
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective coordination of three transition metal ions within a coiled-coil peptide scaffold†
Aimee L.
Boyle
 *a,
Martin
Rabe
b,
Niek S. A.
Crone
a,
Guto G.
Rhys
c,
Nicolas
Soler
d,
Patrick
Voskamp
a,
Navraj S.
Pannu
a and
Alexander
Kros
a
*a,
Martin
Rabe
b,
Niek S. A.
Crone
a,
Guto G.
Rhys
c,
Nicolas
Soler
d,
Patrick
Voskamp
a,
Navraj S.
Pannu
a and
Alexander
Kros
a
aLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: a.l.boyle@chem.leidenuniv.nl
bMax-Planck-Institut für Eisenforschung GmbH, Max-Planck-Straße 1, 40237 Düsseldorf, Germany
cSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
dStructural Biology Unit, Institute of Molecular Biology of Barcelona (IBMB-CSIC), Baldiri Reixac 15, 08028 Barcelona, Spain
First published on 20th June 2019
Abstract
Designing peptides that fold and assemble in response to metal ions tests our understanding of how peptide folding and metal binding influence one another. Here, histidine residues are introduced into the hydrophobic core of a coiled-coil trimer, generating a peptide that self-assembles upon the addition of metal ions. HisAD, the resulting peptide, is unstructured in the absence of metal and folds selectively to form an α-helical construct upon complexation with Cu(II) and Ni(II) but not Co(II) or Zn(II). The structure, and metal-binding ability, of HisAD is probed using a combination of circular dichroism (CD) spectroscopy, analytical ultracentrifugation (AUC), nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography. These show the peptide is trimeric and binds to both Cu(II) and Ni(II) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with the histidine residues involved in the metal coordination, as designed. The X-ray crystal structure of the HisAD-Cu(II) complex reveals the trimeric HisAD peptide coordinates three Cu(II) ions; this is the first example of such a structure. Additionally, HisAD demonstrates an unprecedented discrimination between transition metal ions, the basis of which is likely to be related to the stability of the peptide-metal complexes formed.
:1 ratio with the histidine residues involved in the metal coordination, as designed. The X-ray crystal structure of the HisAD-Cu(II) complex reveals the trimeric HisAD peptide coordinates three Cu(II) ions; this is the first example of such a structure. Additionally, HisAD demonstrates an unprecedented discrimination between transition metal ions, the basis of which is likely to be related to the stability of the peptide-metal complexes formed.
Introduction
A fundamental aim of metalloprotein design is to understand how metal-binding and protein folding affect each other and to use this knowledge to produce novel metallopeptides, often utilising minimal scaffolds.One scaffold that has been widely employed for this purpose is the α-helical coiled coil. Canonical coiled coils are formed when two or more α-helices interact in a specific manner, known as ‘knobs-into-holes’ packing.1 Most coiled coils also have a repeating sequence pattern known as a ‘heptad repeat,’ which is given the nomenclature abcdefg, with hydrophobic residues found at the ‘a’ and ‘d’ positions and polar residues occupying the other positions.2,3 These characteristics have enabled researchers to devise rules that allow coiled coils with prescribed oligomer states and orientations to be reliably designed,4,5 making them attractive candidates for metallopeptide design.
