
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUncovering two kinetic factors in the controlled growth of topologically distinct core–shell metal–organic frameworks†
Fang
Wang
ac,
Sanfeng
He
a,
Hongliang
Wang
a,
Songwei
Zhang
a,
Chunhui
Wu
a,
Haoxin
Huang
a,
Yuqian
Pang
a,
Chia-Kuang
Tsung
b and
Tao
Li
 *a
*a
aSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: litao1@shanghaitech.edu.cn
bDepartment of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, USA
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 27th June 2019
Abstract
Constructing core–shell metal–organic frameworks (MOFs) based on two topologically distinct MOFs is a great way to increase MOF material complexity and explore their new functions. However, such a nucleation process is energetically less favored compared to epitaxial growth due to mismatched unit cell parameters. Here, two kinetic factors, nucleation kinetics and dissolution kinetics, are revealed to be two key factors in overcoming this challenge. Through kinetic control, we demonstrate the growth of 4 types of Zr/Hf-MOF shells uniformly and contiguously on 7 different core MOFs including ZIF-8, an acid labile core. Taking advantage of the modular synthesis of Zr-MOFs, we demonstrate that post-synthetic covalent surface modification on a non-functionalizable MOF surface can be made possible through core–shell construction. We also demonstrated that the size selective catalytic behavior can be systematically tuned through changing either the ligand length or ligand functionality.
Introduction
Hierarchical arrangement of materials into core–shell architectures is a widely implemented strategy to endow materials with new functions. A widely accepted notation for a core–shell material is A@B where B is the shell grown on the core material A.1–3 In many cases, the core possesses the main physical and chemical properties such as localized surface plasmon resonance,4 magnetism,5 photoluminescence,6 catalytic activity,7etc., whereas the shell is responsible for communicating with surrounding media thereby endowing the composite material with good dispersity, stability, targeting specificity, bio-compatibility, etc.5,8–10Metal–organic frameworks (MOFs) are a broad class of porous materials constructed through hierarchical molecular assembly of metal building units and organic linkers. Their high designability paves the way towards many potential applications.11–19 To increase the structural complexity of a MOF, a common method is to combine different functional moieties in a single crystalline phase as exemplified by the concept of “heterogeneity within order”.20–25 Alternatively, hierarchical assembly of MOFs with polymers,26,27 metal nanoparticles,28,29 metal oxides,30 carbon materials,31 covalent organic frameworks (COFs),32 enzymes,33etc. in a core–shell fashion has been repeatedly demonstrated to further extend the functions and utilities of MOFs to a new level.
Core–shell MOFs are a special group of hierarchical MOF composites because the shell layer can enrich the functions of the composite from at least the following three aspects: (1) sort larger guest molecules through a molecular sieving mechanism for selective adsorption or catalysis;34–36 (2) serve as a protective layer to mitigate the inherent instability of the core MOF;34 (3) serve as a transition layer for the covalent attachment of small molecules or polymers to bridge the core MOF and its surroundings.37 Recently, a new concept, domain building blocks (DBBs), put forward by Rosi et al. suggested that multilayer core–shell construction can effectively diversify the functions of MOF materials.38 However, the major bottleneck to achieve such complexity is the lack of synthetic tools. Most of the core–shell MOFs reported to date rely on epitaxial growth which greatly constrains the range of material selection.29,39–44 Recently, Tsung et al. and Li et al. successively reported surfactant mediated and random copolymer (RCP) mediated growth of uniform ZIF-8 shells on various core MOFs.45,46 For large single crystals, Zhou et al. unveiled the principles for the construction of a MOF-on-MOF structure using retrosynthetic principles.47 However, all these examples only allow the growth of shell MOFs with chemically more labile bonds than that of the core MOFs. Using robust Zr MOFs as the shell, although highly desired, has only been reported by Zhou et al. through a kinetically controlled one-step synthesis of large PCN-222@UiO-67 crystals48 and by Tan et al. through Ostwald ripening-mediated growth of UiO-66@PCN-222.49 Yet, the generalizability of this method to other MOFs especially chemically more labile MOFs remains unknown.
Here, we reveal two kinetic factors that are responsible for the construction of uniform and contiguous Zr/Hf-MOF shells on both robust and labile core MOFs. First, we found that boosting the nucleation kinetics by raising the precursor concentration is critical for the rapid formation of densely packed small MOF nuclei around the core MOF. Continued growth of these nuclei sealed the gaps among crystallites resulting in dense and contiguous Zr/Hf-MOF shells grown on a series of robust core MOFs including MOF-801, MIL-96(Al), MIL-101(Cr), UiO-66, MIL-53(Cr) and Ni-MOF-74. To overcome the seemingly impossible task of growing Zr-MOF shells on an acid labile core MOF such as ZIF-8, lowering the dissolution kinetics of the core MOF is key. By reducing the quantity of the Zr-MOF precursors, the dissolution of ZIF-8 was maintained at a very low rate thereby allowing the growth of a dense Zr-MOF shell without compromising the integrity of the ZIF-8 core. By using an amine functionalized Zr-MOF shell, covalent surface modification on a non-functionalizable MOF is made possible through core–shell construction. We also demonstrated that size selective catalysis can be precisely tuned by both ligand substitution and functionalization in the shell MOF.
