
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroscopic, thermodynamic and computational evidence of the locations of the FADs in the nitrogen fixation-associated electron transfer flavoprotein†
Nishya
Mohamed-Raseek
 a,
H. Diessel
Duan
a,
Peter
Hildebrandt
b,
Maria Andrea
Mroginski
b and
Anne-Frances
Miller
*ab
a,
H. Diessel
Duan
a,
Peter
Hildebrandt
b,
Maria Andrea
Mroginski
b and
Anne-Frances
Miller
*ab
aDept. Chemistry, University of Kentucky, 505 Rose Street, Lexington, KY 40506-0055, USA. E-mail: afmill3r2@gmail.com
bMax Volmer Laboratorum für Biophysikalische Chemie, Technische Universität – Berlin, Sekr. PC 14, 135 Straße des 17. Juni, 10623 Berlin, Germany
First published on 28th June 2019
Abstract
Flavin-based electron bifurcation allows enzymes to redistribute energy among electrons by coupling endergonic and exergonic electron transfer reactions. Diverse bifurcating enzymes employ a two-flavin electron transfer flavoprotein (ETF) that accepts hydride from NADH at a flavin (the so-called bifurcating FAD, Bf-FAD). The Bf-FAD passes one electron exergonically to a second flavin thereby assuming a reactive semiquinone state able to reduce ferredoxin or flavodoxin semiquinone. The flavin that accepts one electron and passes it on via exergonic electron transfer is known as the electron transfer FAD (ET-FAD) and is believed to correspond to the single FAD present in canonical ETFs, in domain II. The Bf-FAD is believed to be the one that is unique to bifurcating ETFs, bound between domains I and III. This very reasonable model has yet to be challenged experimentally. Herein we used site-directed mutagenesis to disrupt FAD binding to the presumed Bf site between domains I and III, in the Bf-ETF from Rhodopseudomonas palustris (RpaETF). The resulting protein contained only 0.80 ± 0.05 FAD, plus 1.21 ± 0.04 bound AMP as in canonical ETFs. The flavin was not subject to reduction by NADH, confirming absence of Bf-FAD. The retained FAD displayed visible circular dichroism (CD) similar to that of the ET-FAD of RpaETF. Likewise, the mutant underwent two sequential one-electron reductions forming and then consuming anionic semiquinone, reproducing the reactivity of the ET-FAD. These data confirm that the retained FAD in domain II corresponds the ET-FAD. Quantum chemical calculations of the absorbance and CD spectra of each of WT RpaETF's two flavins reproduced the observed differences between their CD and absorbance signatures. The calculations for the flavin bound in domain II agreed better with the spectra of the ET-flavin, and those calculated based on the flavin between domains I and III agreed better with spectra of the Bf-flavin. Thus calculations independently confirm the locations of each flavin. We conclude that the site in domain II harbours the ET-FAD whereas the mutated site between domains I and III is the Bf-FAD site, confirming the accepted model by two different tests.
Introduction
The term ETF denotes a family of electron transfer flavoproteins that convey electrons between redox enzymes and compounds in energy metabolism.4–7 Their shared heterodimeric structure is described by three domains, as shown in Fig. 1.2,8 The most familiar ‘canonical’ ETFs function in mitochondria receiving electrons from diverse acyl-CoA dehydrogenases and passing them one at a time to the quinone pool via an ETF-quinone oxidoreductase.10 These are the group 1 ETFs.4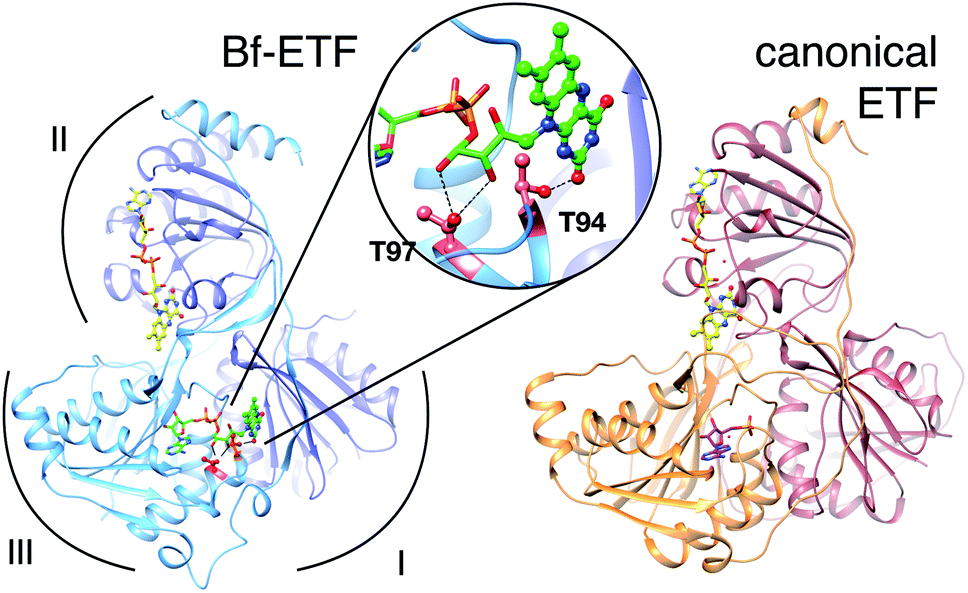 | ||
| Fig. 1 Ribbon diagrams of bifurcating ETF (Bf-ETF, left) and a canonical ETF (right) based on the structures of Acidaminococcus fermentans ETF (AfeETF, 4KPU.pdb)1 and Paracoccus denitrificans ETF (PdeETF, 1EFP.pdb).2 The FAD common to all known ETFs is in yellow stick Cs with CPK colouring of other atoms, whereas the second FAD found only in Bf-ETFs is in green and the AMP that replaces it in canonical ETFs is in magenta. The central inset highlights in red the two Thr residues of RpaEtfS that were replaced by Ala in our T94A.S/T97A.S variant (‘T94/97A ETF’). These Thrs are conserved among Bf-ETFs and shared by AfeETF. Arcs indicate the three domains of the ETF fold. Domain I is formed by the large subunit (EtfL, dark blue) while domain III is formed by the small subunit (EtfS, light blue). Domain II is composed primarily of EtfL but EtfS also contributes its C-terminal helix. Domain II is understood to adopt different orientations relative to the base formed by domains I and III.9 This and other molecular graphics were produced using chimera.11 | ||
Recently, some ETFs have been discovered to possess electron transfer bifurcating activity: ability to accept a pair of electrons from NADH and use one to generate a strong reductant such as flavodoxin hydroquinone (HQ) or reduced ferredoxin. This endergonic electron transfer is paid for by exergonic transfer of the other electron to a strong electron acceptor via a corresponding partner enzyme, such as a quinone reductase in the case of ETFs associated with nitrogen fixation.1,4,9,12–14 The bifurcating ETFs identified so far are all members of group 2.4 Their capacity to redistribute energy among electrons allows proprietor organisms to employ abundant mid-range reductants to drive demanding reactions that require greater reducing power.12 Thus, life-limiting processes such as nitrogen fixation (E°′ = −400 mV (ref. 15)) are made possible based on NADH (E°′ = −320 mV).
A subgroup of Bf-ETFs is found associated with nitrogen fixation and members are therefore named ‘Fix’ (subgroup 2d ETFs4). Although our system of study is the FixAB of Rhodopseudomonas palustris, we retain the ETF notation because our work applies to Bf-ETFs in general.
All known ETFs contain FAD bound in domain II (the domII site, Fig. 1), but the Bf-ETFs possess a second FAD whose AMP portion binds in place of the AMP of canonical ETFs.1 The FMN portion is bound between domains I and III (the domI/III site, Fig. 1).1,9 The protein imbues the two flavins with distinct properties, enabling them to play complementary roles.
In Bf-ETFs, the flavin that accepts a pair of electrons from NADH and sends one to each of two pathways is called the bifurcating flavin (Fig. 1). Besides the activity of pairwise (2-e−) electron transfer reactivity, this flavin has been shown to have a lower reduction midpoint potential (E°).3 The other flavin is believed to mediate single-electron (1-e−) transfers from the Bf-flavin to the electron-accepting quinone reductase of the Fix system,12 or other electron accepting enzyme more generally. Thus, this flavin is known as the electron transfer FAD: ET-FAD.3 Both of the sequential 1-e− transfers have E°s above that of the Bf-flavin.3,16 Because this reactivity resembles that of canonical ETFs, the ET-flavin is believed to occupy the domII site, by analogy with the sole FAD of canonical ETFs.1,17 By process of elimination, the flavin in the domI/III site is believed to be the Bf-flavin. Indeed, NAD+ is seen binding near this FAD in a crystal structure of the Bf-ETF from Acidaminococcus fermentans (AfeETF).1 Moreover this FAD is absent from canonical ETFs, which cannot accept electrons from NADH. Thus it has become accepted that the Bf flavin resides in the domI/III site and the ET-flavin residues in domII. However this highly attractive model1,16 remains to be tested in a hypothesis-driven way.
To do so, we employed site-directed mutagenesis to unambiguously target the domI/III binding site, providing protein-localized perturbations to disrupt binding of one FAD. A combination of spectroscopic and thermodynamic measurements was then used to test the functional identity of the remaining FAD (Bf-FAD vs. ET-FAD). This strategy additionally exploits the finding that the two FADs of RpaETF possess distinct UV-vis absorption and visible CD signatures, besides their differing reactivities.3 Our results assign the ET-FAD to the domII site.
As a second, orthogonal approach, we applied time-dependent density functional theory (TD-DFT) to calculate the electronic spectra of each flavin, using molecular mechanics (MM) to account for the flavin's environment in RpaETF. In each calculation, the identity of the protein site is known, and yields predictions for the spectra expected to result. Our calculations based on the domI/III site matched the spectra of the Bf-FAD better than those of the ET-FAD, whereas the predictions for the domII site were most consistent with the observed data for the ET-FAD. Thus, we provide direct hypothesis-driven tests, by two complementary methods, that 1-e− transfer activity resides in the flavin in domain II while the Bf-flavin is bound in the interface between domains I and III.