The most common examples of metal-binding coiled coils substitute hydrophobic residues at the ‘a’ and ‘d’ positions with amino acids capable of coordinating metal ions, and this often results in the binding of a single metal ion in the hydrophobic core of the coiled coil. The Pecoraro lab in particular have presented multiple designs of this type, utilising their TRI series of peptides,6–8 so called as they form trimeric coiled coils, and variants of α3D;9–12 a single-chain three-helix bundle originally developed by the DeGrado lab.13 The Tanaka lab have also made significant contributions to this field with their trimeric IZ peptide, which contains two histidine residues and binds Co(II), Cu(II), Ni(II) and Zn(II),14 and their tetrameric AM2D and AM2E peptides which also bind Cu(II) and Zn(II).15
Other notable examples of coiled-coil-based peptides which bind a single metal ion include a homodimeric Cd(II)-binding mimic of the protein rubredoxin from the Ogawa lab;16 a Zn(II)-binding heterodimer from the Jerala lab;17 and a Zn(II) binding tetrameric helical bundle from the Kuhlman and Hilvert labs.18 Additionally, the Peacock lab has generated a trimeric coiled-coil peptide that employs asparagine and aspartic acid residues to bind lanthanides.19
While peptides incorporating a single metal-binding site are now designed with relative ease, coordination of multiple metal ions is more challenging. DeGrado and colleagues have had notable success in this endeavour creating Due Ferri, a four-helix bundle of which many variants have been presented;20–23 a notable recent derivative is capable of supporting a tetranuclear Zn(II) cluster.24 The Allen lab has also designed four-helix bundle peptides that are capable of supporting multinuclear metal-binding sites, examples of metal ions bound include both dinuclear Mn(II),25 and Cu(II),26 whilst a similar design which also binds Cu(II) has been presented by the Tanaka lab.27
The Ogawa lab has created a tetrameric coiled-coil peptide which binds a Cu4S4 cluster;28 interestingly the same peptide was dimeric when Cd(II) was bound.16 A second peptide that possessed a multinuclear Cd(II) centre was also developed but the crystal structure revealed a distorted peptide backbone.29 These examples in particular highlight the delicate interplay between peptide folding and metal coordination and how the choice of metal can affect how the peptide assembles.
In addition to the aforementioned four-helix bundles, trimeric peptides have also been employed to bind multiple metal ions: one such structure incorporates two 4Fe–4S clusters;30 and the Tanaka, Pecoraro, and Peacock labs have all designed trimeric peptides capable of simultaneously coordinating two different metals at structurally different sites within the same scaffold.31–34
Some of these designs have also begun to address the issue of metal selectivity; a concept which is critically important for developing effective biosensing or purification devices for example. At the most fundamental level, selectivity can be achieved by designing metal-binding sites with different amino acids, as different metals have preferences for different coordinating ligands. Selectivity can also be achieved by limiting the number of residues available for coordination to the metal of interest, thus selecting for metals that prefer lower coordination numbers.35,36 Alternatively, selectivity can be obtained by choosing which position of the heptad repeat metal-binding residues are incorporated into; it has been demonstrated that Cd(II) produces higher affinity complexes when bound at an ‘a’ position of the heptad repeat.37
In this article we redesign a trimeric coiled-coil scaffold by mutating isoleucine residues within the hydrophobic core to histidine, with the aim of creating a peptide capable of binding transition metal ions. The resulting peptide is unfolded in the absence of metal ions, but folds and self-assembles in the presence of Cu(II) and Ni(II), but not Co(II) or Zn(II). A combination of circular dichroism (CD) spectroscopy, analytical ultracentrifugation (AUC), and nuclear magnetic resonance (NMR) spectroscopy were employed to gain further information regarding peptide secondary structure and stability, oligomeric state, and the nature of the metal-binding site. The X-ray crystal structure of the peptide-Cu(II) complex was solved, which revealed a trinuclear Cu(II) site with the metal ions being coordinated by histidine and glutamic acid residues. To the best of our knowledge, this is the first example of a designed, α-helical, metallopeptide that coordinates three metal ions, and that exhibits a high degree of metal-binding selectivity.