Results and discussion
Nucleation kinetics governed growth of UiO-66 on MIL-101(Cr)
The nucleation of one MOF on another topologically distinct MOF surface is a heterogeneous nucleation process. According to the classic nucleation theory, the critical energy barrier of a heterogeneous nucleation process is equal to the product of the homogeneous nucleation energy barrier and a function of the contact angle (CA) θ (eqn (1)). Since the CA term is always smaller than 1, heterogeneous nucleation is energetically favored over homogeneous nucleation.50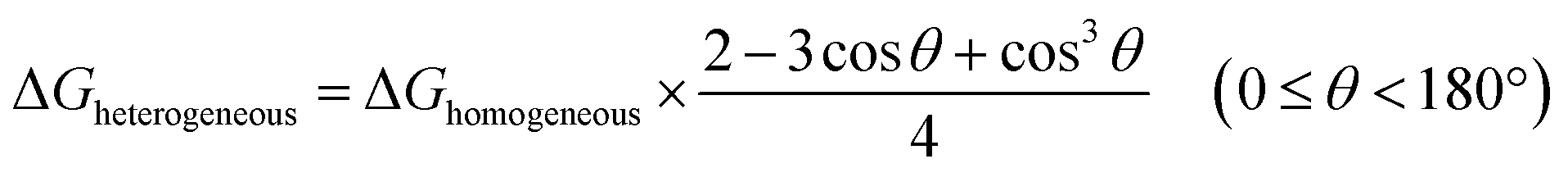 | (1) |
The tendency for this to occur depends on (1) the nucleation kinetics and (2) the surface properties of the core MOF. However, compared to epitaxial growth between two structurally identical MOF analogues, it is energetically less favorable due to mismatched unit cell parameters. To achieve a uniform defect-free MOF shell through non-epitaxial growth, one strategy is to modify the core MOF surface with “nucleation friendly” small molecules or polymers to lower the nucleation energy barrier as demonstrated by Tsung et al. and Li et al. in their efforts to grow a dense ZIF-8 shell on various core MOFs.45,46 Alternatively, increasing the monomer concentration can simultaneously boost the nucleation kinetics and reduce the critical nucleus size to reach the same goal. To verify this hypothesis, a robust core MOF, MIL-101(Cr), was selected for the attempted growth of UiO-66.
MIL-101(Cr) was synthesized according to the method reported by Zhao et al. with slight modification.51 The crystallinity of the MOF was confirmed using both a transmission electron microscope (TEM) image and powder X-ray diffraction (PXRD) patterns (Fig. S1 and S2†). The average particle size is 300 ± 35 nm as determined using a TEM. To investigate the effect of concentration on the nucleation kinetics of UiO-66, MIL-101(Cr) crystals were immersed in three dimethylformamide (DMF) solutions containing 25, 50, and 100 mM of ZrCl4 and terephthalic acid (H2BDC) and heated to 120 °C. After 10 min of growth, a smooth layer of amorphous deposits was observed on the surface of all three MIL-101(Cr) samples as shown in the TEM images (Fig. 1i, v, and ix). Energy dispersion X-ray spectroscopy (EDS) mapping results for Fig. ix reveal that this deposition layer contains Zr (Fig. S3 and S4†). However, no ordering was observed according to the PXRD patterns (Fig. S5†). Therefore, this layer is likely to be an amorphous Zr–ligand complex (AZC). At 20 min, both the 50 and 100 mM grown samples show roughening on the particle surface due to the nucleation of UiO-66. Their PXRD patterns also confirmed such transformations as evidenced by the emergence of the (111) facet diffraction peak of UiO-66 at 7.38° (Fig. 1C). In contrast, the sample grown in 25 mM solution did not start nucleation until 40 min, much slower than the other two samples (Fig. 1iii and S6†). Moreover, the size of the nuclei is also considerably larger leading to a lower surface coverage on MIL-101(Cr). After 1 h, the MIL-101(Cr)@UiO-66 core–shell structure formed in the 100 mM solution exhibit well-defined shell thickness and integrity. Comparably, 25 mM solution only resulted in partial coverage of large UiO-66 crystallites on the core MOF. The sample grown in the 50 mM solution also shows near full coverage of UiO-66 but the crystallite size is relatively large resulting in a rough surface texture (Fig. 1viii). The EDS mapping results of Fig. 1xii reveal the expected layered Zr and Cr distribution confirming the core–shell morphology of MIL-101(Cr)@UiO-66 particle (Fig. 1B). These results suggest that rapid nucleation is critical for the formation of a dense UiO-66 shell on MIL-101(Cr). In addition, despite the rapid nucleation, the shell growth process is relatively slow but steady which allows continuous increase of UiO-66 shell thickness from ∼13 nm at 0.5 h to 69 nm at 2 h (Fig. S7†).