Results
Mutation of the flavin-binding site between domains I and III
With the objective of disrupting FAD binding to one site with minimal effects on the other, we targeted the site between domains I and III because canonical ETFs are stably folded despite lack of a flavin in that site, providing precedent for our goal of a one-flavin ETF. Mutations were chosen that convert amino acids conserved among Bf-ETFs to identities found among canonical ETFs, to further increase the likelihood of a well-behaved variant protein. Nine different mutant fixA and fixB genes were generated and the constructs were transformed as A–B pairs into E. coli. ETFs with mutations in both monomers were not sufficiently well expressed or stable enough to characterize, however the double mutant encoding T94A/T97A EtfS combined with the WT construct encoding EtfL yielded significant quantities of ETF in the soluble fraction (‘T94/97A ETF’, see Fig. 1). In this variant, molecular modelling indicates that our mutations will remove two hydrogen bonds to the ribose and one to the flavin portion of the domI/III FAD. Thus the resulting protein should retain the domII FAD, but bind AMP in domain III in lieu of the domI/III FAD. T94/97A ETF was purified with the aid of His tags on each subunit, with a typical yield of 1.25 mg L−1 of culture. AMP was provided in the buffer used during cell lysis to favour retention or binding of AMP.18 Indeed, Sato et al. demonstrated that prior binding of AMP favours FAD binding to pig ETF.19Cofactor content of T94/97A ETF
The T94A and T97A amino acid substitutions were designed to abrogate binding of one of the two FADs, so total FAD content was quantified by two methods. Based on fluorescence,20 T94/97A ETF contains 0.79 ± 0.05 FAD (n = 2) per dimer. Optical absorbance suggested similar FAD content of 0.86 ± 0.07 (n = 3) per dimer. Because our mutations recreate features of the AMP site of canonical ETFs, we also quantified bound AMP based on the absorbance at 260 nm of cofactors released from the protein, after accounting for the concentration of FAD (ESI Fig. S1†). We find that there are 1.21 ± 0.04 (n = 3) AMP per dimer. According to Sato et al., AMP can also bind to the FAD binding site of canonical ETFs,18,19 thus, suprastoichiometric AMP could have occupied a small fraction of the domII FAD-binding sites, however these will not contribute to the visible range of our optical spectra.Visible spectrum of T94/97A ETF resembles that of the ET-FAD of RpaETF
The absorption spectrum of T94/97A ETF was weaker in the visible range than that of WT RpaETF, consistent with lower FAD content (above) but the signal also had a different shape, with a much smaller dip between the two bands (Fig. 2). Although the long-wavelength band peaked at 450 nm, as for free FAD, the shorter-wavelength band peaked at 394 nm, red-shifted by 18 nm from the λmax of free FAD but similar to the λmax near 400 nm observed for ET-FAD in other Bf-ETFs.1,17 However the FAD released from T94/97A ETF upon protein denaturation was indistinguishable from authentic FAD (Fig. 2) indicating that the unusual spectral signature is not a consequence of covalent modification.21,22 Indeed, barely separated bands have been observed in the Bf-ETFs from A. fermentans and Megasphaera elsdenii.1,17 Thus the unusual signature of T94/97A ETF-bound flavin must be attributed to the protein environment, and moreover an environment that may be characteristic of Bf-ETFs.17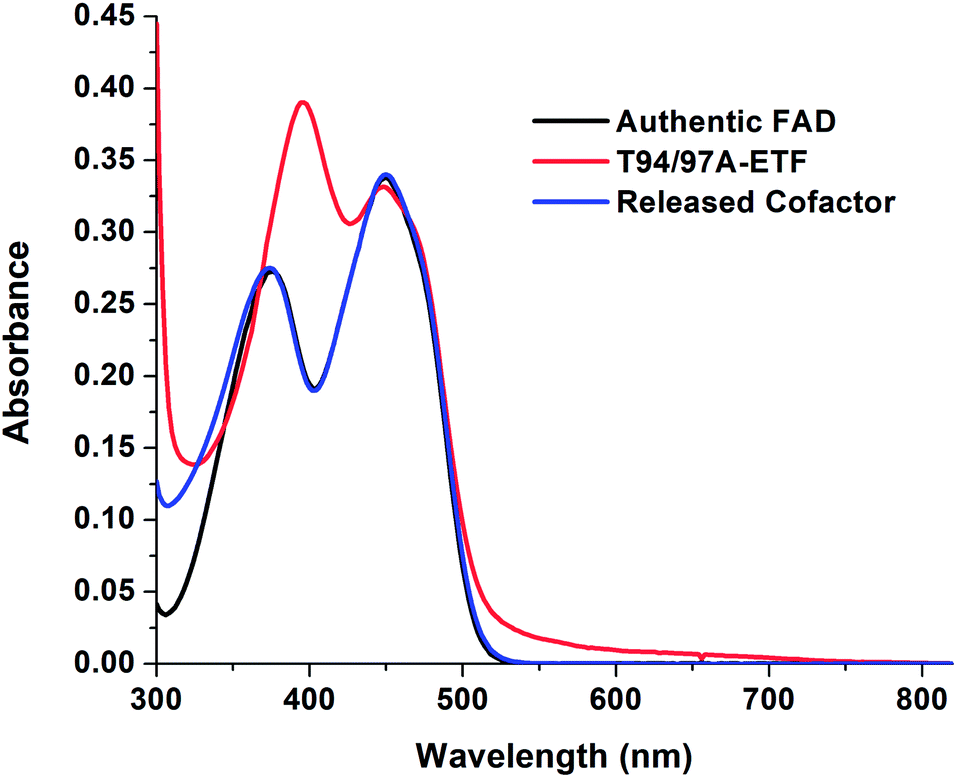 | ||
| Fig. 2 Visible spectra of T94/97A ETF and cofactors released from it, showing that the released flavin has a spectrum similar to authentic FAD whereas its signal when bound in purified 37.6 μM T94/97A ETF protein is perturbed, reflecting interactions with the protein environment. | ||
The barely resolved flavin signal of M. elsdenii ETF (MelETF) corresponds to the presumed ET-FAD17 as does the similar signal from AfeETF because it is reduced at higher E°.1 This suggests that T94/97A's FAD is the ET-FAD. Because we disrupted the domI/III site, this in turn suggests that the ET-FAD resides in the domII site.
Circular dichroism spectrum of T94/97A ETF resembles that of ET-FAD of WT RpaETF
Although the amount of FAD present in T94/97A ETF is compatible with 80% occupancy of one of the two sites, it is also possible that the FAD is distributed among both sites at 40% occupancy. To learn whether the FAD was predominantly in one of the two sites, rather than distributed in both, we exploited circular dichroism (CD) signals from the bound flavins.23 Free FAD exhibits very weak CD despite its strong visible absorption23,24 but can display significant CD in the range 300–500 nm when bound in protein sites,24 and the CD signals vary widely depending on the identity of the protein. Indeed, the two FADs in WT RpaETF display readily distinguishable CD signals.23 Equal distribution of FAD between the two sites in T94/97A ETF should produce a CD spectrum similar to that of WT RpaETF but weaker, whereas occupation of just one of the two sites should reproduce one or the other of the signals from single sites. Indeed, the visible CD spectrum of T94/97A ETF closely matches that of the ET-FAD (Fig. 3), at two different pHs (ESI Fig. S2†). Thus, T94/97A ETF retains predominantly the ET-FAD, and we confirm that ET-FAD is located in the domII site because the other site was mutated to favour binding of AMP over FAD.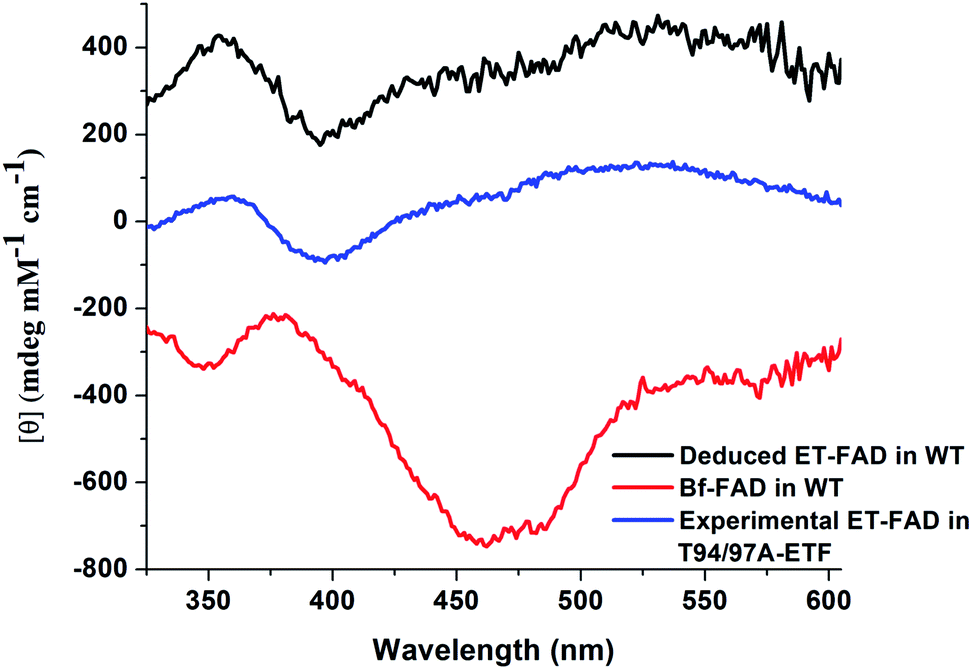 | ||
| Fig. 3 Visible CD spectrum of T94/97A ETF (in blue) in the oxidized (OX) state, compared with control spectra. These are (in red) the spectrum of WT RpaETF in which only the Bf-FAD is OX (the ET-FAD is HQ and therefore makes very little contribution), and (in black) the CD signal of the ET-FAD from WT RpaETF. The latter was obtained by collecting the CD spectrum of fully OX RpaETF and then subtracting away the contribution of Bf-FAD (red) to reveal the contribution of the ET-FAD.3 The CD spectrum of the FAD in T94/97A ETF shows better agreement with the ET-FAD CD signal vs. the Bf-FAD CD signal. Note that vertical offsets of ±250 were applied to the two WT RpaETF signals in order to avoid overlap with the spectrum of T94/97A ETF. | ||
Consistent with possession of only ET-FAD, T94/97A ETF was not reduced upon addition of NADH (ESI Fig. S3†).16 Similarly, NADH does not reduce canonical ETFs,16 although WT RpaETF is rapidly reduced by NADH demonstrating an aspect of bifurcation activity that is attributed to the Bf-FAD.3,16 Thus, failure to react with NADH indicates absence of the Bf-FAD from T94/97A ETF, consistent with the CD signal and FAD quantitations, and confirms that the domI/III site would normally contain the Bf-FAD.