Results and discussion
Peptide design
We utilized the homotrimeric coiled-coil CC-Tri,4 as the starting point for our metallopeptide design. Histidine residues were placed at hydrophobic positions within the central heptads, generating three mutants: HisA, HisD, and HisAD, Table 1. The central heptads were chosen as the optimal sites for mutation as it has previously been shown that substituting isoleucine for polar residues within the terminal heptads leads to a loss of oligomer state specificity.38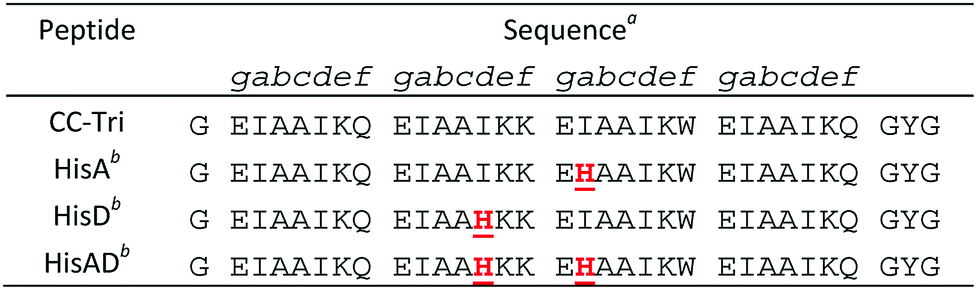
HisA, HisD, and HisAD contain a histidine, (His, H), residue in place of an isoleucine, (Ile, I), at either the ‘d’ position of the second heptad (HisD), the ‘a’ position of the third heptad (HisA), or at both of these positions (HisAD); it is anticipated that metal ions will be coordinated by these His residues. Glutamic acid residues may also play a role in metal coordination, as has been observed for other designs.15,26
Secondary structure characterization in solution
The peptides were initially analysed using CD spectroscopy to examine their secondary structure in solution, both in the presence and absence of a variety of transition metal ions. HisA and HisD were both still folded as α-helical species in the absence of metal ions, (Fig. S1†), although the ellipticity value at 222 nm was reduced compared to CC-Tri (−17024 deg cm2 dmol res−1 and −23190 deg cm2 dmol res−1 for HisA and HisD respectively, compared to −31482 deg cm2 dmol res−1 for CC-Tri, Fig. S1†). This decrease is not surprising given that there is an intrinsic energy penalty associated with including a polar residue in the hydrophobic core, especially for de novo designed sequences.38 Addition of Co(II), Cu(II), Ni(II), or Zn(II) did not alter the folding of these peptides, (Fig. S1†).