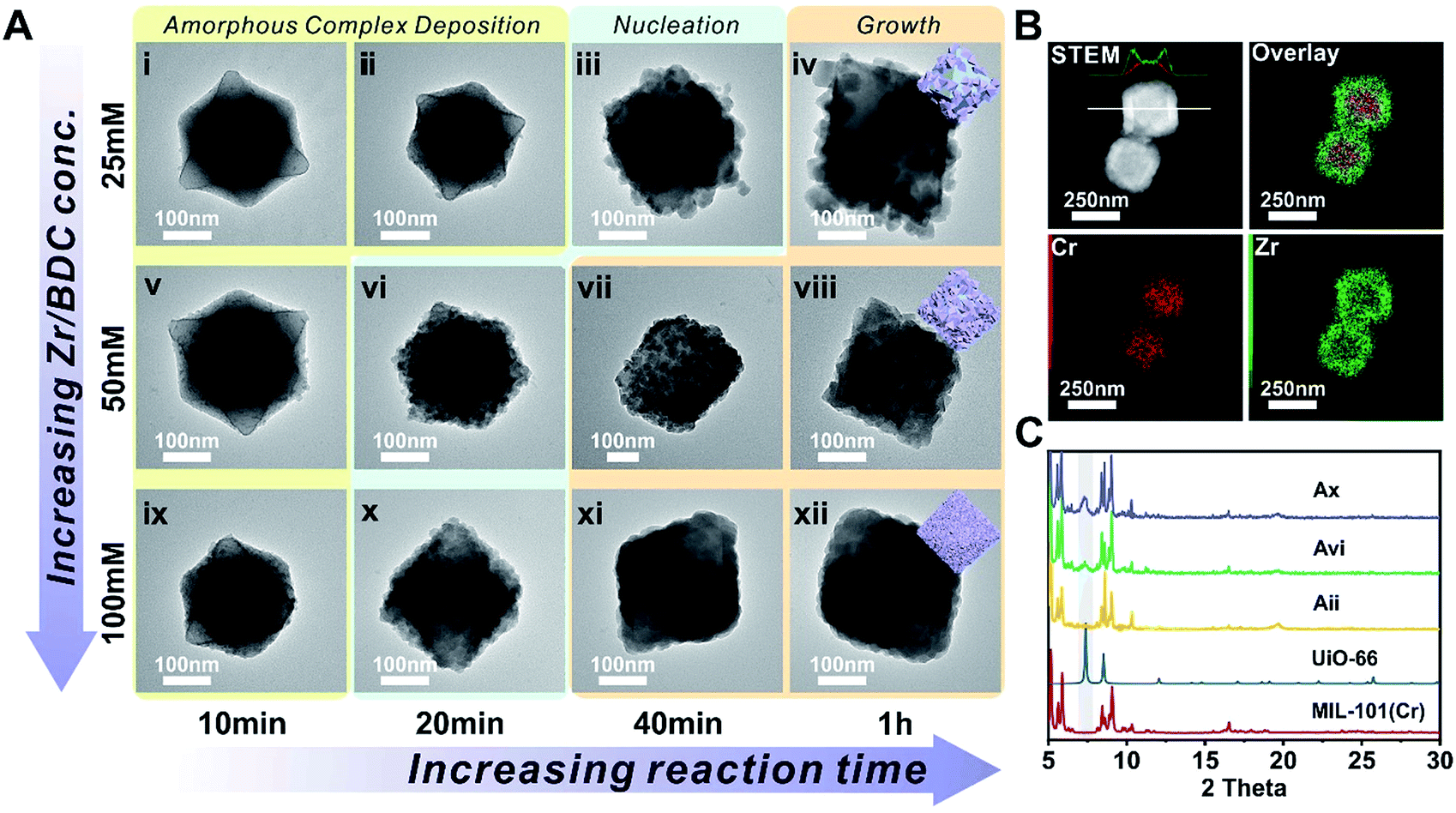 | ||
| Fig. 1 (A) TEM images of MIL-101(Cr)@UiO-66 at different growth stages; (B) STEM and EDS elemental mapping images of MIL-101(Cr)@UiO-66 (Axii); (C) PXRD patterns of MIL-101(Cr) (red, simulated), UiO-66 (blue, simulated), and MIL-101(Cr)@UiO-66 (Aii, yellow; Avi, green; Ax, purple). The grey zone highlights the characteristic diffraction peak of the (111) facet of UiO-66. | ||
Growth of Zr/Hf-MOF shells on various core MOFs
Next, we extended this strategy to the growth of a series of Zr/Hf-MOF shells on various core MOFs. Fig. 2A–H show the TEM images of MOF-801@UiO-66, MIL-96(Al)@UiO-66-NH2, MIL-53(Cr)@UiO-66, MIL-101(Cr)@UiO-66(Hf), MIL-101(Cr)@UiO-66-NH2, Ni-MOF-74@UiO-66-NH2, UiO-66@MOF-801 and MIL-101(Cr)@MOF-801, respectively. All the samples exhibit dense and uniform shell layers with thickness in the range of 10 to 82 nm. The PXRD patterns of these core–shell samples all clearly exhibit combinations of two crystalline phases (Fig. S8 to S15†).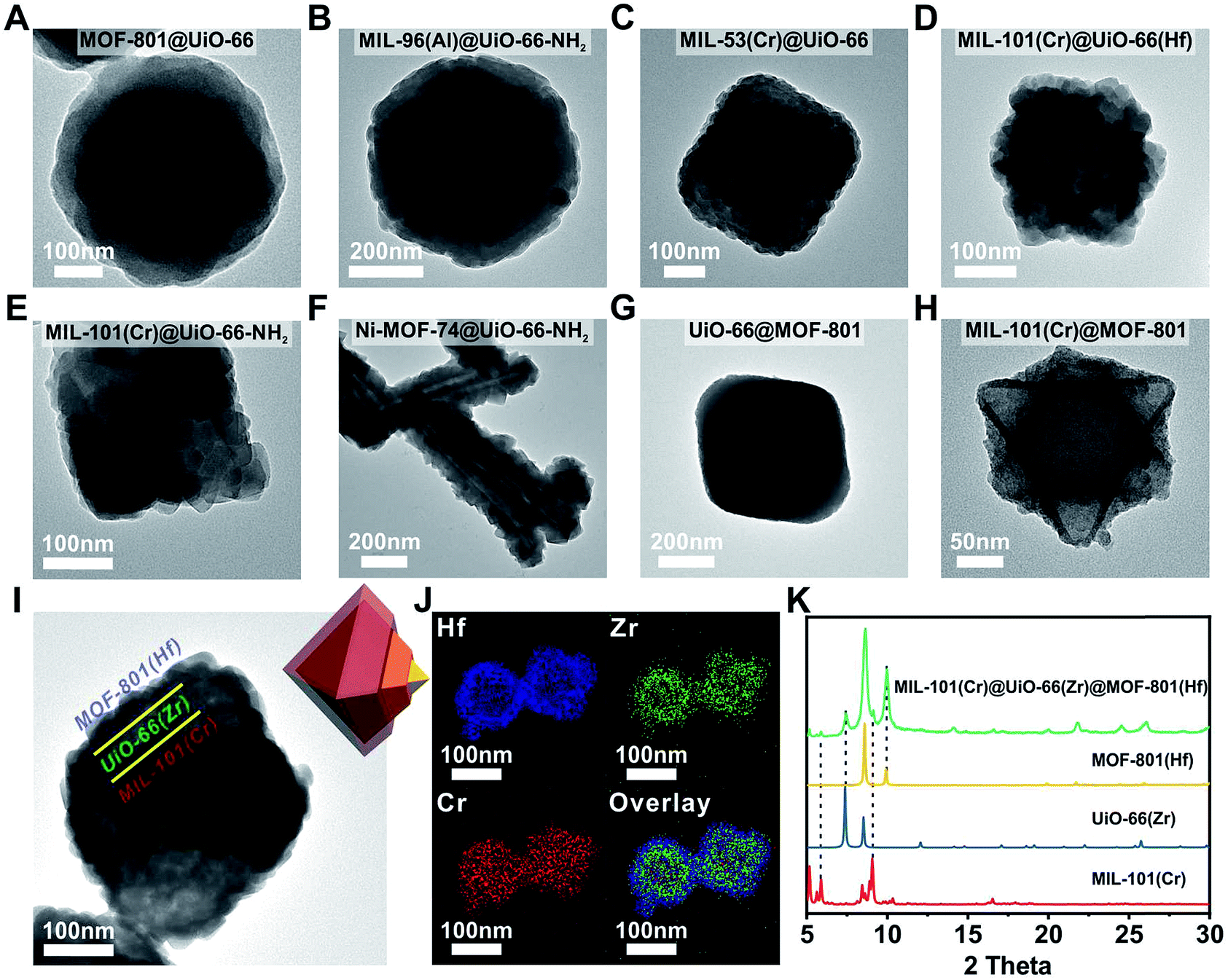 | ||
| Fig. 2 TEM images of (A) MOF-801@UiO-66, (B) MIL-96(Al)@UiO-66-NH2, (C) MIL-53(Cr)@UiO-66, (D) MIL-101(Cr)@UiO-66(Hf), (E) MIL-101(Cr)@UiO-66-NH2, (F) Ni-MOF-74@UiO-66-NH2, (G) UiO-66@MOF-801, (H) MIL-101(Cr)@MOF-801 and (I) MIL-101(Cr)@UiO-66(Zr)@MOF-801(Hf); (J) EDS elemental mapping images of MIL-101(Cr)@UiO-66(Zr)@MOF-801(Hf); (K) PXRD patterns of MIL-101(Cr) (red, simulated), UiO-66(Zr) (blue, simulated), MOF-801(Hf) (yellow, simulated) and MIL-101(Cr)@UiO-66(Zr)@MOF-801(Hf) (green). | ||
Considering the generalizability of this shell growth method, it is possible to increase the complexity of core–shell MOF composites by adding more DBBs, in other words, growing more than one shell layer. As a proof of principle, a double-shell composite, MIL-101(Cr)@UiO-66(Zr)@MOF-801(Hf) was synthesized. The TEM micrograph (Fig. 2I) unambiguously shows that a MIL-101(Cr) particle was uniformly coated with a ∼40 nm UiO-66 shell wrapped in another ∼30 nm MOF-801(Hf) shell. Further structural information was provided using EDS mapping images of Cr, Zr, and Hf (Fig. 2J) and the PXRD pattern (Fig. 2K). The unusual spreading of the Cr EDS signal across the shell layers is likely due to leaching of Cr from the core under acidic conditions. Recent studies by Liu et al. also suggested that despite the well-known robustness of MIL-101(Cr), the center of the MIL-101(Cr) particles is less crystalline and more labile than the exterior and thus these particles are prone to dissolution.52 Nevertheless, the three-layered morphology of MIL-101(Cr)@UiO-66(Zr)@MOF-801(Hf) is clearly demonstrated. This example suggests that the DBB concept can be further extended to MOFs with arbitrary unit cell parameter combinations.
In addition to Zr/Hf-MOFs, this rapid nucleation approach can also be applied to the growth of the ZIF-8 shell. At a lower precursor concentration, ZIF-8 cannot be grown on the UiO-66 surface. By increasing the concentration of 2-methylimidazole (MIM) from 0.576 M to 1.344 M, the UiO-66@ZIF-8 core–shell MOF was successfully synthesized (Fig. S16†).