Redox activity of T94/97A ETF
While the CD signal indicates that the flavin experiences a similar protein environment in T94/97A ETF as the ET-FAD of WT RpaETF, we also tested its functionality. We expected that the FAD of T94/97A ETF should have reactivity resembling that of the ET-FAD of WT RpaETF, but perturbed reactivity would be evidence of long-range coupling between the two flavin sites. Specifically, the ET-FAD is expected to accept electrons one at a time, forming an ASQ before adopting a HQ state.Fig. 4 shows that the domII FAD of T94/97A ETF retains the characteristic 1-e− activity of the ET-FAD, undergoing reduction by dithionite or reducing equivalents from xanthine oxidase (via a mediator) to form ASQ, as indicated by growth in intensity near 374 nm and at wavelengths longer than 500 nm. Further reduction caused diminution of intensity at 374 nm. Thus, dithionite titration of T94/97A ETF showed a biphasic titration profile (Fig. 4 inset). The protein's instability upon full reduction to HQ resembles behaviour reported for the ET-FAD in MelETF17 Thus, the domII FAD of T94/97A ETF retains the type of reactivity expected for ET-FAD upon mutation of the domI/III site.
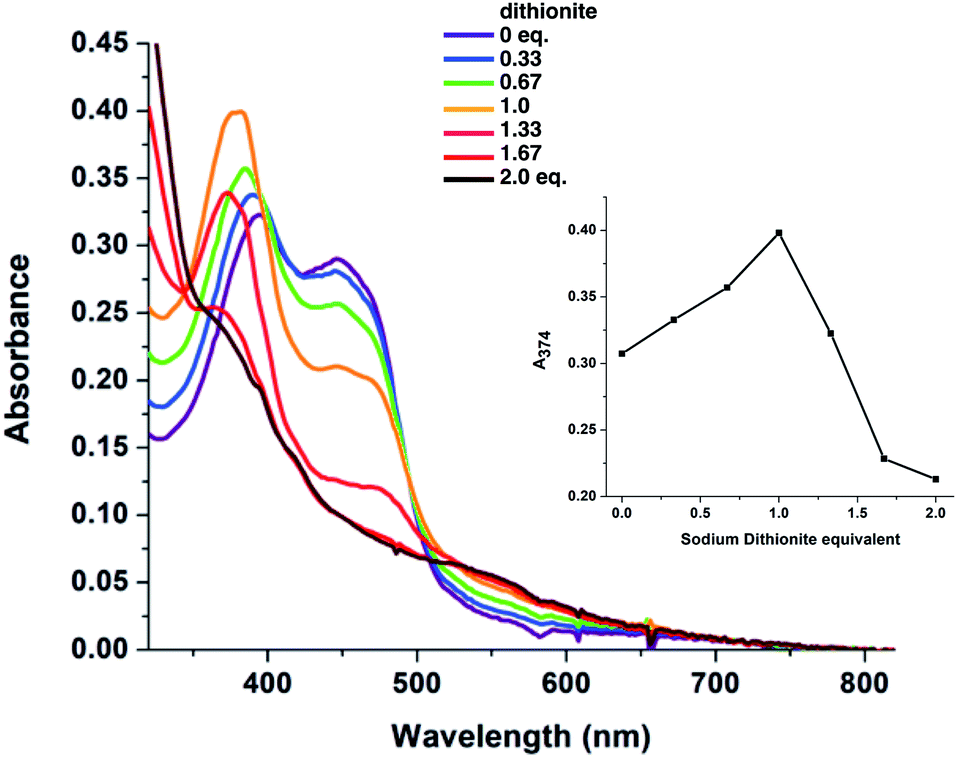 | ||
| Fig. 4 Visible spectra observed in the course of stepwise reduction of T94/97A ETF with dithionite, and plot of the absorbance (A374) illustrating the change in the nature of the reaction after dithionite exceeded 1 equiv. In the first phase of reduction A374 increases, whereas in the second it decreases. The concentration of ETF-bound FAD was 34 μM. | ||
Reduction midpoint potentials of T94/97A ETF
We quantified ET-FAD reactivity in T94/97A ETF by measuring the E° associated with reduction of the oxidized (OX) state to the anionic semiquinone (ASQ),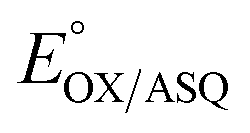 . In titrations of WT RpaETF, the optical signals of the two FADs are overlapped so the spectra did not reveal which flavin was undergoing reduction. However this ambiguity is absent from the T94/97A ETF because only the domII FAD is present.
. In titrations of WT RpaETF, the optical signals of the two FADs are overlapped so the spectra did not reveal which flavin was undergoing reduction. However this ambiguity is absent from the T94/97A ETF because only the domII FAD is present. 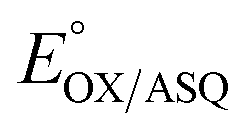 was determined using anaerobic spectro-electrochemical titrations and xanthine/xanthine oxidase to deliver reducing equivalents in the presence of mediator and the reference dye methylene blue (MB).25Fig. 5 shows that the log of the ratio of the OX to reduced (RED) fractions of dye adheres to a linear correlation vs. the analogous log ratio for the FAD
was determined using anaerobic spectro-electrochemical titrations and xanthine/xanthine oxidase to deliver reducing equivalents in the presence of mediator and the reference dye methylene blue (MB).25Fig. 5 shows that the log of the ratio of the OX to reduced (RED) fractions of dye adheres to a linear correlation vs. the analogous log ratio for the FAD 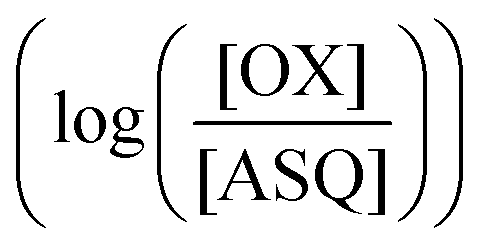 , as expected from the Nernst equation. The slope of 0.49 agrees well with the theoretical value of 0.5 expected for 1-e− reduction of FAD consistent with the appearance of ASQ features in the optical spectra. The intercept of the line yielded an
, as expected from the Nernst equation. The slope of 0.49 agrees well with the theoretical value of 0.5 expected for 1-e− reduction of FAD consistent with the appearance of ASQ features in the optical spectra. The intercept of the line yielded an 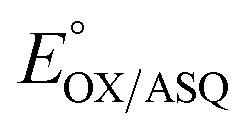 of −7 ± 4 mV at pH 8.0 (average over three separate experiments), based on an E° of −19 mV for MB at this pH.26 This is higher than the corresponding
of −7 ± 4 mV at pH 8.0 (average over three separate experiments), based on an E° of −19 mV for MB at this pH.26 This is higher than the corresponding 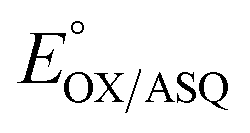 of WT RpaETF of −47 mV,3 revealing a long–range effect of the amino acid substitutions and/or absence of a flavin in the domI/III site.
of WT RpaETF of −47 mV,3 revealing a long–range effect of the amino acid substitutions and/or absence of a flavin in the domI/III site.
