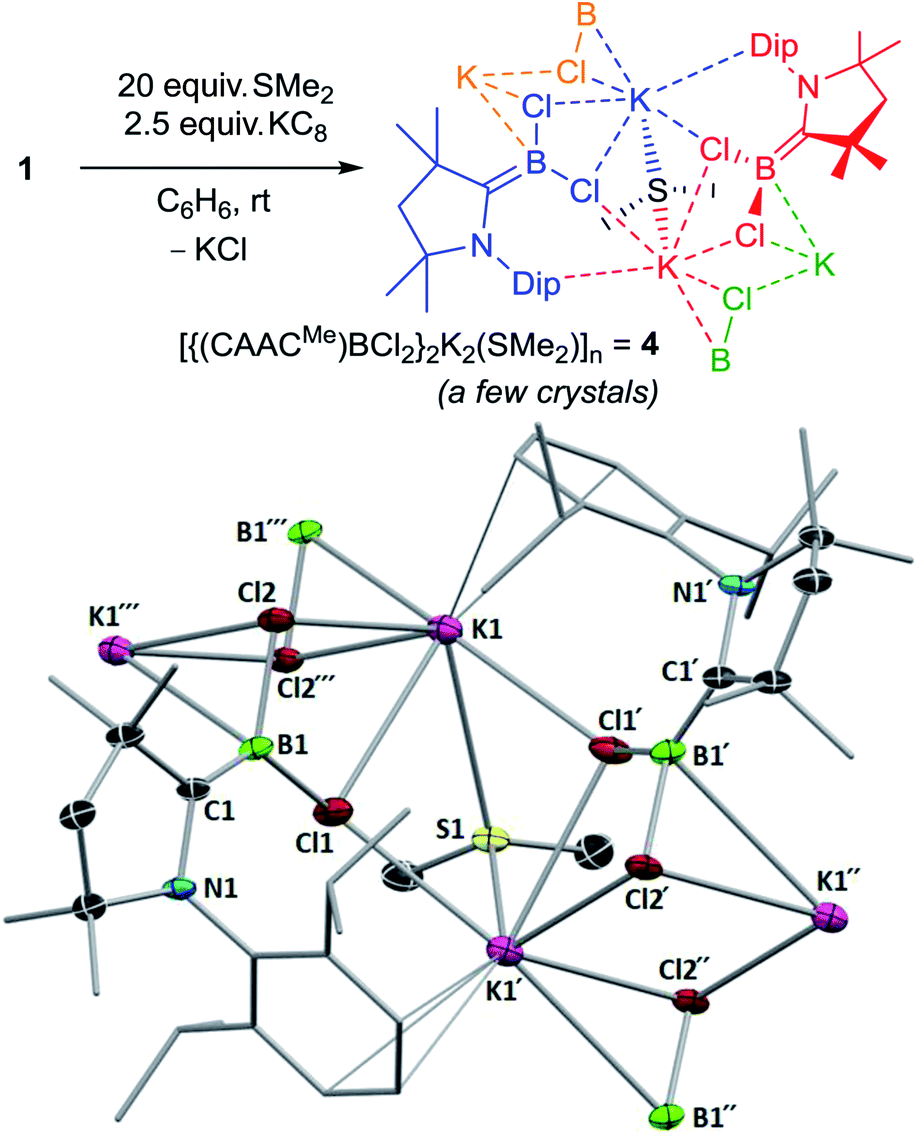 | ||
| Fig. 4 (Top) Synthesis of polymeric 4 showing the connectivity of adjacent boryl anion units in different colours. (Bottom) Crystallographically-derived molecular structure of 4 showing the connectivity of two boryl anion units. Thermal ellipsoids drawn at 50% probability level. Ellipsoids on the CAACMe ligand periphery and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): N1–C1 1.442(2), B1–C1 1.427(3), B1–Cl1 1.839(2), B1–Cl2 1.852(2), Cl1–K1 3.2351(6), Cl1′1–K1 3.1617(8), Cl2–K1 3.1866(6), Cl2′1–K1 3.1489(7), K1–S1 3.4032(7), Σ(∠B1) 359.95(14). | ||
A comparison of all six 3-L chloroborylenes shows an increasing upfield shift in the 11B NMR resonances in the following order (Table 2): CAACMe (18.7 ppm) > SIMes (11.9 ppm) > IMes (8.4 ppm) > PEt3 (5.6 ppm) ≈ IMeMe (5.5 ppm) > PMe3 (2.8 ppm). This correlates well with the trend of the overall electron-donating nature of the ligands L, i.e. their combined σ donor and π acceptor abilities, usually determined using the Tolman electronic parameter (TEP),27c which is defined as the ν(CO) stretching frequency of Ni(CO)3L complexes: the lower the TEP, the less electron-donating L is overall. While the TEP of IMeMe is yet to be determined experimentally,27d the trend in this table suggests a value close to 2062 cm−1 and similar electronic properties to PEt3.