Growth of a Zr-MOF shell on an acid labile core MOF through dissolution kinetics control
Among these core–shell examples, it is worth noting that Ni-MOF-74 is regarded as a more labile MOF than UiO-66-NH2. Nevertheless, the growth conditions of UiO-66-NH2 were mild enough to be tolerated by Ni-MOF-74 nanorods thereby successfully achieving a well-defined core–shell structure. This observation drove us to pursue the possibility of growing Zr-MOF shells on more labile core MOFs.A key challenge for growing Zr-MOFs on an acid labile core MOF is that the core typically cannot withstand the aggressive synthetic conditions in the presence of acidic precursors such as Zr4+ and H2BDC thus leading to dissolution. Such a phenomenon has been observed by Zhong et al. in the case of growing UiO-66 on MOF-5.53 During our early attempt to grow UiO-66-NH2 on ZIF-8 using a rapid nucleation approach, we found that UiO-66-NH2 crystallites nucleated around ZIF-8 crystallites to form a continuous hollow capsule (Fig. 3Bi) where ZIF-8 served as a sacrificial template and was completely etched away by acidic precursors. Interestingly, when large ZIF-8 particles with a size of 1.0 ± 0.1 μm were used as the core, ZIF-8 residues were observed in the hollow UiO-66-NH2 shells resembling a yolk–shell structure (Fig. S17†). This implies that by manipulating the dissolution kinetics of ZIF-8, it is possible to grow a Zr-MOF shell without significant etching on the ZIF-8 core. Therefore, we gradually reduced the ZIF-8/UiO-66-NH2 growth solution ratio54 from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:2, 1:4, 1:6, 1:8 and 1:10 while keeping the precursor concentration at 100 mM to maintain the rapid nucleation kinetics (Fig. S18†). After 2 h of growth, there is a clear trend suggesting that a small amount of growth solution results in slower ZIF-8 dissolution. In particular, at a 1:1 growth solution ratio, no apparent dissolution of ZIF-8 was observed. Meanwhile, a uniform layer of deposition appeared on ZIF-8. EDS mapping results show that Zr is present in this layer suggesting the formation of an AZC layer similar to that of MIL-101(Cr) at an early growth stage (Fig. 1Aix). After digesting ZIF-8 using acetic acid, a ∼5 nm AZC layer was revealed in the form of a slightly collapsed hollow cube. Repeating this growth step one more time with fresh growth solution led to a ZIF-8@UiO-66-NH2 core–shell structure containing a ∼15 nm UiO-66-NH2 crystalline shell uniformly coated on the ZIF-8 core. After digesting the ZIF-8 core using acid, the hollow polycrystalline UiO-66-NH2 shell exhibits good structural integrity according to both the PXRD pattern and TEM image (Fig. 3C and Biii, inset). The N2 adsorption isotherm of ZIF-8@UiO-66-NH2 falls in between that of ZIF-8 and UiO-66-NH2 indicating that the crystallinity and porosity of both MOFs are well maintained (Fig. 3D).