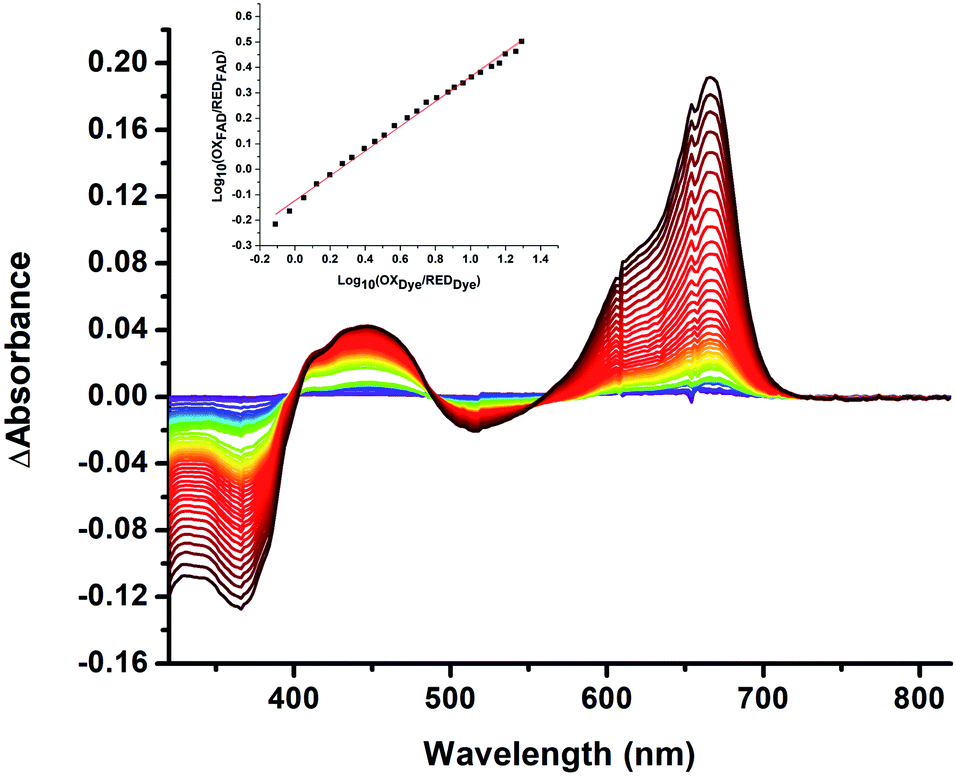 | ||
| Fig. 5 Difference spectra observed in the course of reductive titration of T94/97A ETF (374 and 450 nm) and methylene blue (664 nm). Each difference spectrum shown is an observed spectrum minus the spectrum at the end of the first phase of reduction (Fig. 4). The difference spectra thus emphasize changes associated with the reduction of OX to ASQ. Inset: plot of log([oxidized FAD]/[reduced FAD]) for the flavin of T94/97A ETF vs. analogous quantity for the dye illustrating linear behaviour consistent with the Nernst equation. The slope near 0.5 is consistent with a 1-e− event. The best fit is y = 0.491x − 0.126. R2 = 0.996. | ||
Assignment of protein site identity based on density functional theory computations
The distinct absorbance and CD signals from the two flavins in WT RpaETF suggested that these spectra reflect the different protein environments of the two sites, and therefore could be used to know which of the two sites binds the Bf-FAD. To test the ability of the protein environment to account for the distinct signals, we calculated the visible CD and absorption spectra predicted by each of the two flavin sites, using a quantum mechanical/molecular mechanical (QM/MM) approach in which the flavin itself and a few nearby hydrogen bond partners received quantum mechanical treatment whilst the rest of the protein was described by an electrostatic reaction field presented as an array of charges.27,28 No crystal structure is available for RpaETF so we employed a model built on the structure of AfeETF as a template, exploiting the 40% amino acid sequence identity and 60% similarity of these two ETFs (see Experimental section). The calculations were calibrated based on the absorption spectra. Although the calculated vertical transition energies bore systematic offsets of 0.33 to 0.35 eV, similar to findings of other studies29–32 (Fig. 6), the calculations were successful in reproducing the distinct shapes of the optical signals of the two flavins. Specifically, the calculations predicted two well-resolved bands in the visible range for the flavin bound in the domI/III site, but barely resolved transitions for the flavin bound in the domII site (Fig. 6A). The same predictions were obtained for two sizes of basis set and for various choices of quantum mechanical region (ESI Fig. S4†). These results are consistent with the observed signals of the two flavins in WT RpaETF3 and confirm assignment of the barely resolved signature associated with ET-flavin to the domain II site vs. the more typical flavin signature associated with the Bf-flavin to the domI/III site.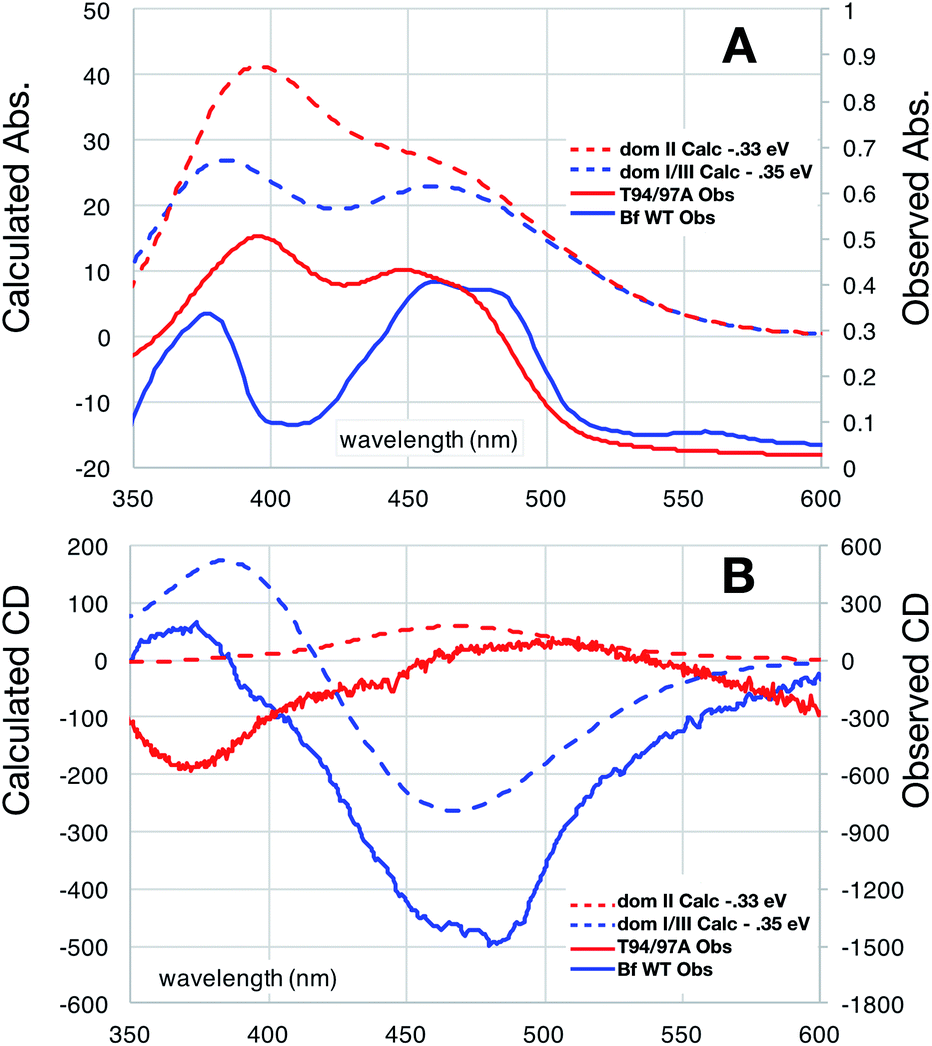 | ||
| Fig. 6 Comparison of observed vs. computed spectra. Absorbance data are in panel (A) whereas CD spectra are in (B). Spectra of OX T94/97A ETF (red) are compared with those of the Bf-FAD (blue) collected on WT RpaETF in which contributions from the ET-FAD were suppressed by chemical reduction (i.e. only the Bf-FAD remains in the strongly absorbing OX state). Calculated spectra are based on a TD-DFT computations using B3LYP and the 6-31++G(d,p) basis with quantum treatment of the domII active site (55 atoms, ESI Fig. S5†) or the domI/III active site (50 atoms, ESI Fig. S5†). See ESI Fig. S6† for an example of the individual transitions contributing to these envelopes and the effects of different sizes of quantum zones. | ||
Calculated CD spectra were also compared with those of the two flavins (Fig. 6B). The CD spectrum calculated for the domI/III flavin agrees only with that observed for the Bf-flavin.3 The signs of the bands as well as the relative energies at which they occurred were robust to changes in basis set and quantum region size, indicating that they genuinely reflect the effect of the protein environment on the flavin electronics.
The spectrum calculated for the flavin in domain II is distinguished by being much weaker, in which regard it agrees best with the observed CD of the ET-flavin, in both the WT RpaETF and T94/97A ETF. The sign of the longest-wavelength band is calculated to be positive as is also true for the observed signal (Fig. 3 and 6), but our calculations fail to predict the ellipticity at shorter-wavelengths and are not independent of the parameters used for the calculation. However all our computed CD spectra for the domII site are incompatible with the Bf-flavin data (Fig. 6 and S4†), which instead agrees well with the domI/III site. Therefore we conclude that the Bf-flavin occupies the domI/III site.
The calculations possess the strategic advantage of each being based on a known site in the protein. Thus they provide independent confirmation of the finding based on mutagenesis, that the ET-FAD is bound in domII whereas the Bf-flavin is bound in the domI/III site. Overall, we have provided two independent tests of the model of FAD functionality placement in Bf-ETFs. We have thereby also strengthened the functional inferences and structural homologies with canonical ETFs upon which the model was based. Furthermore we have demonstrated the capacity of QM/MM methods to distinguish between the two flavin sites in Bf-ETFs, thereby opening the door to parsing the contributions of individual active site residues to the distinct reactivities of the two flavins, and understanding the unusual optical signature of the ET-FAD.
Discussion
RpaETF is a group 2 ETF, along with all the ETFs known to bifurcate and to contain two FADs.1,4,17 This and the close association of the fixA and fixB genes with a gene apparently encoding an ETF-quinone oxidoreductase as well as genes for nitrogen fixation33 indicate that the RpaETF is a Bf-ETF.4Our work builds on elegant experiments by Shiga's team wherein replacement of the more tightly-bound flavin with a covalently modified analog was used to demonstrate that electron transfer from NADH flows to that FAD and thence to the other one (‘FAD-2’).16 This implied that FAD-2 was the ET-FAD. In complementary work Chowdhury et al. observed the adenine portion of NAD+ bound near the domI/III FAD suggesting that it is the one that accepts hydride, consistent with the fact that it is the flavin whose presence distinguishes Bf-ETFs from canonical ETFs.