| a Torsion angles. b C1–B1–C1′. c The asymmetric unit contains two structurally distinct borylene molecules. d Torsion angle (N3,C21,B1,Cl1). e C1–B1–P1. | ||||||
|---|---|---|---|---|---|---|
| 3-L, L = | CAACMe | IMesc | SIMes | IMeMe | PEt3 | PMe3 |
| B1–C1 | 1.530(2) | 1.437(7), 1.450(7) | 1.481(3) | 1.440(3) | 1.456(3) | 1.449(2) |
| B1–Cl1 | 1.843(3) | 1.884(5), 1.879(6) | 1.854(2) | 1.855(2) | 1.855(2) | 1.8516(17) |
| B1–C21 | — | 1.575(7), 1.570(7) | 1.550(3) | 1.578(3) | — | — |
| B1–P1 | — | — | — | — | 1.912(2) | 1.9114(17) |
| C1–N1 | 1.389(2) | 1.429(5), 1.428(6) | 1.408(2) | 1.432(2) | 1.421(2) | 1.4294(18) |
| Σ(∠B1) | 359.97(15) | 360(4), 359.9(4) | 359.58(15) | 359.94(17) | 359.56(14) | 359.7(11) |
| C1–B1–C21 | 133.9(2)b | 132.1(4) | 128.49(17) | 122.96(18) | 129.06(16)e | 131.54(12)[e] |
| (N1,C1,B1,Cl1)a | 14.51(16) | 4.9(8), 1.3(7) | 3.2(3) | 2.8(3) | 3.0(3) | 0.6(2) |
| (N2,C21,B1,Cl1)a | — | 57.7(5), 49.2(6) | 44.4(2)d | 79.3(2) | — | — |
Se
Moreover, for L = carbene, the 11B NMR shifts of 3-L decrease with the 77Se NMR shifts for the corresponding LSe adducts, which are a measure of the π acidity of the corresponding carbene: the more downfield the 77Se NMR chemical shift, the more π-accepting the carbene.28 Consequently, the 11B NMR shift of 3-L represents a reliable measure of the relative electron-donating ability of L.
Another measure of the relative σ donor and π acceptor abilities of L is provided by a comparison of the X-ray structural data of 3-L (Fig. 3, Table 1). For the carbene ligands, this is linked to the degree in which the CAACMe and L π frameworks, respectively, are rotated out of the plane of the borylene core, represented by the torsion angles (N1,C1,B1,Cl1) and (N2,C21,B1,Cl1), respectively, as well as the relative B–CCAAC and B–CL bond lengths. In the C2-symmetric 3-CAACMe the (N1,C1,B1,Cl1) torsion angles are only 14.51(16)°, which enables a large overlap between the borylene lone pair and the empty pz orbitals on both carbene carbon atoms. As a result, the borylene lone pair is fully delocalised over the C–B–C π orbital, which is further confirmed by the B–C bond lengths of 1.530(2) Å, which are intermediate between a single and double bond. While the (N1,C1,B1,Cl1) torsion angle remains very small for all the other derivatives (<5°), allowing for excellent π overlap of CAACMe with the borylene lone pair, the (N2,C21,B1,Cl1) torsion angle increases in the following order: CAACMe (14.5(2)°) < SIMes (44.4(2)°) < IMes (53.5(6)° = avg. of the two molecules present in the asymmetric unit) < IMeMe (79.3(2)°). This is in agreement with the decrease in π acidity in this ligand series (see Table 2). As the (N2,C21,B1,Cl1) torsion angle grows closer to orthogonality, π overlap decreases until it becomes negligible for L = IMeMe. This is also apparent in the lengthening of the B–CL bond from 1.530(2) Å in 3-CAACMe to 1.578(3) Å in 3-IMeMe, which suggests a typical dative bond, concomitant with a shortening of the B–CCAAC bond to 1.440(3) Å in the IMeMe analogue, indicating a typical BC double bond,25 as the entire π electron density from the borylene lone pair is employed in π backbonding from boron to CAACMe. A similar trend can also be observed in the structural data from Bertrand's (CAAC)LBH hydroborylenes, I13 and IV-L (Fig. 1)21 for which the torsion angles between the borylene plane and π framework of L increase in the order of CAACCy < BIiPr < BACiPr, which fits with the decreasing π acidity of L, concomitant with a shortening of the B–CCAAC bonds and a lengthening of the B–CL bonds. For the two 3-PR3 derivatives, in which the phosphines act as pure σ donors, the B–C bond lengths of 1.456(2) (R = Et) and 1.449(2) (R = Me) Å indicate a BC double bond,25 similarly to the 3-IMeMe derivative. In the only other structurally characterised CAAC–phosphine-stabilised borylene, compound II-PEt3 (Fig. 1), the B–C bond is slightly longer (1.484(6) Å) due to a small amount of π backbonding to the π-acidic cyano ligand.18
DFT calculations by the group of Bertrand have shown that the π-type HOMO of the (CAAC)LBH hydroborylenes I and IV-L is delocalised over the CCAAC–B–CL moiety, with the extent of delocalisation increasing with the π acidity of L.13,21 Given the structural and electronic similarities of these compounds to our 3-CAACMe and 3-NHC derivatives, we limited our DFT analysis to the bonding and electronic structure of 3-PMe3, in which the phosphine ligand acts as a pure σ donor. As expected, the HOMO of 3-PMe3 corresponds largely to the B–C π bond, with minor C1–N1 and B–Cl π antibonding interactions (Fig. 5b). The calculated B–C Wiberg bond index of 1.53 indicates a full BC double bond (Fig. 5a). This is also borne out by natural resonance theory (NRT), which gives the phosphonium alkylidene borate as the major resonance form (38.3%, see Fig. S45 in the ESI†), as well as natural population analysis (NPA), which places a large positive charge of +1.25 on P1 and small negative charges of −0.07 and −0.19 on B1 and C1, respectively (Fig. 5a).