HisAD, in contrast, was largely unfolded in the absence of metal species, (Fig. 1). This indicates that mutating two residues within the hydrophobic core of CC-Tri is highly destabilising, and the peptide can no longer self-assemble. Upon addition of an equimolar concentration of different transition metal ion species, it was discovered that both Cu(II) and Ni(II) could induce HisAD to fold into a helical structure, but Zn(II) and Co(II) could not. This selective, metal-induced, folding is highly intriguing as all four metal species have previously been shown to coordinate to coiled-coil peptides containing a similar arrangement of His ligands.14 This selectivity is not likely to be a result of geometric preferences as, although each metal has preferred coordination geometries, all four metals have been shown to adopt a range of geometries in metalloproteins.39,40 It is possible that this selectivity is a reflection of the stabilizing effects of the transition metal ions; Ni(II) and Cu(II) species form the most stable metal–ligand complexes according to the Irving–Williams series.41
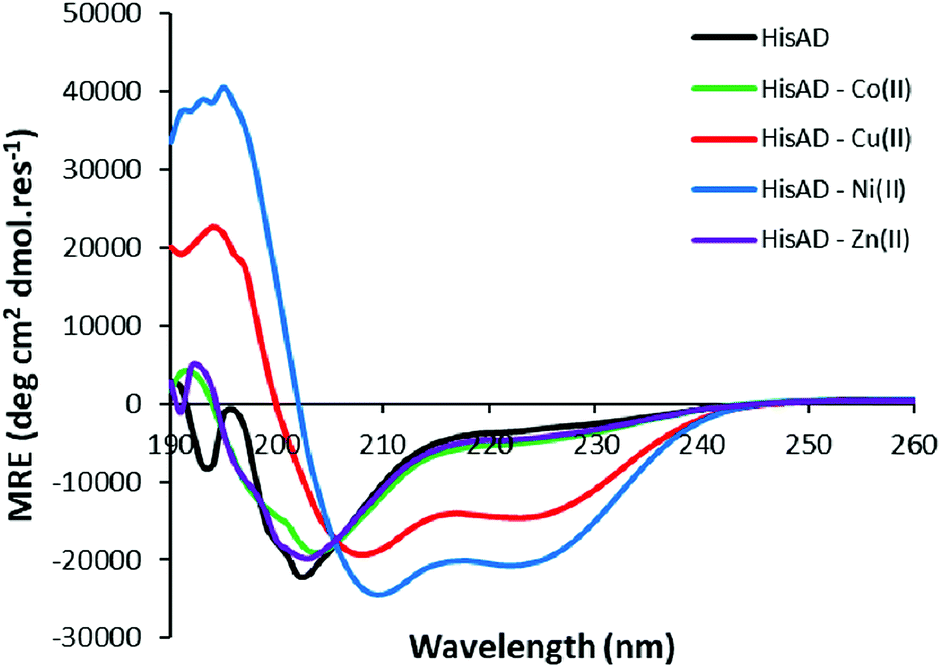 | ||
| Fig. 1 CD spectra of HisAD in the absence and presence of divalent transition metal ions. Conditions: [peptide] = 100 μM, [metal] = 100 μM, 10 mM phosphate buffer pH 7.4, 20 °C. | ||
NMR confirms histidine residues are involved in Cu(II) and Ni(II) coordination
To verify that the histidine residues were involved in the coordination of the metal ions, as designed, 1H NMR spectra of HisAD were recorded in the absence and presence of both Cu(II) and Ni(II).In the absence of metal, signals from the histidine protons were evident at 6.96 and 7.89 ppm, (Fig. 2, black lines, and Fig. S2†). These were assigned as being from histidine, as they were not evident in the NMR spectra of CC-Tri (Figs. S5–7†). Upon addition of 1 equivalent of Cu(II), the complete disappearance of the histidine ε-CH proton signal at 7.89 ppm was observed, (Fig. 2A, red line and Fig. S3†). HisAD also contains tryptophan (Trp, W) and tyrosine (Tyr, Y) residues, and the paramagnetic nature of Cu(II) leads to significant line broadening and overlapping peaks, therefore it was difficult to determine whether the signal from the histidine δ-CH, at 6.96 ppm, had also disappeared. A mutant of HisAD without Trp and Tyr, HisAD-WY, was therefore synthesised and the addition of 1 equivalent of Cu(II) lead to the disappearance of both His resonances (Fig. S8 and S9†).