:1 to 1:2, 1:4, 1:6, 1:8 and 1:10 while keeping the precursor concentration at 100 mM to maintain the rapid nucleation kinetics (Fig. S18†). After 2 h of growth, there is a clear trend suggesting that a small amount of growth solution results in slower ZIF-8 dissolution. In particular, at a 1:1 growth solution ratio, no apparent dissolution of ZIF-8 was observed. Meanwhile, a uniform layer of deposition appeared on ZIF-8. EDS mapping results show that Zr is present in this layer suggesting the formation of an AZC layer similar to that of MIL-101(Cr) at an early growth stage (Fig. 1Aix). After digesting ZIF-8 using acetic acid, a ∼5 nm AZC layer was revealed in the form of a slightly collapsed hollow cube. Repeating this growth step one more time with fresh growth solution led to a ZIF-8@UiO-66-NH2 core–shell structure containing a ∼15 nm UiO-66-NH2 crystalline shell uniformly coated on the ZIF-8 core. After digesting the ZIF-8 core using acid, the hollow polycrystalline UiO-66-NH2 shell exhibits good structural integrity according to both the PXRD pattern and TEM image (Fig. 3C and Biii, inset). The N2 adsorption isotherm of ZIF-8@UiO-66-NH2 falls in between that of ZIF-8 and UiO-66-NH2 indicating that the crystallinity and porosity of both MOFs are well maintained (Fig. 3D).
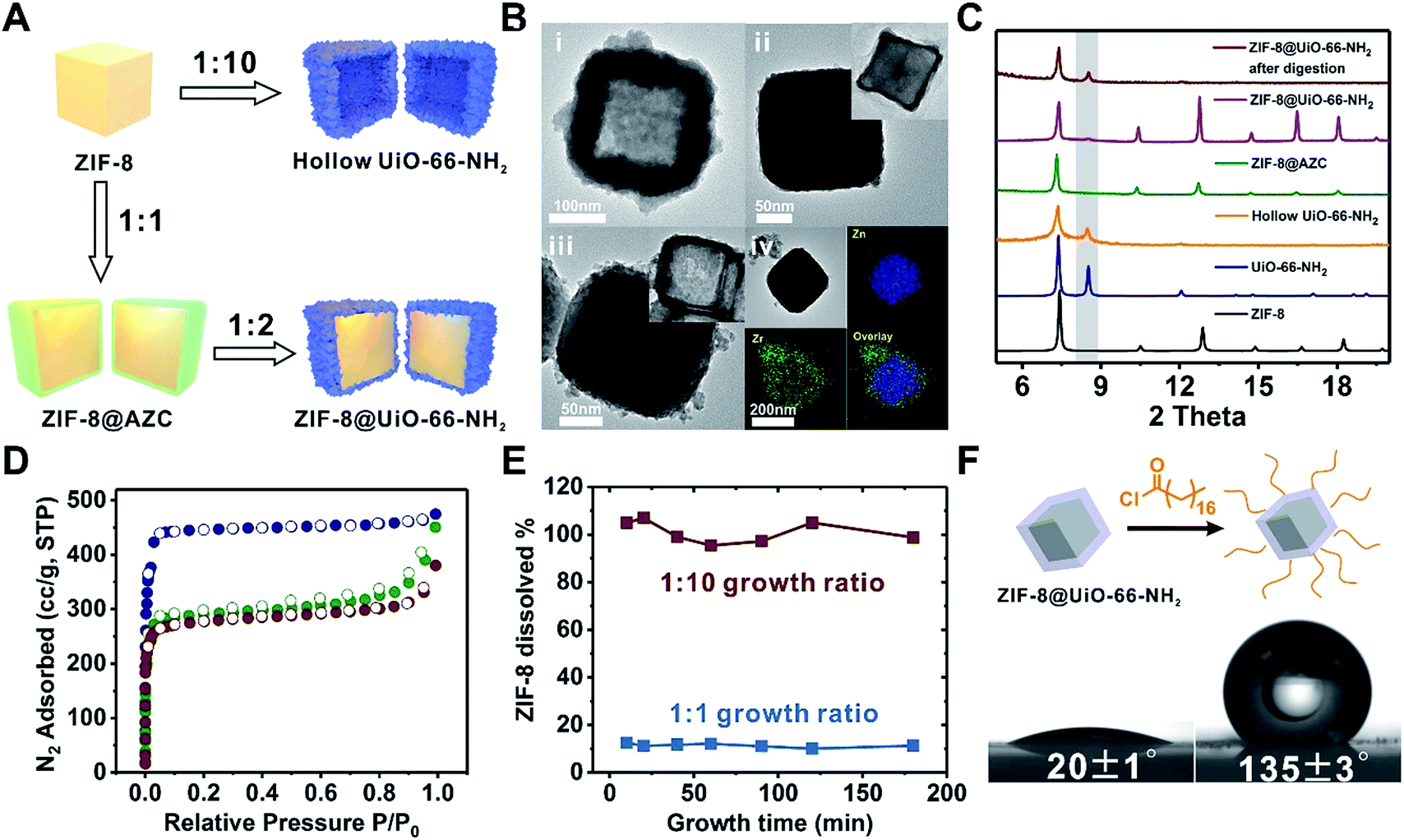 | ||
| Fig. 3 (A) Schematic illustration of the growth process of UiO-66-NH2 on ZIF-8. (B) TEM images of hollow UiO-66-NH2 (i), ZIF-8@AZC (ii), and ZIF-8@UiO-66-NH2 (iii), and EDS elemental mapping images of ZIF-8@AZC (iv). The insets in ii and iii are the TEM images of the core–shell particles after acid digestion. (C) PXRD patterns of ZIF-8 (black, simulated), UiO-66-NH2 (blue, simulated), hollow UiO-66-NH2 (yellow), ZIF-8@AZC (green), ZIF-8@UiO-66-NH2 (purple), and ZIF-8@UiO-66-NH2 after acid digestion (red). (D) N2 adsorption–desorption isotherms (77 K) of ZIF-8 (blue), ZIF-8@UiO-66-NH2 (green), and UiO-66-NH2 (red). (E) The dissolution kinetics of ZIF-8 from ZIF-8@UiO-66-NH2 during the shell growth process. (F) Water contact angle measurements for non-functionalized ZIF-8@UiO-66-NH2 (left) and alkane modified ZIF-8@UiO-66-NH2 (right). | ||
To understand the dissolution kinetics, the supernatants of the growth mixtures were obtained at different time points and the Zn2+ concentration was monitored by inductively coupled plasma optical emission spectrometry (ICP-OES). Fig. 3E shows that complete ZIF-8 dissolution was observed in the 1:10 growth solution after 10 min of reaction. In contrast, only ∼11% ZIF-8 was dissolved and leached out for the 1:1 growth sample. With the reaction time extended to 3 h, no further ZIF-8 dissolution was observed. It is clear that by reducing the amount of Zr-MOF precursors, there are fewer acidic species available in the solution thereby lowering the dissolution kinetics of ZIF-8 at an early stage of shell growth. The rapid deposition of an AZC layer further passivated the ZIF-8 surface and therefore effectively suppressed the diffusion of acidic species toward ZIF-8 and the dissolved species toward the solution, consequently stopping the dissolution of ZIF-8.