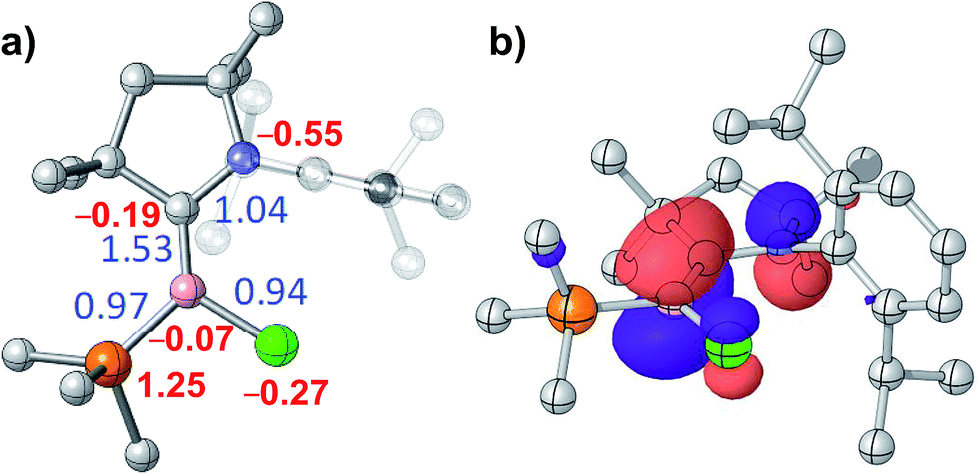 | ||
| Fig. 5 (a) Optimised structure of 3-PMe3: Wiberg bond indices in blue, NPA charges in red. (b) Plot of the HOMO of 3-PMe3 (isovalues ±0.05a0−3/2). | ||
With the exception of the two phosphine derivatives, compounds 3-L were intensely coloured (Fig. 6, top).¶ Furthermore, whereas the NHC and phosphine derivatives were highly air-sensitive, solutions of 3-CAACMe exposed to air retained their colour for several hours at room temperature, thus demonstrating the unusual stability of this species. An overlay of the UV-vis spectra of all 3-L compounds (Fig. 6, bottom) shows that the wavelength of the most red-shifted absorbance maximum decreases in the order of L = CAACMe (556 nm, purple-blue) > IMes (550 nm, purple-pink) > SIMes (538 nm, pink) > IMeMe (470 nm, orange-red) > PMe3 (350 nm, pale yellow) > PEt3 (336 nm, pale yellow). A TD-DFT study of our tetrameric cyanoborylene, [(CAACMe)B(CN)]4, had shown that its most red-shifted UV-vis absorption bands correspond to transitions from the four π-bonding borylene-centred molecular orbitals (MOs) to four π*-antibonding MOs centred on the π system of the cyano ligands and the aromatic Dip substituents of the CAAC ligand.18 By analogy, the highest wavelength absorption bands of 3-L likely correspond to π–π* transitions from the borylene-centred HOMO to the LUMO distributed over the π system of the ligands. A red-shift of this transition, i.e. a decrease in the HOMO–LUMO gap, can be correlated with an increase in (a) the σ-donor capacity of L, which can destabilise the HOMO and/or (b) the π-acceptor ability of L and/or (c) the amount of π conjugation within the molecule, where both (b) and (c) can stabilise the LUMO. In this case, from the alkylphosphine derivatives, which present no π system on L (λmax < 350 nm), via the IMeMe derivative with its conjugated imidazole-2-ylidene ring (λmax = 470 nm), to the CAAC and (aryl)NHC derivatives with their extensive π systems (λmax > 530 nm), the red-shift is seen to increase with the extent of the π system on L, as well as the amount of π delocalisation of the borylene lone pair.