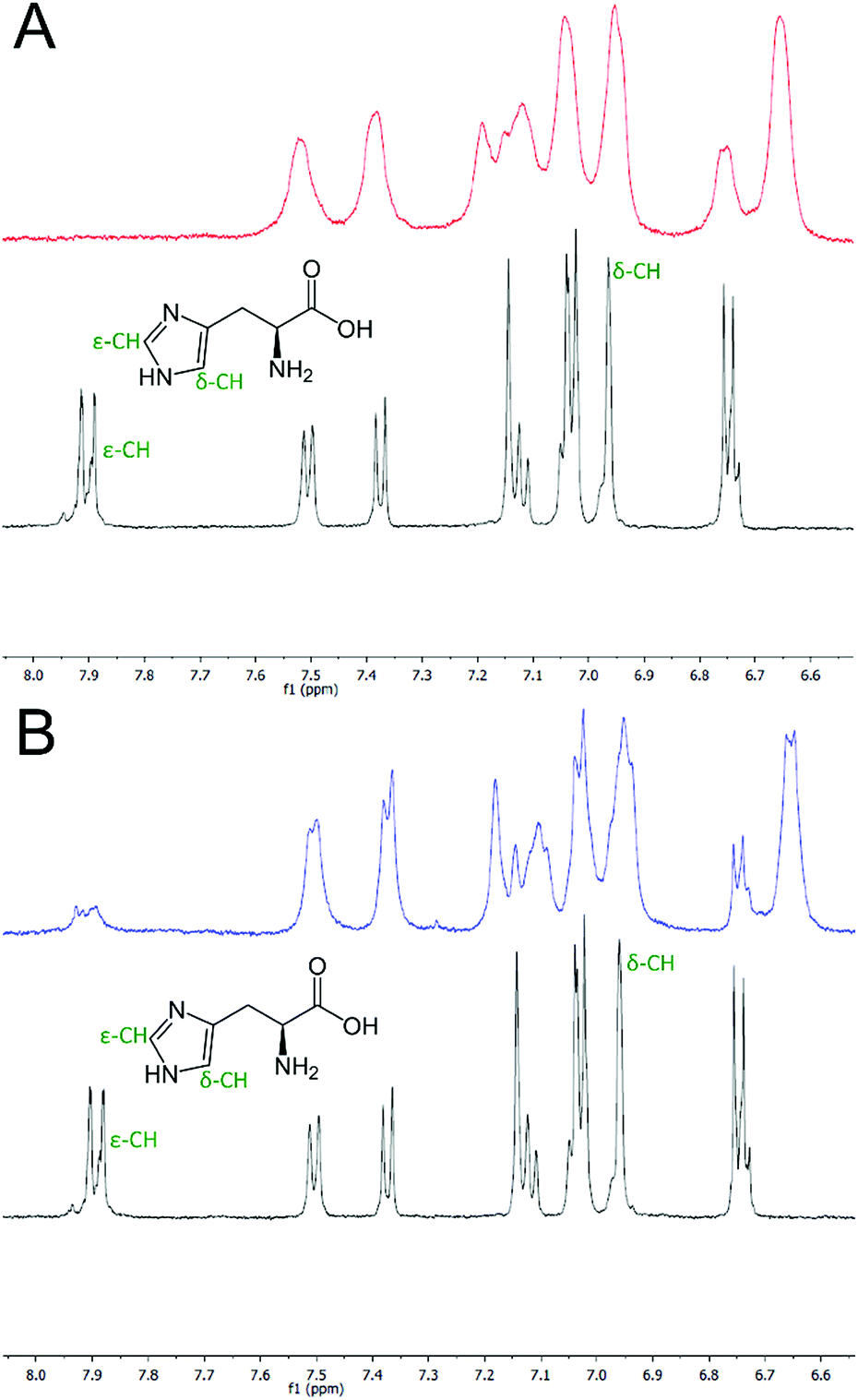 | ||
| Fig. 2 One-dimensional 1H NMR spectra of the aromatic region of HisAD. (A) HisAD in the absence (black line) and presence (red line) of 1 equivalent of Cu(II). (B) HisAD in the absence (black line) and presence (blue line) of 1 equivalent of Ni(II). The signals corresponding to the histidine protons are indicated. Conditions: [peptide] = 500 μM, [metal] = 500 μM, 10 mM deuterated phosphate buffer, pH 7.4. | ||
Addition of Ni(II) produced a similar spectrum, (Fig. 2B, blue line and S4†). The line broadening here was not as pronounced as the electron relaxation time of Ni(II) is 1–3 orders slower than for Cu(II).42
Whilst these spectra do not preclude the involvement of residues other than histidine coordinating to the metal ions, they do confirm that both His residues are indeed involved in coordinating both Cu(II) and Ni(II), as designed.
Cu(II) and Ni(II) peptide complexes are trimeric in solution
The coordination of metals to peptides has been known to affect the peptides oligomeric state,28,43,44 highlighting the delicate interplay between peptide folding and metal-coordination. AUC experiments were therefore performed to probe the oligomerization state of HisAD in the presence of Cu(II) and Ni(II). As HisAD is largely unfolded in the absence of metals, AUC experiments with apo-HisAD were not performed.Both sedimentation velocity (SV) and sedimentation equilibrium (SE) experiments showed monodisperse assemblies were formed; only one peak was observed in the SV experiments and the SE curves fitted well to a single, ideal-species model, (Fig. S10A–D†). These data indicate that HisAD forms a trimeric species in the presence of both Cu(II) and Ni(II). These results show that HisAD forms the same trimeric oligomer state in solution as the parent peptide CC-Tri, meaning that the binding of Cu(II) and Ni(II) do not influence the designed, intended, oligomeric state of HisAD.