With the amine functionalized UiO-66-NH2 as the shell, it is possible to covalently attach small molecules to the core–shell composite for the surface modification of ZIF-8. As a proof of concept, ZIF-8@UiO-66-NH2 was reacted with stearoyl chloride in tetrahydrofuran (THF). The extraction experiment shows that alkane modified ZIF-8@UiO-66-NH2 can be extracted to the ethyl acetate phase whereas the non-functionalized ZIF-8@UiO-66-NH2 prefers to stay in the aqueous phase (Fig. S19†). Furthermore, the water contact angle of ZIF-8@UiO-66-NH2 increased from 20 ± 1° to 135 ± 3° after surface modification suggesting that the ZIF-8@UiO-66-NH2 surface was successfully modified by hydrophobic alkyl molecules (Fig. 3F).
Manipulating the MOF shell for size selective catalysis
To manifest the tunability of Zr-MOFs, we constructed a series of Pt NP-containing core–shell catalysts to showcase their ability for size selective hydrogenation through changing either the ligand length or ligand functionality. On the other hand, size selective catalysis is also a great way to verify the uniformity of the as-grown shell layers. To start with, ∼2 nm polyvinylpyrrolidone (PVP) coated platinum nanoparticles (Pt NPs) were synthesized according to a reported method55 (Fig. S20†). They were then adsorbed on to the core MOF MIL-101(Cr) or UiO-66 through electrostatic interaction to give MIL-101(Cr)@Pt (1) and UiO-66@Pt (Fig. 4B and S21†). Then, UiO-66, UiO-66-NH2 and MOF-801 shells were grown on MOF@Pt to give three core–shell composite catalysts: MIL-101(Cr)@Pt@UiO-66, MIL-101(Cr)@Pt@UiO-66-NH2, and UiO-66@Pt@MOF-801. For convenience, they are denoted as 2, 3, and 4, respectively. The TEM images show the sandwiched structural hierarchy where Pt NPs are located at the core–shell interface. Three nitro compounds, nitrobenzene (NB), 2,3-dimethyl nitrobenzene (DMNB), and 1-nitronaphthalene (NN), with increasing molecular sizes were selected to demonstrate the size selectivity of the catalysts. After 150 min of the hydrogenation reaction in a H2 atmosphere, 100% conversion of all three nitro compounds was achieved with catalyst 1. With a 30 ± 2 nm UiO-66 coating, 2 successfully accomplished full conversion of the smaller NB and DMNB, but only managed to convert 62% NN indicating a size selective behavior. With UiO-66-NH2 and MOF-801 coatings, both 3 and 4 exhibited appreciable (∼50%) conversion of NB but very little conversion (∼4%) of NN indicating a clear size cutoff for the larger species due to the shortening of the linker length (MOF-801) or the introduction of the steric functional group (UiO-66-NH2). Interestingly, despite the similar reactivity of 3 and 4 towards NB and NN, 4 exhibits much higher conversion (26%) of DMNB than 3 (6.4%). This demonstrates that the size selective catalytic behavior can be further fine-tuned through incorporation of steric functional groups such as –NH2. Given the availability of various functional groups in Zr-MOFs and synthetic tools such as post-synthetic modification, ligand exchange, etc., it is reasonable to believe that molecular recognition in catalysis can be potentially achieved through core–shell construction.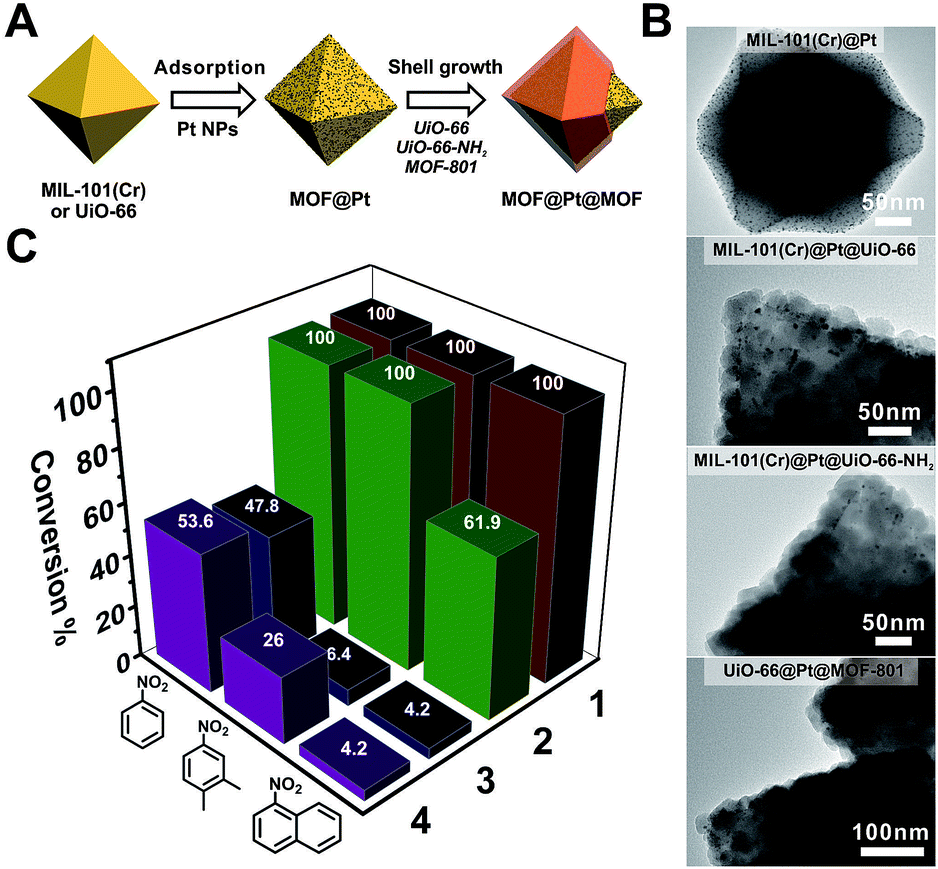 | ||