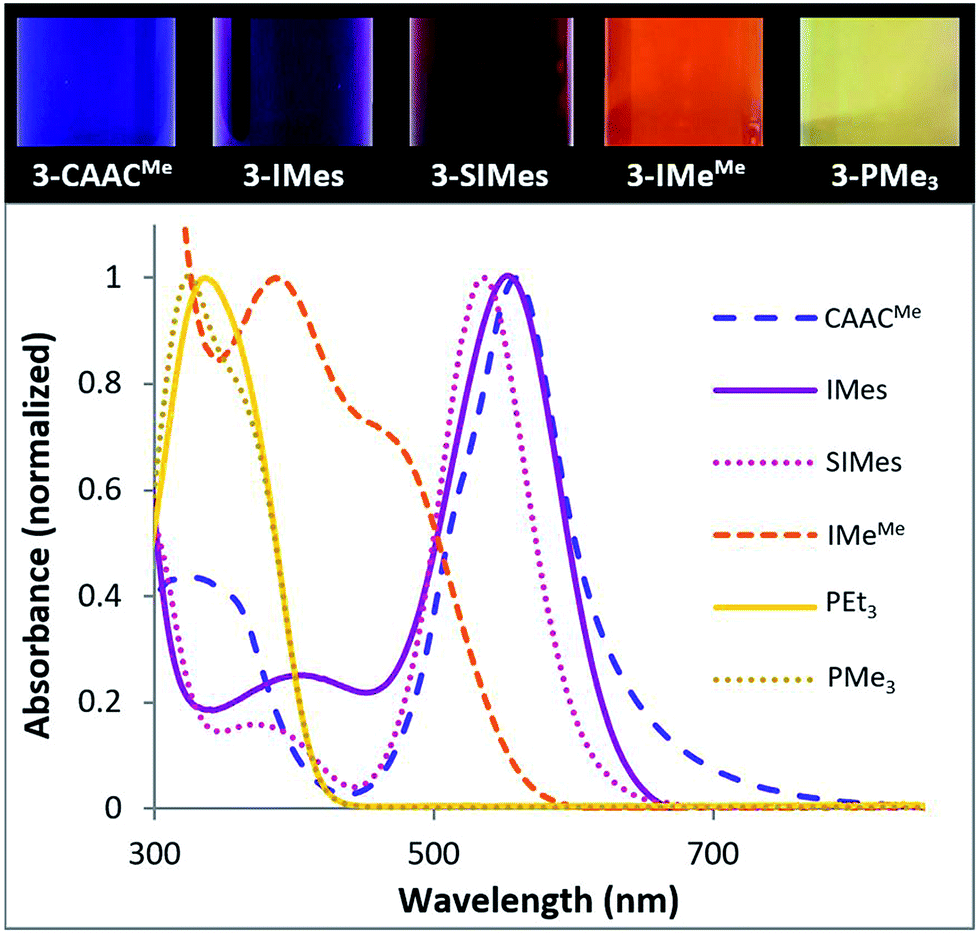 | ||
| Fig. 6 (Top) Colours of 1.2 mM benzene solutions of 3-L. (Bottom) Superposition of the UV-vis absorption spectra (highest absorbance normalised to 1) of 3-L. | ||
Cyclovoltammetric experiments performed on the all-carbene-supported 3-L borylenes (solvent: THF, supporting electrolyte: [n-Bu4N][PF6]) showed a first semi- or fully reversible oxidation wave between E1/2 = −1.00 V (3-CAACMe) and E1/2 = −1.26 V (3-IMeMe, vs. (Fc+/Fc), see Fig. S35–S38 in the ESI†). Although the potential differences are relatively small, it is apparent that E1/2 becomes slightly more negative as L becomes less π-accepting, i.e. as electron density increases at the borylene centre, which is to be expected. Furthermore, 3-CAACMe and 3-SIMes show irreversible reduction waves at Epc = −2.68 V and Epc = −3.17 V, respectively, whereas 3-IMes and 3-IMeMe show no reduction wave down to −3.30 V. Here again, the trend correlates with the overall electron-donating ability of L: the less electron-rich the borylene centre, the more facile its reduction.||
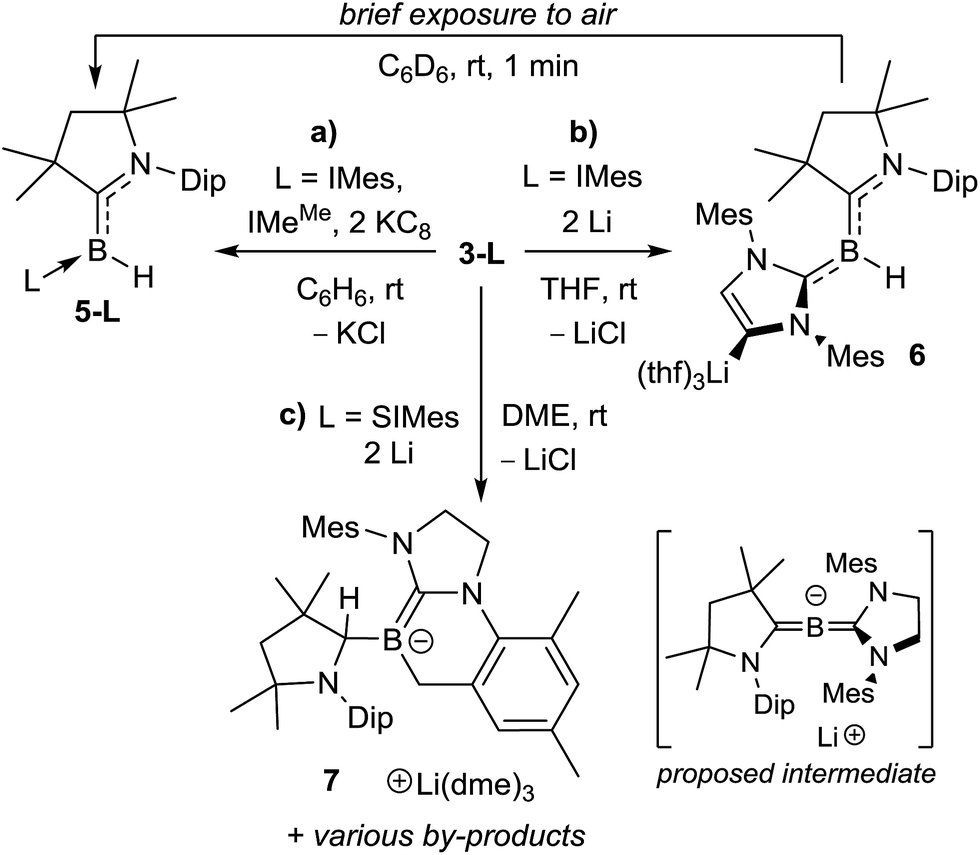
These results prompted us to attempt to reduce these compounds chemically. The room temperature reduction of either 3-IMes or 3-IMeMe with KC8 in benzene resulted in conversion to new red-coloured species displaying broad 11B NMR resonances at 1.4 and −3.0 ppm respectively (Scheme 6a). These were identified by NMR spectroscopy as the corresponding hydroborylenes, 5-IMes and 5-IMeMe.19a The reduction of 3-CAACMe with KC8 in benzene led to the formation of ca. 40% 5-CAACMe, which displays an 11B NMR shift similar to that of borylene I (δ11B 12.5 ppm),13 as well as a second unidentified species with a broad 11B NMR downfield shift at 54 ppm. The identity of 5-CAACMe was further confirmed by X-ray crystallographic analysis (see Fig. S42 in the ESI†).