The X-ray crystal structure of the HisAD-Cu(II) complex reveals three Cu(II) ions are coordinated by the HisAD trimer
Intrigued by the metal-binding selectivity exhibited by HisAD, in combination with the knowledge that the trimeric structure of the parent peptide is retained upon binding Cu(II) and Ni(II), detailed structural information was highly desirable. Crystals of HisAD with both Cu(II) and Ni(II) were obtained, however only the Cu(II)-containing crystals diffracted to a sufficient resolution to allow a dataset to be obtained and a structure to subsequently be solved, (Fig. 3 and Table 2).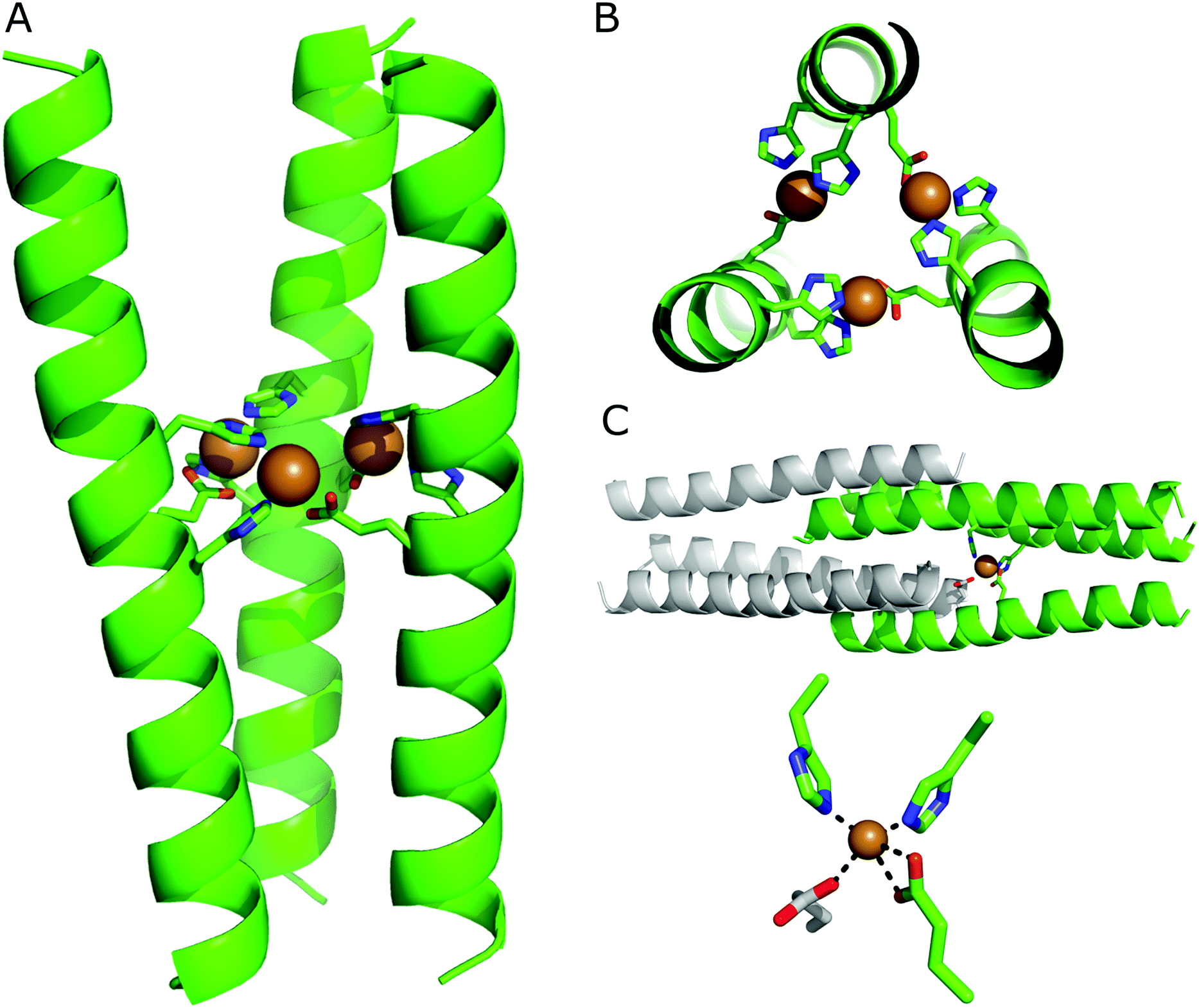 | ||
| Fig. 3 X-ray crystal structure of the HisAD-Cu(II) complex, PDB ID: 6I1J. (A) Side view and; (B) top-down view. The peptide backbone is depicted in cartoon format, residues coordinating the copper ions are shown as sticks and the Cu(II) ions are rendered as spheres. In (B) the helical backbone is partially cut away for clarity. (C) In the crystal structure each Cu(II) ion is coordinated by two histidine residues and one glutamic acid residue from one trimer, and an additional glutamic acid from a symmetry-related trimer. The top panel shows the entire structure, with only one Cu(II) ion shown for clarity. The symmetry-related trimer is coloured grey. The lower panel provides a close-up of the coordination of the Cu(II) centre. All images are generated with PyMol, for the residues shown as sticks colouring is as follows: carbon, green; oxygen, red; nitrogen, blue. Cu(II) ions are shown as orange spheres. | ||
| Beamline | ID30B |
|---|---|
| Detector | Pilatus3_6M |
| Frames | 2400 |
| Oscillation (°) | 0.15 |
| Exposure time per frame, total (s) | 0.025, 60 |
| Space group | P 63 22 |
|
|
| Unit-cell parameters | |
| α, β, γ | 30.52, 30.52, 106.04 |
| a, b, c, | 90, 90, 120 |
| Resolution range (A), (outer resolution shell) | 26.51–2.35, (2.54–2.35) |
| Observations | 44183 (9096) |
| Unique reflections | 1493 (293) |
| Completeness (%) | 99.9 (100) |
| CC (1/2) | 1.00 (0.92) |
| R pim (%) | 0.011 (0.424) |
| 〈I/σ(I)〉 | 65.3 (1.9) |
| Multiplicity | 29.6 (31.0) |
| R factor (%) | 26.7 |
| R complete (%) | 25.5 |
| Bond lengths (Å) | 0.015 |
| Bond angles (°) | 1.705 |
| Ramachandran preferred regions | 100 |
| Allowed regions | 0 |
| Outliers | 0 |
The crystal structure of the HisAD-Cu(II) complex was solved at 2.35 Å resolution. This structure revealed that the trimeric HisAD peptide coordinates three Cu(II) ions, (Fig. 3). This is the first example of a designed α-helical peptide that coordinates three metal ions.