| Fig. 4 (A) Schematic illustration of the fabrication process of Pt NP-containing core–shell MOF catalysts. (B) TEM images of MIL-101(Cr)@Pt, MIL-101(Cr)@Pt@UiO-66, MIL-101(Cr)@Pt@UiO-66-NH2, and UiO-66@Pt@MOF-801. (C) The conversion of nitrobenzene, 2,3-dimethyl nitrobenzene, and 1-nitronaphthalene over different catalysts after 150 min of the hydrogenation reaction. For clear visualization of Pt NPs, core–shell particles with very thin shell layers were selected for imaging. More representative images can be found in Fig. S22–S24.† | ||
Conclusions
In summary, we unveiled two key kinetic factors that govern the growth of uniform Zr/Hf-MOF shells on a series of core MOFs. The first kinetic factor, nucleation kinetics, controls the nucleus size of the shell MOF, and consequently its surface coverage and uniformity on the core MOF. The second kinetic factor, dissolution kinetics, dictates how fast the core MOF dissolves during the growth process which is a non-negligible factor when an acid labile core MOF such as ZIF-8 is involved. We believe that these principles for constructing core–shell MOFs will guide the rational design of more sophisticated hierarchical MOF composite materials for various applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work made use of the resources of the Instrumental Analysis Center of the SPST at ShanghaiTech University. This work was supported by the National Natural Science Foundation of China (Grant No. 21701110), Shanghai Young Oriental Scholar program and the start-up funding from ShanghaiTech University. This work was partially supported by the CℏEM of the SPST at ShanghaiTech under grant No. EM02161943.Notes and references
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D. L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- J. F. Li, Y. J. Zhang, S. Y. Ding, R. Panneerselvam and Z. Q. Tian, Chem. Rev., 2017, 117, 5002–5069 CrossRef CAS PubMed.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- K. Khaletskaya, J. Reboul, M. Meilikhov, M. Nakahama, S. Diring, M. Tsujimoto, S. Isoda, F. Kim, K.-i. Kamei, R. A. Fischer, S. Kitagawa and S. Furukawa, J. Am. Chem. Soc., 2013, 135, 10998–11005 CrossRef CAS PubMed.
- E. Feng, T. Zheng, X. He, J. Chen and Y. Tian, Sci. Adv., 2018, 4, 3494 CrossRef PubMed.
- G. H. Carey, A. L. Abdelhady, Z. Ning, S. M. Thon, O. M. Bakr and E. H. Sargent, Chem. Rev., 2015, 115, 12732–12763 CrossRef CAS PubMed.
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- H. Wang, S. He, X. Qin, C. Li and T. Li, J. Am. Chem. Soc., 2018, 140, 17203–17210 CrossRef CAS PubMed.
- C. K. Choi, J. Li, K. Wei, Y. J. Xu, L. W. Ho, M. Zhu, K. K. To, C. H. Choi and L. Bian, J. Am. Chem. Soc., 2015, 137, 7337–7346 CrossRef CAS PubMed.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- H. C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- L. Sun, M. G. Campbell and M. Dinca, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- Q. Liu, H. Cong and H. Deng, J. Am. Chem. Soc., 2016, 138, 13822–13825 CrossRef CAS PubMed.