 | ||
| Scheme 6 Reduction of 3-L, L = IMes, SIMes, IMeMe. | ||
The reduction of 3-IMes with excess Li sand in THF also proceeded cleanly to a red-orange compound presenting a broad 11B NMR resonance at 2.8 ppm (compound 6, Scheme 6b). X-ray crystallographic analysis of 6 revealed a planar hydroborylene (Σ(∠B1) 359.8(8)°) stabilised on the one side by a strongly π-accepting CAACMe ligand (B1–C1 1.454(4) Å) and on the other side by a σ-donating IMes ligand (B1–C21 1.583(4) Å) lithiated at the backbone C4 position, the coordination sphere of the lithium atom being completed by three THF residues (Fig. 7, top). Such metalation of unsaturated NHC backbones in the presence of strong, non-nucleophilic bases or reducing agents has been well documented over the last decade.29 Brief exposure of a C6D6 solution of 6 to air led to an immediate colour change from red to orange and NMR spectra showed clean conversion to 5-IMes (Scheme 6c). Interestingly, this route is more reliable and selective than the reduction of 3-IMes with KC8 (Scheme 6b) or the reduction of (CAACMe)BHX2 (X = Cl, Br) in the presence of one equiv. IMes, which besides 5-IMes also yields the dihydrodiborene (CAACMe)2B2H2.26c The boron-bound hydrides in 5-L and 6 presumably arise from hydrogen abstraction from the reaction solvent by intermediate dicoordinate boron-centred radicals.**
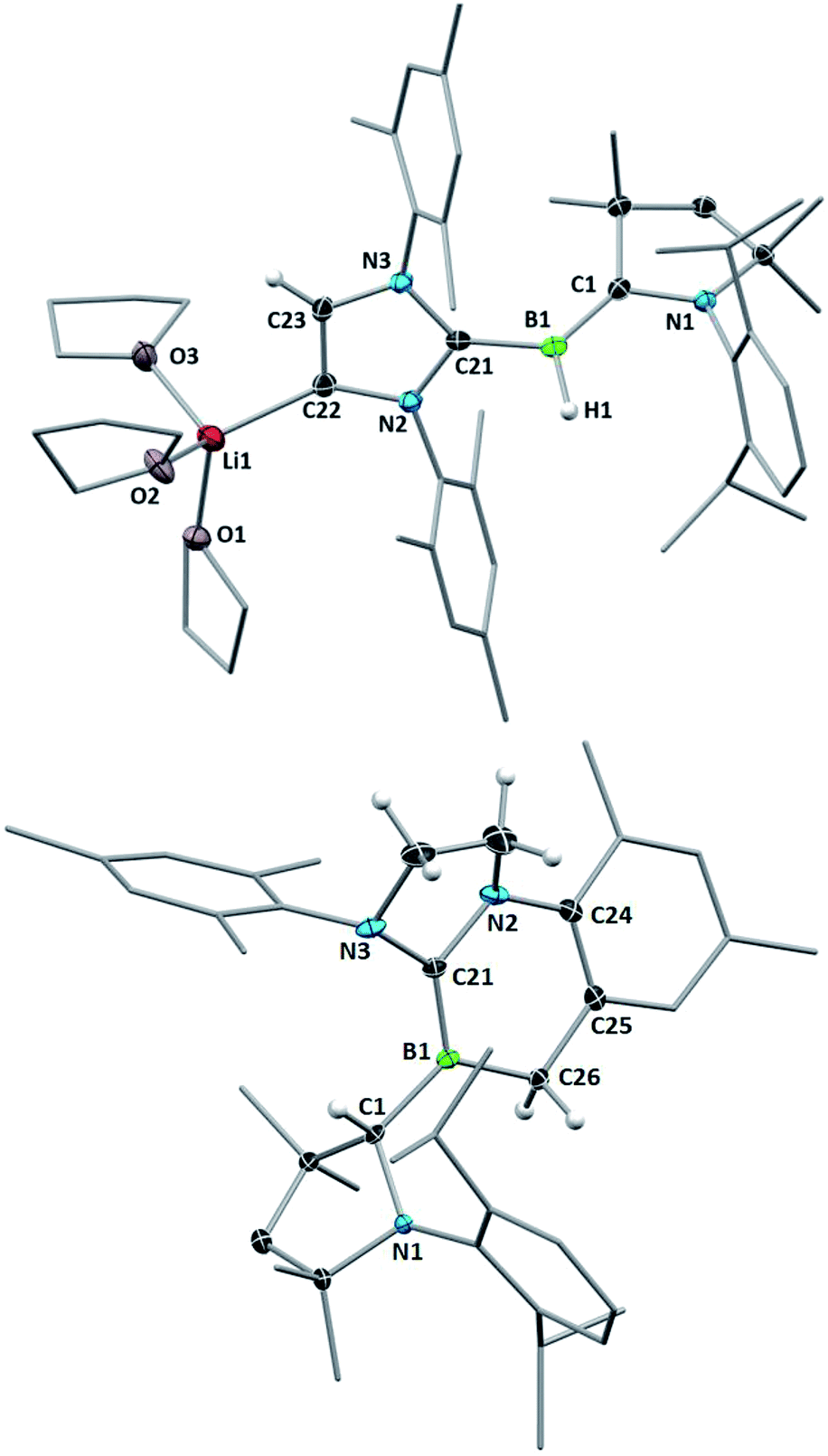 | ||
| Fig. 7 Crystallographically-derived molecular structures of 6 (top) and 7 (bottom). Thermal ellipsoids drawn at 30% probability level. Ellipsoids on the CAACMe ligand periphery and hydrogen atoms omitted for clarity, except for the boron-bound and IMes backbone hydrogens. Selected bond lengths (Å) and angles (°) for 6: N1–C1 1.446(3), B1–C1 1.454(4), B1–H1 1.24(2), B1–C21 1.583(4), C22–C23 1.359(3), C22–Li1 2.100(5), 3.1617(8), Σ(∠B1) 359.8(8), torsion angle (N1,C1,B1,H1) 2.6(11), torsion angle (N2,C21,B1,H1) 51.0(11); for 7: N1–C1 1.500(3), B1–C1 1.615(3), B1–C21 1.435(3), B1–C26 1.606(3), Σ(∠B1) 359.96(2). | ||
The reduction of 3-SIMes with KC8 in benzene or Li in THF or 1,2-dimethoxyethane (DME) did not proceed selectively, as evidenced by the appearance of multiple new 11B NMR resonances. Recrystallisation of the product mixture of the Li-based reduction in DME, however, yielded a few red crystals of the alkylideneborate 7 with a Li(dme)3 countercation (Scheme 6d, Fig. 7, bottom). The boron centre is planar (Σ(∠B1) 359.96(2)°) and displays a B1–C1 single bond to the now sp3-hybridised C1 atom (B1–C1 1.615(3); C1–N1 1.500(3) Å) and a formal B1–C21 double bond (1.435(3) Å) to the former SIMes ligand. Alkylidene borates are typically generated by α-deprotonation of a suitable sp2-borane precursor with a strong, non-nucleophilic base,25a by nucleophilic quaternisation of a neutral alkylidene borane,25b