Herein, we have confirmed experimentally that the FAD bound in domII, corresponding to the single FAD of canonical ETFs2,8 is indeed the one with the higher E°s characterizing sequential 1-e− events,3 the ET-FAD.1,3,34 Because our T94/97A ETF retains the very weak CD signal and barely resolved absorbance spectrum of ET-FAD, these too are confirmed to reflect the domII site. The domI/III FAD whose binding site we perturbed must therefore be the Bf-FAD with a single lower E° describing a 2-e− reaction.3,14,16 Thus, our experiments confirm the accepted model for Bf-ETFs.1,3,12,13,16
Our mutagenesis strategy sought to produce a system resembling the canonical ETFs, allowing us to test the analogies that had been used to infer ET function for the domII site of Bf-ETFs. We targeted conserved amino acid residues in the domain I/III flavin binding site: T97, T94, V232, G127, D93 in the EtfS protein, residues whose identities are differently conserved in canonical vs. Bf-ETFs, along with R165 in EtfL. Unfortunately, many of the double mutants containing substitutions in both proteins were not stable enough for us to be confident that no FAD would be released in the course of redox titrations, or were poorly expressed. However, ETFs bearing substitutions in only EtfS proved more stable and of those, T94/97A ETF was the successful candidate with respect to protein expression and stability. We speculate that many of our other substitutions had adverse effects on the interface between the two monomers.
We exploited the very different CD signals of the two FADs in RpaETF, and demonstrated that the signal retained by T94/97A ETF is the one previously assigned to the ET-FAD.3 Because the CD signal of T94/97A ETF is very similar to that of the ET-FAD of WT RpaETF, which also resembles the CD signal of Methylophilus methylotrophus ETF35 our CD results further confirm the analogy between the ET-FADs in the different groups of ETFs. Based on the similarity of the CD signal of T94/97A ETF to that of the ET-FAD in WT RpaETF, it appears that the domII site does not experience altered electrostatic anisotropy due to the amino acid substitutions and lack of flavin in the domI/III site, consistent with the distance between the two sites and their locations in different domains.
T94/97A ETF has a weaker absorbance spectrum than does the WT, as expected based on its lower FAD content. Its spectrum resembles that of the ET-FAD of AfeETF and MelETF with respect to shape (Fig. 2 and 6), displaying peaks at 394 and 450 nm and lacking a prominent dip between two bands.1,17 Although 8-formyl flavin is formed in many canonical ETFs,21,36 such a modification appears not to be responsible for T94/97A ETF's unusual spectrum, as the flavin released from T94/97A ETF does not display the optical signature of 8-formyl flavin.
NADH proved unable to reduce our T94/97A ETF, instead producing aggregation, although the corresponding WT RpaETF underwent full reduction by NADH.3 This confirms the absence of Bf-FAD in the T94/97A ETF. T94/97A ETF nevertheless underwent stepwise reduction with the 1-e− donor dithionite to form ASQ (evident at 374 nm and 550 nm) and then HQ, retaining the reaction pattern of wild type ET-FAD.3,16 In WT RpaETF changes at 550 were complicated by loss of a CT band attributed to Bf-FAD that absorbs in that region.3 The relative instability of the HQ of T94/97A ETF is consistent with earlier findings in bifurcating MelETF, where reduced ET-FAD dissociates.17 Indeed, T94/97A ETF is much more stable in the ASQ state than in the HQ state, as for canonical ETFs.37,38 Thus, our mutations have produced an ETF with many biophysical properties in common with canonical ETFs, by replacing only two residues and preventing acquisition of the Bf-FAD.
Our 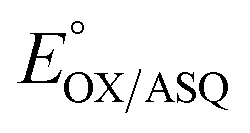 value of −7 ± 4 mV is in the range of those observed for ET-flavins of Bf-ETFs (−47 to +81 mV),3,14,16 but at the negative end of the range of values reported so far for canonical ETFs of +196 to −14.3,38 It was suggested that charge-transfer between reduced ET-FAD and oxidized Bf-FAD produced a long-wavelength signal in RpaETF.3 This, and proposed conformational interactions1,9 could influence the E°s of the ET-FAD depending on the status of the Bf-flavin.37 As predicted by the charge-transfer hypothesis, the long-wavelength signal is absent in the T94/97A ETF. Thus the higher
value of −7 ± 4 mV is in the range of those observed for ET-flavins of Bf-ETFs (−47 to +81 mV),3,14,16 but at the negative end of the range of values reported so far for canonical ETFs of +196 to −14.3,38 It was suggested that charge-transfer between reduced ET-FAD and oxidized Bf-FAD produced a long-wavelength signal in RpaETF.3 This, and proposed conformational interactions1,9 could influence the E°s of the ET-FAD depending on the status of the Bf-flavin.37 As predicted by the charge-transfer hypothesis, the long-wavelength signal is absent in the T94/97A ETF. Thus the higher 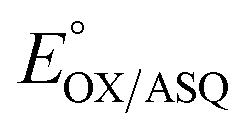 of T94/97A ETF compared to WT RpaETF could reflect effects of absence of a flavin in the domI/III site. In particular, the ASQ state of ET-FAD might be more favourable in T94/97A ETF due to there being one less phosphate nearby (≈13 Å) because the Bf-FAD is replaced by AMP.
of T94/97A ETF compared to WT RpaETF could reflect effects of absence of a flavin in the domI/III site. In particular, the ASQ state of ET-FAD might be more favourable in T94/97A ETF due to there being one less phosphate nearby (≈13 Å) because the Bf-FAD is replaced by AMP.
The qualitative agreement between the computed and observed optical signatures was notable, despite the overestimated transition energies also reported in other work.31,32 This is a testimony to the overall quality of the structural model produced39 and validates the calculations, although experimentally-determined crystal structures remain an important goal for further work on RpaETF. Our structural model likely limits the quality of our predictions, since ETFs are known to undergo conformational changes that significantly alter the exposure of the domII flavin9,40 and the conformation of T94/97A ETF observed in solution may differ from that of the crystallized AfeETF that served as the template for our model used in the calculations. Nevertheless the low accuracy of our spectral predictions is offset by very high location accuracy, and the calculations produced clearly-distinct predictions for the two sites, permitting attribution of individual spectra to specific flavins in the protein. Thus the calculations provide a second and independent test of which flavin is located where.
The calculated absorption spectrum of the domI/III flavin displays two resolved bands as is typical of free flavin. It is notable that the calculations correctly predict significant ellipticity for flavin bound in this site, which contrasts with the very weak CD observed and calculated for free flavin (ESI Fig. S6,† note vertical axis scale). Thus, QM/MM and our structural model replicated the ellipticity produced by the domI/III environment.
For the domII site, the calculations correctly predicted less resolved transitions and the overall shape of the absorbance band, as well as the considerably weaker CD.
Overall, our ability to model the effects on the flavin of different protein environments paves the way for more computational investigations of the significance of nearby amino acids and their protonation states in explaining the origin of the different optical signatures of flavins, and eventually their reactivities. We speculate that the location of the Bf-flavin between domains I and III, contributed by the two subunits of ETF, makes the Bf-flavin's activity amenable to modulation by movement of one domain relative to the other.
Experimental
Molecular biology
Mutations were introduced into the genes for wild type RpaETF: fixA and fixB in the plasmids pMCSG28 (carbenicillin resistant) and pMCSG21 (spectinomycin resistant), respectively.3 At the level of the protein, we adopt the letters S and L to identify the small and large subunit of the heterodimer, respectively, because these are homologous to the S and L subunits of other ETFs. However due to different gene orders in the fix operon the large FixAB subunit is FixB whereas the large ETF subunit is EtfA. Our proposal to adopt an ‘S’ and ‘L’ notation instead seeks to circumvent the confusion engendered by EtfAB's correspondence to FixBA. We retain the traditional notation for genes in order to retain correspondence with extensive annotation in databases.Back-to-back orientation primers encoding the desired amino acid substitutions were designed using NEB base changer (ESI Table S1†) and employed according to the vendor's recommendations to introduce the mutations via polymerase chain reactions using Q5 High Fidelity DNA polymerase (New England Biolabs, Ipswich MA). A pair of constructs encoding EtfS and EtfL were then used to transform competent Nico21(DE3) Escherichia coli (New England Biolabs, Ipswich MA), to permit protein expression.
Protein expression and purification
The T97A/94A ETF variant of RpaETF was grown in Terrific Broth medium supplemented with 20 mg L−1 riboflavin, 20 mg L−1 adenosine 5-monophosphate (AMP), 2 mM MgSO4, 100 μg mL−1 of carbenicillin and 100 μg mL−1 of spectinomycin. Cultures were grown at 37 °C, with shaking at 250 rpm until they reached an optical density (OD) of ≈1 and then cooled to 18–20 °C. Gene expression was induced with 0.1 mM IPTG and the cultures were grown for 20 hours at 18–20 °C. Cells were harvested by centrifugation at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900×g at 4 °C for 10 min.
900×g at 4 °C for 10 min.
Harvested cells (10 g L−1 culture) were washed with PBS (phosphate buffered saline at pH 7.4 containing 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4) and then lysed by resuspending in 80 mL of BugBuster (primary amine free, Millipore) supplemented with 1 mM AMP, 2 μl/80 mL of benzonase nuclease HC (250 U μL−1), 2 μL/80 mL of lysozyme (30 kU μL−1), 1 mM TCEP (tris(2-carboxyethyl)phosphine) and 1 mM AEBSF (4-(2-aminoethyl)benzenesulfonyl fluoride) hydrochloride protease inhibitor and incubated at 4 °C with stirring for 2 hours. The insoluble fraction was removed by centrifugation at 15000g for 45 min at 4 °C.
Clarified cell lysate was mixed with 1.5 mL of Co-nitrilotriacetate (NTA) resin (Thermo Fisher) that had been pre-equilibrated with 3 mL of equilibration buffer containing 20 mM Tris pH 7.4, 500 mM KCl, 10% w/v glycerol and 1 mM TCEP. After a binding interval of 30 min with gentle stirring at 4 °C, protein-bound resin was loaded into a column (2.5 × 30 cm) and washed with 20 bed volumes of wash buffer containing 20 mM Tris pH 7.4, 500 mM KCl, 10% w/v glycerol, 1 mM TCEP and 20 mM imidazole. Protein was eluted with 2 bed volumes of elution buffer containing 20 mM Tris pH 7.4, 500 mM KCl, 10% w/v glycerol, 1 mM TCEP and 100 mM imidazole. Eluate was subjected to buffer exchange by gel filtration on 10-DG desalting column (Bio Rad) in order to remove the imidazole and transfer the protein to the working buffer containing 20 mM Bis-Tris propane (pH 8.0), 200 mM KCl and 10% w/v glycerol.