- C. Liu, C. Zeng, T. Y. Luo, A. D. Merg, R. Jin and N. L. Rosi, J. Am. Chem. Soc., 2016, 138, 12045–12048 CrossRef CAS PubMed.
- Q. Pang, B. Tu and Q. Li, Coord. Chem. Rev., 2019, 388, 107–125 CrossRef CAS.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882–885 CrossRef CAS PubMed.
- H. Furukawa, U. Muller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS PubMed.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- S. He, H. Wang, C. Zhang, S. Zhang, Y. Yu, Y. Lee and T. Li, Chem. Sci., 2019, 10, 1816–1822 RSC.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- W. W. Zhan, Q. Kuang, J. Z. Zhou, X. J. Kong, Z. X. Xie and L. S. Zheng, J. Am. Chem. Soc., 2013, 135, 1926–1933 CrossRef CAS PubMed.
- S. J. Yang and C. R. Park, Adv. Mater., 2012, 24, 4010–4013 CrossRef CAS PubMed.
- Y. Peng, M. Zhao, B. Chen, Z. Zhang, Y. Huang, F. Dai, Z. Lai, X. Cui, C. Tan and H. Zhang, Adv. Mater., 2018, 30, 1705454 CrossRef PubMed.
- P. Li, S.-Y. Moon, M. A. Guelta, L. Lin, D. A. Gómez-Gualdrón, R. Q. Snurr, S. P. Harvey, J. T. Hupp and O. k. Farha, ACS Nano, 2016, 10, 9174–9182 CrossRef CAS PubMed.
- T. Li, J. E. Sullivan and N. L. Rosi, J. Am. Chem. Soc., 2013, 135, 9984–9987 CrossRef CAS PubMed.
- F. K. Shieh, S. C. Wang, C. I. Yen, C. C. Wu, S. Dutta, L. Y. Chou, J. V. Morabito, P. Hu, M. H. Hsu, K. C. Wu and C. K. Tsung, J. Am. Chem. Soc., 2015, 137, 4276–4279 CrossRef CAS PubMed.
- K. Hirai, S. Furukawa, M. Kondo, H. Uehara, O. Sakata and S. Kitagawa, Angew. Chem., Int. Ed., 2011, 50, 8057–8061 CrossRef CAS PubMed.
- K. A. McDonald, J. I. Feldblyum, K. Koh, A. G. Wong-Foy and A. J. Matzger, Chem. Commun., 2015, 51, 11994–11996 RSC.
- T. Y. Luo, C. Liu, X. Y. Gan, P. F. Muldoon, N. A. Diemler, J. E. Millstone and N. L. Rosi, J. Am. Chem. Soc., 2019, 141, 2161–2168 CrossRef CAS PubMed.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS PubMed.
- J. A. Boissonnault, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2017, 139, 14841–14844 CrossRef CAS PubMed.
- S. Choi, T. Kim, H. Ji, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2016, 138, 14434–14440 CrossRef CAS PubMed.
- H. Ji, S. Lee, J. Park, T. Kim, S. Choi and M. Oh, Inorg. Chem., 2018, 57, 9048–9054 CrossRef CAS PubMed.
- K. Koh, A. G. Wong-Foy and A. J. Matzger, Chem. Commun., 2009, 6162–6164 RSC.
- H. J. Lee, Y. J. Cho, W. Cho and M. Oh, ACS Nano, 2013, 7, 491–499 CrossRef PubMed.
- J. Zhuang, L. Y. Chou, B. T. Sneed, Y. Cao, P. Hu, L. Feng and C. K. Tsung, Small, 2015, 11, 5551–5555 CrossRef CAS PubMed.
- F. Wang, H. Wang and T. Li, Nanoscale, 2019, 11, 2121–2125 RSC.
- L. Feng, S. Yuan, J. L. Li, K. Y. Wang, G. S. Day, P. Zhang, Y. Wang and H. C. Zhou, ACS Cent. Sci., 2018, 4, 1719–1726 CrossRef CAS PubMed.
- X. Yang, S. Yuan, L. Zou, H. Drake, Y. Zhang, J. Qin, A. Alsalme and H. C. Zhou, Angew. Chem., Int. Ed., 2018, 57, 3927–3932 CrossRef CAS PubMed.
- Y. Liu, Y. Yang, Y. Sun, J. Song, N. G. Rudawski, X. Chen and W. Tan, J. Am. Chem. Soc., 2019, 141, 7407–7413 CrossRef CAS PubMed.
- G. Cao, Nanostructures and Nanomaterials: Synthesis, Properties and Applications, 2004, vol. 1, p. 94 Search PubMed.
- T. Zhao, F. Jeremias, I. Boldog, B. Nguyen, S. K. Henninger and C. Janiak, Dalton Trans., 2015, 44, 16791–16801 RSC.
- W. Liu, J. Huang, Q. Yang, S. Wang, X. Sun, W. Zhang, J. Liu and F. Huo, Angew. Chem., Int. Ed., 2017, 56, 5512–5516 CrossRef CAS PubMed.
- H. Huang, J.-R. Li, K. Wang, T. Han, M. Tong, L. Li, Y. Xie, Q. Yang, D. Liu and C. Zhong, Nat. Commun., 2015, 6, 8847 CrossRef CAS PubMed.
- Note: the 1:X (1≤ X ≤ 10) growth ratio is defined as the mass ratio between the core MOF and the shell MOF based on the assumption that the yield of the shell MOF is 100%.
- M. J. Hossain, H. Tsunoyama, M. Yamauchi, N. Ichikuni and T. Tsukuda, Catal. Today, 2012, 183, 101–107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02576f |
| This journal is © The Royal Society of Chemistry 2019 |