or by tautomerisation of intramolecular frustrated Lewis pair systems involving an α-proton transfer from a highly electron-withdrawing CHn–B(C6F5)2 moiety to the Lewis basic site.25d In contrast, the formation of 7 presumably results from the 2e− reduction of 3-SIMes to a highly reactive dicoordinate [(CAACMe)(SIMes)B]− anion (proposed intermediate in Scheme 6), which undergoes intramolecular ortho-methyl C–H activation of the mesityl substituent, followed by a 1,2-hydride migration from the boron centre to the adjacent CAAC carbene carbon atom. Similar mesityl-CH3-activations have been observed previously in the reduction of IMes-stabilised or mesityl-substituted boranes,30 as has the 1,2-hydride migration in CAAC-supported hydroboranes.31 Unfortunately, the other products of this reduction could not be cleanly isolated. No tractable products were obtained from various reduction attempts of the phosphine derivatives 3-PMe3 and 3-PEt3.
Conclusions
To conclude, we have described a facile synthetic route towards a series of metal-free haloborylenes, (CAAC)LBCl, by twofold reduction of a (CAAC)BCl3 precursor with KC8 in the presence of a donor ligand L. Two likely intermediates in these reduction reactions were isolated in the form of a [(CAAC)BCl2]˙ radical and a [(CAACMe)BCl2]− anion, the potassium countercation of which is coordinated by L. Variation of L from a highly π-accepting CAAC ligand via moderately π-accepting NHCs to purely σ-donating phosphines provides a series of borylenes displaying more or less sterically shielded and electron-rich boron centres, as determined by X-ray crystallographic analyses, NMR and UV-vis spectroscopy and cyclic voltammetry. The PMe3 derivative undergoes facile phosphine–NHC ligand exchange with a small NHC but not with the more sterically demanding ones. DFT calculations show that in the absence of possible π backbonding to L, the borylene lone pair is stabilised solely by π backbonding to the CAAC ligand. Finally, the room-temperature reduction of the CAAC and NHC derivatives led to the isolation of several hydroborylenes and the product of an intramolecular C–H activation, suggesting the formation of radical and anionic dicoordinate “[(CAAC)(NHC)B]−” reduction intermediates. We are currently studying the reactivity of these compounds, in particular 3-PMe3, towards Lewis acids and bases, unsaturated small molecules and electrophilic substitution, and will be reporting our results in a follow-up study.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (HB) for financial support. Quantum-chemical calculations were performed at the Centre for Scientific Computing (CSC) Frankfurt on the FUCHS and LOEWE-CSC high-performance computer clusters.Notes and references
- M. Soleihavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282 CrossRef PubMed.