Cofactor quantification
545 Da.3 400 μL of 28 μM ETF in working buffer was used per assay, in individual 1.5 mL microcentrifuge tubes wrapped in aluminium foil to exclude light and prevent photochemical transformation of cofactors. Cofactors were released by denaturing the ETF by heating at 100 °C for 10 min after which denatured protein was removed by centrifugation at 14000×g for 10 min after cooling the samples. Supernatant was transferred to a quartz cuvette and the optical spectrum was recorded on a HP 8453 spectrophotometer. Absorbance at 450 nm was used to determine the released flavin concentration (ε450 = 11.3 mM−1 cm−1).16 From this and FAD's ε260 of 35.9 mM−1 cm−1 we calculated the absorbance due to FAD at 260 nm. The excess A260 was used to determine the concentration of AMP (ε260 = 15.0 mM−1 cm−1).42
Circular dichroism
CD spectra were recorded using a JASCO J-800 series spectropolarimeter from 600 to 300 nm at 4 °C in a 1.0 cm path length quartz cuvette with the following parameters: scan speed = 100 nm min−1, bandwidth = 2.00 nm, scanning speed = 100 nm min−1, and three accumulations. The molar ellipticity was calculated using the equation, [θ] = θ/(c × l), in which θ is the ellipticity in millidegrees, c is the concentration in mM, and l is the cell path length in cm.000×g for 10 min after cooling. The supernatant was transferred to a microcentrifuge tube and diluted 10-fold with working buffer. The emission spectrum of the solution was recorded at 20 °C using a Thermo Fisher Lumina fluorimeter with the following parameters: excitation at λex = 450 nm, emission measured over λem = 480–600 nm, scan speed = 100 nm min−1, excitation slit 5 nm, emission slit 5 nm, gain: ‘high’, data interval: 0.5 nm, instrument zeroing conditions, emission shutter: closed. After collection of an initial spectrum, 2 μl of 6 mU phosphodiesterase solution was added (Abnova, cat no. P5263) to convert FAD into FMN and AMP, thereby releasing the flavin from adenine-mediated fluorescence quenching.20 Emission spectra were recorded each minute after mixing, until no further changes are observed using the same settings as given above.20 The maximum emission value was converted to an FMN concentration based on a calibration curve of emission amplitude at 524 nm vs. [FMN], generated using authentic FMN (14H0610, 70% purity, Sigma).
Reductive titration of T94/97A RpaETF by NADH and sodium dithionite
Reductive titrations were performed in inert atmosphere, monitored using a HP 8452A spectrophotometer (Agilent technologies) equipped with an OLIS controller, inside a glove box (Belle Technology, Waymouth, UK), using a 1 cm path length self-masking quartz cuvette at room temperature. ETF was reduced by small aliquots amounting to 2 μM NADH each after dilution into the reaction (ε340 = 6.22 mM−1 cm−1)16 or 5.6 μM sodium dithionite (ε315 = 8.04 mM−1 cm−1).43 This low concentration provides sufficient driving force to fully reduce the FAD at the pHs of 8 and 9 that we used.44Reduction midpoint potential determination
Potentiometric titrations were also monitored optically using a HP 8452A spectrophotometer (Agilent technologies) equipped with an OLIS controller in an inert atmosphere afforded by a glove box (Belle Technology, Waymouth, UK) using a 1 cm path length self-masking quartz cuvette at room temperature. Xanthine oxidase in combination with xanthine was used to provide a slow continuous delivery of reducing equivalents.45 The reaction mixture contained 400 μM xanthine, 5 μM methylviologen, 16.2 μM T94/97A-RpaETF, and 3 μM methylene blue (MB, as a reference dye, E° = −19.0 mV, pH 8.0) in working buffer. The reaction was initiated by addition of 10 nM xanthine oxidase, and spectra were recorded every 1 min. The reduction midpoint potential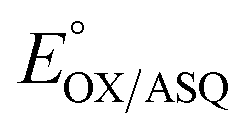 of the reduction of OX FAD to ASQ was calculated by relating the extent of reduction of FAD with the extent of concurrent reduction of the reference dye. Conversion of OX to ASQ was quantified based on absorbance changes at 388 nm, the dye isosbestic, while the reduction of the MB dye was quantified based on loss of absorbance at 664 nm.
of the reduction of OX FAD to ASQ was calculated by relating the extent of reduction of FAD with the extent of concurrent reduction of the reference dye. Conversion of OX to ASQ was quantified based on absorbance changes at 388 nm, the dye isosbestic, while the reduction of the MB dye was quantified based on loss of absorbance at 664 nm.
The point in the titration corresponding to the maximum ASQ concentration was identified and treated as the endpoint of the first 1-e− reduction, and assigned to full conversion to ASQ based on the agreement between the shape of the spectrum and that of authentic samples of 100% ASQ. Each spectrum up to that point in the titration was converted to a difference spectrum by subtracting the endpoint spectrum, to reveal the remaining OX population (Fig. 5). The first point in the titration was not 100% OX, as the RpaETF undergoes slow spontaneous reduction in the glove box. However the amount of ASQ in the spectrum could be estimated by subtracting a weighted spectrum of purely ASQ RpaETF in such a way as to produce a flat zero baseline at wavelengths > 500 nm where ASQ is positive but OX generally does not absorb. The weighting required to accomplish this revealed what fraction, f, of the total starting absorbance corresponded to ASQ, and thus the magnitude of the residual population, 1 − f, that underwent reduction in the course of the measurement. We obtained 0.05 < f <0 .10 in three titrations. The difference spectra were thus interpreted to reflect the reaction of the (1 − f) portion of the sample and for the example of the midpoint in the change, f + (1 − f)/2 = 0.5 + f/2 of the sample is ASQ while 0.5 − f/2 is OX. Absorbance at 388 nm (dye isosbestic) was used to find the absorbance change due to reduction of FAD and thereby to calculate to calculate the fraction of OX converted to ASQ. [FADOX]/[FADASQ] at each point in the titration were calculated from the fractional populations. A664 was used to calculate the extent to which oxidized dye, DYEOX, was converted to reduced dye, DYERED. The ratio of oxidized to reduced dye along with that of the FAD were employed in the Nernst equation,
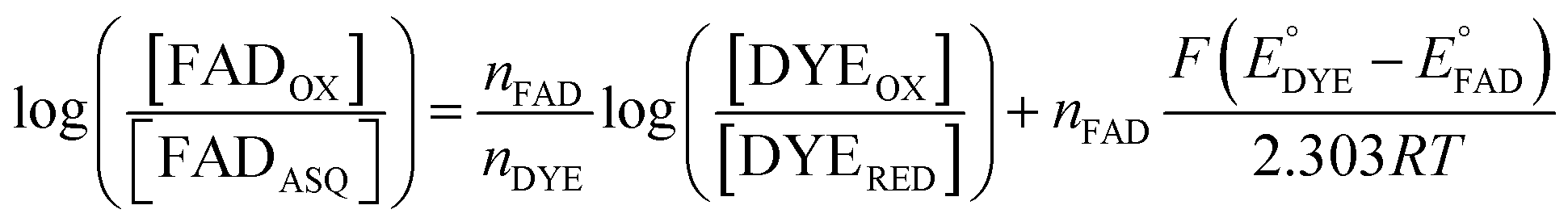 | (1) |
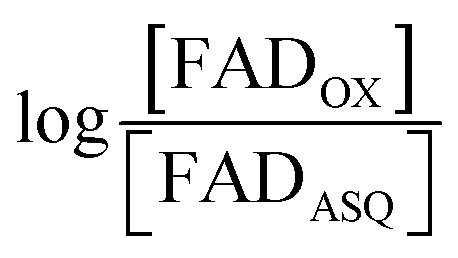 versus
versus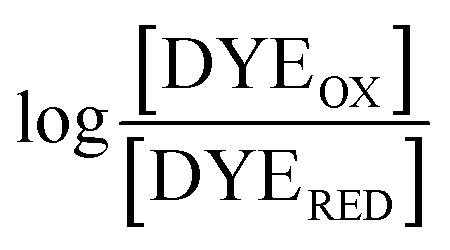 , was used to determine nFAD from the known value of nDYE = 2.26 The intercept of the plot then permits calculation of
, was used to determine nFAD from the known value of nDYE = 2.26 The intercept of the plot then permits calculation of 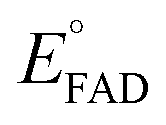 for the ETF FADOX/ASQ.
for the ETF FADOX/ASQ.
Computation
Models for the structure of RpaETF were produced using Swiss Modeler46 based on the structure of AfeETF 4KPU.pdb.1 Sequence identities are 39% (57% similar) for EtfL and 44% (63% similar) for EtfS. The model RpaETF structure accommodated the FAD cofactors as present in the AfeETF structure with only a few small clashes with amino acid side chains. These and a few clashes between side chains at the interfaces between chains L and S were resolved by energy minimization as implemented in Chimera.11 Protons were added in Chimera, and each His side chain was examined for interactions with neighbouring side chains to identify the most reasonable choice of protonation state.The structure of the protein was minimized in a cube of explicit water (95 Å edge length) and allowed to relax via molecular dynamics as the temperature was increased to 300 K (3 ps) and then equilibrated at 300 K (5 ps) using the CHARMM36 force field.47 We implemented a quantum description of the active site flavin head group and a selection of the most closely-interacting protein residues (ESI Fig. S5†) in a QM/MM27 optimization executed by Chemshell,48 with QM energies and gradients furnished by Turbomole49 without application of any symmetry constraints, as described previously.28 All protein atoms, those of both FADs and atoms of waters within a 40 Å radius sphere centered at N5 of the FAD of interest (ESI Fig. S7†) were subjected to geometry optimization in which the QM fragment was described with density functional theory (DFT) using the B3LYP functional50–52 and either the 6-31G(d) or the 6-31++G(d,p) basis,53 while the balance of the atoms were treated using MM with the CHARMM36 empirical force field.47 Coupling between the QM and MM regions was accomplished via electrostatic embedding using the charge-shift scheme.54 For atoms in the QM region with covalent bonds to atoms outside, the latter were replaced with hydrogen link-atoms, a charge shift scheme was applied at the QM/MM boundary55 and van der Waals interactions between QM atoms and MM atoms were treated using force field parameters.