- W. Loehte-Holtgreven and E. S. van der Vleugel, Z. Phys., 1931, 70, 188 CrossRef.
- M. Krasowska and H. F. Bettinger, J. Am. Chem. Soc., 2012, 134, 17094 CrossRef CAS PubMed.
- P. L. Timms, J. Am. Chem. Soc., 1967, 89, 1629 CrossRef CAS.
- (a) P. L. Timms, Acc. Chem. Res., 1973, 6, 118 CrossRef CAS; (b) P. L. Timms, J. Am. Chem. Soc., 1968, 90, 4585 CrossRef CAS.
- (a) D. Maeder, Helv. Phys. Acta, 1943, 16, 503 CAS; (b) A. G. Maki, F. J. Lovas and R. D. Suenram, J. Mol. Spectrosc., 1982, 91, 424–429 CrossRef CAS; (c) A. G. Massey and J. J. Zwolenik, J. Chem. Soc., 1963, 5354 RSC.
- P. Hassanzadeh and L. Andrews, J. Phys. Chem., 1993, 97, 4910 CrossRef CAS.
- See for example: (a) M. Nomoto, T. Okabayashi, T. Klaus and M. Tanimoto, J. Mol. Struct., 1997, 413–414, 471 CrossRef CAS; (b) J. A. Coxon and S. Naxakis, Chem. Phys. Lett., 1985, 117, 229 CrossRef CAS; (c) K. K. Irikura, J. Phys. Chem. Ref. Data, 2007, 36, 389 CrossRef CAS.
- H. Braunschweig and T. Wagner, Angew. Chem., Int. Ed., 1995, 34, 825 CrossRef CAS.
- (a) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197 RSC; (b) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924 CrossRef CAS PubMed; (c) H. Braunschweig, C. Kollann and F. Seeler, Transition Metal Borylene Complexes, in Contemporary Metal Boron Chemistry I. Structure and Bonding, ed. T. B. Marder and Z. Lin, Springer, Berlin, Heidelberg, 2008, vol. 130, pp. 1–27 Search PubMed.
- (a) K. Yuvaraj, M. Bhattacharyya, R. Prakash, V. Ramkumar and S. Ghosh, Chem.–Eur. J., 2016, 22, 8889 CrossRef CAS PubMed; (b) D. Vidovic and S. Aldridge, Angew. Chem., Int. Ed., 2009, 48, 3669 CrossRef CAS PubMed; (c) H. Braunschweig, M. Colling, C. Hu and K. Radacki, Angew. Chem., Int. Ed., 2002, 41, 1359 CrossRef CAS.
- (a) X. Wang, B. O. Roos and L. Andrews, Chem. Commun., 2010, 46, 1646 RSC; (b) X. Wang, B. O. Roos and L. Andrews, Angew. Chem., Int. Ed., 2010, 49, 157 CrossRef CAS PubMed.
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610 CrossRef CAS PubMed.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 CrossRef CAS PubMed.
- M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282 CrossRef CAS PubMed.
- S. Morisako, R. Shang, Y. Yamamoto, H. Matsui and M. Nakano, Angew. Chem., Int. Ed., 2017, 56, 15234 CrossRef CAS PubMed.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159 CrossRef CAS PubMed.
- M. Arrowsmith, D. Auerhammer, R. Bertermann, H. Braunschweig, G. Bringmann, M. A. Celik, R. D. Dewhurst, M. Finze, M. Grüne, M. Hailmann, T. Hertle and I. Krummenacher, Angew. Chem., Int. Ed., 2016, 55, 14464 CrossRef CAS PubMed.
- (a) J. Böhnke, M. Arrowsmith and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 10368 CrossRef PubMed; (b) J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, K. Hammond, T. Kramer, J. O. C. Jiménez-Halla and J. Mies, Angew. Chem., Int. Ed., 2015, 54, 13801 CrossRef PubMed; (c) J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2015, 54, 4469 CrossRef PubMed.
- (a) M. Nutz, B. Borthakur, C. Pranckevicius, R. D. Dewhurst, M. Schäfer, T. Dellermann, F. Glaab, M. Thaler, A. K. Phukan and H. Braunschweig, Chem.–Eur. J., 2018, 24, 6843 CrossRef CAS PubMed; (b) H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802 CrossRef CAS PubMed; (c) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327 CrossRef CAS PubMed.
- D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837 RSC.