The resulting atomic coordinates and array of point charges embodying an electrostatic description of the protein environment were then used in quantum chemical calculations of molecular properties and spectroscopic observables using Gaussian 16.56 For adjacent residues of unknown protonation state, both possibilities were computed, and the results remain consistent with experimental spectra after shifting the calculated spectra to align them with the λmax of the band near 450 nm (ESI Fig. S8†). Results of time-dependent density functional theory (TD-DFT)57–59 and symmetry-adapted cluster – configuration interaction SAC-CI60,61 calculations executed in Gaussian were compared and TD-DFT was found to provide better agreement with experimental transition energies for our system, using B3LYP50–52 and 6-31G* (also see ref. 62). To test the robustness of TD-DFT results, calculations were repeated with two sizes of basis set commonly used for these calculations:63 6-31G* and 6-31++G**31,64–66 (ESI Fig. S9 and S4†), and four different functionals (ESI Fig. S10†). B3LYP50–52 is commonly-used and widely accepted for flavins and other pigments,63,67–69 but other functionals recommended for such calculations include PBE0,70 wB97xD,71 and LC-wHPBE.72 We compared these four with the 6-31++G(d,p) basis, and found that B3LYP affords the best description of lumiflavin (ESI Fig. S10†). Our results agree with those of a particularly insightful comparison of computational methods for simulating the vibrationally-resolved electronic spectrum of three flavin variants, that found that B3LYP's overall performance in replicating the spectrum of riboflavin was on par with more elaborate methods and the other functionals tried.31
The computed excitation energies and oscillator strengths were used to calculate optical spectra for comparison with experimental data. TD-DFT derived vertical transition energies were too high by 0.10 to 0.55 eV, consistent with published assessments of this method on comparable molecules.29,30 However the deviations from experiment were systematic, so that shifts in the energy of a transition as a function of substitution were well reproduced.30,73 The TD-DFT method is known to be unsuitable for transitions with a large amount of charge transfer character74 however the transitions in the visible range did not display much CT character,62 for either of our flavins.
The apparent blue shift (hypsochromic shift) of the calculated vertical transition energies may also reflect that neither the experimental absorption maximum nor the calculations reflect a simple 0–0 excitation,31,75,76 as room temperature experiments will include additional features due to population of vibrational substates of the electronic ground state. However even when vibronic structure is taken into account,31,75 Ai et al. obtained energy offsets of −0.47 eV between computed and observed optical band positions (450 nm band).32 Thus treatment of vibrational structure is not expected to fully resolve our discrepancies, and will be assessed in future work. Consequently, we do not draw conclusions from the individual transition energy values, but restrict ourselves to interpreting shifts in the transitions resulting from modification of the environment.
Calculated transition energies and transition moment amplitudes were used to simulate absorption spectra with Gaussian lines 0.5 eV wide at half height (i.e. half-bandwidth at 1/e peak height of Δσ = 0.3 eV) for each transition up to a transition energy of σ = 6.2 eV (wavelengths ≥ 200 nm) for smaller QM zones, or up to σ = 5 eV (λ ≥ 250 nm) for larger QM zones, using eqn (2) where σi is the energy of the ith transition in eV, fi is the oscillator strength of the ith transition, N, c, me have their usual meanings and ε1(σ) is the extinction coefficient due to the ith transition as a function of photon energy σ.77 Plots of total ε vs. wavelength were obtained by summing the contributions from all transitions with oscillator strengths ≥ 1% of the maximum value obtained for that calculation, and converting transition energies σ to wavelengths λ using λ = hc/σ where h is Planck's constant.
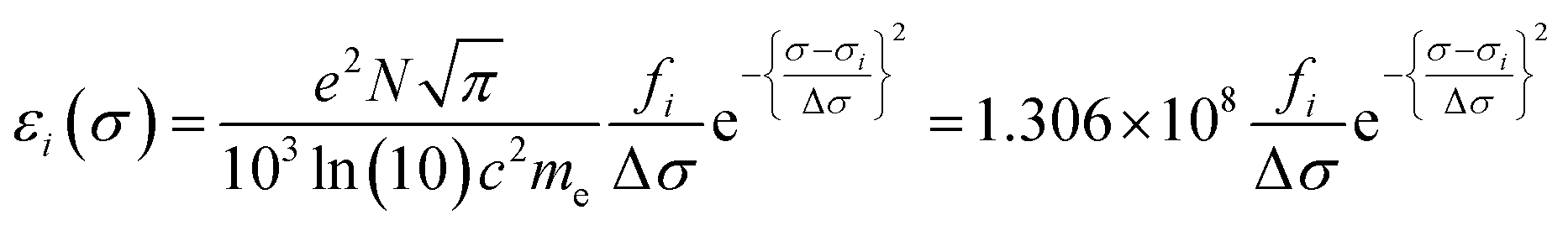 | (2a) |
 | (2b) |
Calculated rotatory strengths obtained using dipole-length gauge78,79 were used to simulate the electronic CD spectra within the same energy ranges and line width using eqn (3), where Ri is the rotatory strength of the ith transition (in 10−40 erg-esu-cm/Gauss) and Δεi(σ) is the optical activity (difference extinction coefficient) as a function of photon energy σ.63,80 Plots of Δε vs. wavelength were obtained by summing the contributions from all transitions with rotatory strengths ≥ 1% of the maximum obtained for that calculation, and converting transition energies σ to wavelengths λ as above.80,81
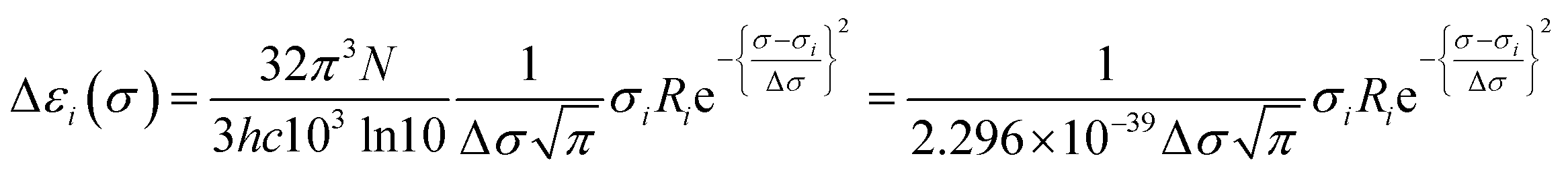 | (3) |
Conclusions
Complementary spectroscopy, ab initio calculations and assessment of chemical reactivity were used to determine which of RpaETF's flavin-based activities were retained after mutagenic disruption of FAD binding in the site between domains I and III. Thus we established that the ET-FAD shared with canonical ETFs is retained in domain II. It follows that the Bf-flavin is the one bound at the interface between domains I and III, in agreement with the accepted model. Our hypothesis-driven tests of the model furthermore demonstrate the ability of QM/MM calculations to reproduce distinctions between the two flavin sites, paving the way to understanding what features of the flavins' environments are responsible for the flavins' sharply contrasting activities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported as part of the Biological and Electron Transfer and Catalysis (BETCy) EFRC, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Basic Energy Sciences under Award DE-SC0012518, the National Sciences Foundation, Chemistry of Life Processes CHE-1808433, and the German Research Foundation to the ‘Unifying Concepts in Catalysis” Cluster of Excellence UniCat-EXC314. Additional support was provided by the Research Challenge Trust fund of Kentucky (NMR) and the Technische Universität-Berlin (MAM, PH and AFM). AFM thanks the Odenwaldstraße Society for hospitality while executing calculations, Mssrs. S. Kraus and J. Krauss for excellent IT support and Prof. E. C. Glazer for access to a spectropolarimeter.Notes and references
- N. P. Chowdhury, A. M. Mowafy, J. K. Demmer, V. Upadhyay, S. Koelzer, E. Jayamani, J. Kahnt, M. Hornung, U. Demmer, U. Ermler and W. Buckel, J. Biol. Chem., 2014, 289, 5145–5157 CrossRef CAS PubMed.
- D. L. Roberts, D. Salazar, J. P. Fulmer, F. E. Frerman and J. J. Kim, Biochemistry, 1999, 38, 1977–1989 CrossRef CAS PubMed.
- H. D. Duan, C. E. Lubner, M. Tokmina-Lukaszewska, G. H. Gauss, B. Bothner, P. W. King, J. W. Peters and A. F. Miller, J. Biol. Chem., 2018, 293, 4688–4701 CrossRef CAS PubMed.
- A. M. Garcia Costas, S. Poudel, A.-F. Miller, G. J. Schut, R. N. Ledbetter, K. Fixen, L. C. Seefeldt, M. W. Adams, C. S. Harwood, E. S. Boyd and J. W. Peters, J. Bacteriol., 2017, 199(21), e00440-17 CrossRef PubMed.
- H. S. Toogood, D. Leys and N. S. Scrutton, FEBS J., 2007, 274, 5481–5504 CrossRef CAS PubMed.
- S. Ghisla and C. Thorpe, Eur. J. Biochem., 2004, 271, 494–508 CrossRef CAS PubMed.
- K. L. Crane and H. Beinert, J. Biol. Chem., 1956, 218, 717–731 Search PubMed.
- D. L. Roberts, F. E. Frerman and J. J. Kim, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14355–14360 CrossRef CAS PubMed.
- J. K. Demmer, N. P. Chowdhury, T. Selmer, U. Ermler and W. Buckel, Nat. Commun., 2017, 8 CAS.
- N. J. Watmough and F. E. Frerman, Biochim. Biophys. Acta, 2010, 1797, 1910–1916 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- R. N. Ledbetter, A. M. Garcia Costas, C. E. Lubner, D. E. Mulder, M. Tokmina-Lukaszewska, J. H. Artz, A. Patterson, T. S. Magnuson, Z. J. Jay, H. D. Duan, J. Miller, M. H. Plunkett, J. P. Hoben, B. M. Barney, R. P. Carlson, A.-F. Miller, B. Bothner, P. W. King, J. W. Peters and L. C. Seefeldt, Biochemistry, 2017, 56, 4177–4190 CrossRef CAS PubMed.
- W. Buckel and R. K. Thauer, Chem. Rev., 2018, 118 Search PubMed.
- G. J. Schut, N. R. Raseek, M. Tokmina-Lukaszewska, D. E. Mulder, D. M. N. Nguyen, G. L. Lipscomb, J. P. Hoben, A. Patterson, C. E. Lubner, P. W. King, J. W. Peters, B. Bothner, A. F. Miller and M. W. W. Adams, J. Biol. Chem., 2019, 293, 3271–3283 CrossRef PubMed.
- J. M. Chan, J. Christiansen, D. R. Dean and L. C. Seefeldt, Biochemistry, 1999, 38, 5779–5785 CrossRef CAS PubMed.
- K. Sato, Y. Nishina and K. Shiga, J. Biochem., 2013, 153, 565–572 CrossRef CAS PubMed.
- K. Sato, Y. Nishina and K. Shiga, J. Biochem., 2003, 134, 719–729 CrossRef CAS PubMed.
- K. Sato, Y. Nishina and K. Shiga, J. Biochem., 1993, 114, 215–222 CrossRef CAS PubMed.
- K. Sato, Y. Nishina and K. Shiga, J. Biochem., 1992, 112, 804–810 CrossRef CAS PubMed.
- A. Aliverti, B. Curti and M. A. Vanoni, in Methods in Molecular Biology, ed. S. K. Chapman and G. A. Reid, Humana Press Inc., Totowa, NJ, 1999, vol. 131, pp. 9–23 Search PubMed.
- P. Augustin, M. Toplak, K. Fuchs, E. C. Gerstmann, R. Prassl, A. Winkler and P. Macheroux, J. Biol. Chem., 2018, 293, 2829–2840 CrossRef CAS PubMed.
- K. Yorita, T. Matsuoka, H. Masaki and V. Massey, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13039–13044 CrossRef CAS PubMed.
- A. W. Munro, S. M. Kelly and N. C. Price, in Flavoprotein Protocols, ed. S. K. Chapman and G. A. Reid, Humana Press, Totowa, NJ, 1999, pp. 111–123, DOI:10.1385/1-59259-266-x:111.
- G. Tollin and D. E. Edmondson, Biochemistry, 1971, 10, 113–124 CrossRef CAS PubMed.
- C. E. Lubner, D. P. Jennings, D. W. Mulder, G. J. Schut, O. A. Zadvornyy, J. P. Hoben, M. Tokmina-Lukaszewska, L. Berry, D. M. Nguyen, G. L. Lipscomb, B. Bothner, A. K. Jones, A. F. Miller, P. W. King, M. W. W. Adams and J. W. Peters, Nat. Chem. Biol., 2017, 13, 655–659 CrossRef CAS PubMed.
- B. Norden and F. Tjerneld, Biopolymers, 1982, 21, 1713–1734 CrossRef CAS PubMed.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed. Engl., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- M. A. Mroginski, F. Mark, W. Thiel and P. Hildebrandt, Biophys. J., 2007, 93, 1885–1894 CrossRef CAS PubMed.
- D. Jacquemin, V. Wathelet, E. A. Perpete and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef CAS PubMed.
- J. Fabian, Dyes Pigm., 2010, 84, 36–53 CrossRef CAS.
- B. Karasulu, J. P. Götze and W. Thiel, J. Chem. Theory Comput., 2014, 10, 5549–5566 CrossRef CAS PubMed.
- Y.-J. Ai, G. Tian, R.-Z. Liao, Q. Zhang, W.-H. Fang and Y. Luo, ChemPhysChem, 2011, 12, 2899–2902 CrossRef CAS PubMed.
- K. B. Pechter, L. Gallagher, H. Pyles, C. S. Manoil and C. S. Harwood, J. Bacteriol., 2015, 198, 867–876 CrossRef PubMed.
- N. P. Chowdhury, J. Kahnt and W. Buckel, FEBS J., 2015, 282, 3149–3160 CrossRef CAS PubMed.
- C.-Y. Yang, PhD thesis, Ohio State University, 2006.
- T. C. Lehman and C. Thorpe, Arch. Biochem. Biophys., 1992, 292, 594–599 CrossRef CAS PubMed.
- F. Talfournier, A. W. Munro, J. Basran, M. J. Sutcliffe, S. Daff, S. K. Chapman and N. S. Scrutton, J. Biol. Chem., 2001, 276, 20190–20196 CrossRef CAS PubMed.
- C. M. Byron, M. T. Stankovich, M. Husain and V. L. Davidson, Biochemistry, 1989, 28, 8582–8587 CrossRef CAS PubMed.
- K. Arnold, L. Bordoli, J. Kopp and T. Schwede, Bioinformatics, 2006, 22, 195–201 CrossRef CAS PubMed.
- J. K. Demmer, J. Bertsch, C. Oppinger, H. Wohlers, K. Kayastha, U. Demmer, U. Ermler and V. Muller, FEBS Lett., 2018, 592, 332–342 CrossRef CAS PubMed.
- H.-K. Ku, H.-M. Lim, K.-H. Oh, H.-J. Yang, J.-S. Jeong and S.-K. Kim, Anal. Biochem., 2013, 434, 178–180 CrossRef CAS PubMed.
- K. Sato, Y. Nishima and K. Shiga, J. Biochem., 1997, 121, 477–486 CrossRef CAS PubMed.
- F. K. Yousafzai and R. R. Eady, J. Biol. Chem., 2002, 277, 34067–34073 CrossRef CAS PubMed.
- S. G. Mayhew, Eur. J. Biochem., 1978, 85, 535–547 CrossRef CAS PubMed.
- V. Massey, in Flavins and flavoproteins, ed. B. Curti, S. Ronchi and G. Zanetti, Walter de Gruyer, Berlin, 1991, pp. 59–66 Search PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2014, 46, W296–W303 CrossRef PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell Jr, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, J. Mol. Struct.: THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- TURBOMOLE 6.3, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, http://www.turbomole.com/.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS.
- D. Bakowies and W. Thiel, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef CAS.
- A. H. de Vries, P. Sherwood, S. J. Collins, A. M. Rigby, M. Rigutto and G. J. Kramer, J. Phys. Chem. B, 1999, 103, 6133–6141 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- T. Nakajima and H. Nakatsuji, Chem. Phys. Lett., 1997, 280, 79–84 CrossRef CAS.
- J. Hasegawa, S. Bureekaew and H. Nakasuji, J. Photochem. Photobiol., A, 2007, 189, 205–210 CrossRef CAS.
- O. Falklöf, B. Durbeej and P. Norman, J. Phys. Chem., 2015, 119 Search PubMed.
- A. E. Nugroho and H. Morita, J. Nat. Med., 2014, 68, 1–14 CrossRef CAS PubMed.
- M. Francl, W. Pietro, W. J. Hehre, J. Binkley, M. A. Gordon, D. DeFrees and J. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- V. Barone, A. Baiardi and J. Bloino, Chirality, 2014, 26, 588–600 CrossRef CAS PubMed.
- I. M. M. Wijaya, T. Domratcheva, T. Iwata, E. D. Getzoff and H. Kandori, J. Am. Chem. Soc., 2016, 138, 4368–4376 CrossRef CAS PubMed.
- O. Dmitrenko, C. Thorpe and R. D. Bach, J. Phys. Chem. B, 2003, 107, 13229–13236 CrossRef CAS.
- A. Vdovin, A. Slenczka and B. Dick, Chem. Phys., 2013, 422, 195–203 CrossRef CAS.
- S. Salzmann and C. M. Marian, Chem. Phys. Lett., 2008, 463, 400–404 CrossRef CAS.
- D. Jacquemin, M. Bouhy and E. A. Perpête, J. Chem. Phys., 2006, 124, 1–9 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- D. Jacquemin, E. A. Perpete, G. Scalmani, M. J. Frisch, K. Kobayashi and C. Adamo, J. Chem. Phys., 2007, 126, 1–12 Search PubMed.
- M. Guillaume, B. Champagne and F. Zutterman, J. Phys. Chem., 2006, 110, 13007–13013 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- B. Klaumünzer, D. Kröner and P. Saalfrank, J. Phys. Chem. B, 2010, 114, 10826–10834 CrossRef PubMed.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- Gaussian, Creating UV/Visible plots from the results of excited states calculations, http://gaussian.com/uvvisplot/, accessed 23 May 2018, 2018 Search PubMed.
- C. Diedrich and S. Grimme, J. Phys. Chem. A, 2003, 107, 2524–2539 CrossRef CAS.
- R. Pecul, K. Ruud and T. Helgaker, Chem. Phys. Lett., 2004, 388, 110–119 CrossRef.
- P. J. Stephens and N. Harada, Chirality, 2010, 22, 229–233 CAS.
- T. D. Crawford, M. C. Tam and M. L. Abrams, J. Phys. Chem. A, 2007, 111, 12057–12068 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: One table and 10 figures documenting genetic constructs, spectroscopic controls and details of the computations. See DOI: 10.1039/c9sc00942f |
| This journal is © The Royal Society of Chemistry 2019 |