- (a) H. Wang, J. Zhang, H. K. Lee and Z. Xie, J. Am. Chem. Soc., 2018, 140, 3888 CrossRef CAS PubMed; (b) H. Wang, L. Wu, Z. Lin and Z. Xie, J. Am. Chem. Soc., 2017, 139, 13680 CrossRef CAS PubMed.
- (a) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896 CrossRef PubMed; (b) P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- (a) M. M. Olmstead, P. P. Power, K. J. Weese and R. J. Doedens, J. Am. Chem. Soc., 1987, 109, 2541 CrossRef CAS; (b) M. Pilz, J. Allwohn, P. Willershausen, W. Massa and A. Berndt, Angew. Chem., Int. Ed., 1990, 29, 1030 CrossRef; (c) C.-W. Chiu and F. P. Gabbai, Angew. Chem., Int. Ed., 2007, 46, 6878 CrossRef CAS PubMed; (d) J. Möbus, G. Kehr, C. G. Daniliuc, R. Fröhlich and G. Erker, Dalton Trans., 2014, 43, 632 RSC.
- (a) D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590 CrossRef CAS PubMed; (b) M. Arrowsmith, D. Auerhammer, R. Bertermann, H. Braunschweig, M. A. Ali Celik, J. Erdmannsdörfer, I. Krummenacher and T. Kupfer, Angew. Chem., Int. Ed., 2017, 56, 11263 CrossRef CAS PubMed; (c) M. Arrowsmith, J. D. Mattock, J. Böhnke, I. Krummenacher, A. Vargas and H. Braunschweig, Chem. Commun., 2018, 54, 4669 RSC; (d) M. Arrowsmith, J. D. Mattock, S. Hagspiel, I. Krummenacher, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 15272 CrossRef CAS PubMed.
- (a) U. S. D. Paul, C. Sieck, M. Haehnel, K. Hammond, T. B. Marder and U. Radius, Chem.–Eur. J., 2016, 22, 11005 CrossRef CAS PubMed; (b) R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485 CrossRef CAS PubMed; (c) C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS; (d) D. G. Gusev, Organometallics, 2009, 28, 6458 CrossRef CAS.
- (a) M. Tretiakov, Y. G. Shermolovich, A. P. Singh, P. P. Samuel, H. W. Roesky, B. Niepötter, A. Visscher and D. Stalke, Dalton Trans., 2013, 42, 12940 RSC; (b) K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2416 CrossRef CAS.
- (a) M. Uzelac and E. Hevia, Chem. Commun., 2018, 54, 2455 RSC; (b) J. B. Waters and J. M. Goicoechea, Coord. Chem. Rev., 2015, 293–294, 80 CrossRef CAS; (c) Y. Wang, Y. Xie, M. Y. Abraham, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2010, 132, 14370 CrossRef CAS PubMed.
- (a) A. Hermann, J. Cid, J. D. Mattock, R. D. Dewhurst, I. Krummenacher, A. Vargas, M. J. Ingleson and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 10091 CrossRef CAS PubMed; (b) N. Arnold, H. Braunschweig, R. D. Dewhurst, F. Hupp, K. Radacki and A. Trumpp, Chem.–Eur. J., 2016, 22, 13927 CrossRef CAS PubMed; (c) D. P. Curran, A. Boussonniere, S. J. Geib and E. Lacote, Angew. Chem., Int. Ed., 2012, 51, 1602 CrossRef CAS PubMed; (d) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044 CrossRef CAS PubMed; (e) Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069 CrossRef CAS PubMed.
- (a) D. Auerhammer, M. Arrowsmith, H. Braunschweig, R. D. Dewhurst, J. O. C. Jiménez-Halla and T. Kupfer, Chem. Sci., 2017, 8, 7066 RSC; (b) S. Wuertemberger-Pietsch, H. Schneider, T. B. Marder and U. Radius, Chem.–Eur. J., 2016, 22, 13032 CrossRef CAS PubMed; (c) M. R. Momeni, E. Rivard and A. Brown, Organometallics, 2013, 32, 6201 CrossRef CAS; (d) G. D. Frey, J. D. Masuda, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 9444 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR, EPR, UV-vis, CV and X-ray crystallographic data, details of the computational analyses. CCDC 1893541–1893552. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01039d |
| ‡ Geometry optimisations were performed at the ωB97XD/SDD* level of density functional theory; bonding analyses reported are based on wave functions obtained from subsequent M06-2X/6-311G(d,p) single-point calculations. See ESI† for full details. |
| § These attempted ligand exchange reactions had to be carried out at room temperature as 3-PMe3 started decomposing in solution at 60 °C. |
| ¶ 3-PMe3 and 3-PEt3 display near identical solution colours at 0.12 mM. |
| || The cyclovoltammograms of 3-PMe3 and 3-PEt3 are virtually identical (see Fig. S39 and S40 in the ESI) and show multiple irreversible redox events, presumably due to rapid phosphine dissociation upon oxidation or reduction. |
| ** NMR-spectroscopic monitoring of the reduction of 3-IMes with Li in THF showed that 5-IMes is an intermediate in the formation of 6. Since the latter is quantitative, hydrogen abstraction from the ligand backbone can be excluded as source of the BH functionality. |
| This journal is © The Royal Society of Chemistry 2019 |