It is evident from the packing of the helices that the metal ions drive the association and organization of this complex. At the C-terminus of HisAD, the packing of the helices resembles that of the parent peptide, CC-Tri, (Fig. S11†). At the N-terminus however, the structure is splayed and the hydrophobic core is not as tightly packed; therefore SOCKET, a program that searches for knobs-into-holes interactions characteristic of coiled coils,45 only finds such interactions at the C-terminal side of the binding site. The reason for this disruption of the helix packing is due to the need for both His13 and His17 to be oriented towards the Cu(II) ion, and it is at this point that a deviation from the CC-Tri supercoiling is apparent, (Fig. S11B†).
In addition to the nitrogen atoms of His13 and His17, each Cu(II) ion is additionally coordinated by both oxygen atoms of a glutamic acid residue (Glu16) from a second helix. This leads to the formation of a ‘daisy-chain’ like arrangement of ligands and metal ions, (Fig. 3B). The coordination sphere is completed by a carboxylate from an N-terminal glutamic acid (Glu2) of a symmetry-related molecule, (Fig. 3C); such a phenomenon has been observed for other helical metallopeptides.46 Combined, these ligands form a distorted tetrahedral coordination sphere around the Cu(II) ion, (Fig. 3C); bond lengths and angles are provided in Table S1.†
The HisAD-Ni(II) complex is the most thermodynamically stable
To probe the thermodynamic properties of the peptide:metal complexes, Cu(II) or Ni(II) were titrated into a solution of HisAD and the change in secondary structure of the peptide was monitored by CD spectroscopy. Fitting of these titration data allows binding energies and dissociation constants of the complexes to be determined.Titrations with Cu(II) or Ni(II) both resulted in the same trend; an increase in helicity was observed as the amount of metal added to the peptide solution increased, with maximal binding being observed around a 1:1 peptide:metal ratio, (Fig. 4A & B), which reflects the stoichiometry of the HisAD-Cu(II) crystal structure. The 1:1 HisAD-Ni(II) complex was more folded than HisAD-Cu(II), (−19,701 deg cm2 dmol res−1 and −13758 deg cm2 dmol res−1 respectively), which indicates that Ni(II) is more stabilising than Cu(II). This was supported by analysis of thermal unfolding data that determined the melting temperature (TM) at 100 μM of the HisAD-Cu(II) complex to be 32 °C, whereas HisAD-Ni(II) had a TM of 45 °C, (Fig. S12†). It should be noted that both these metallopeptide complexes are less stable that CC-Tri, the peptide HisAD is based on, as CC-tri does not completely unfold, even at elevated temperatures.4
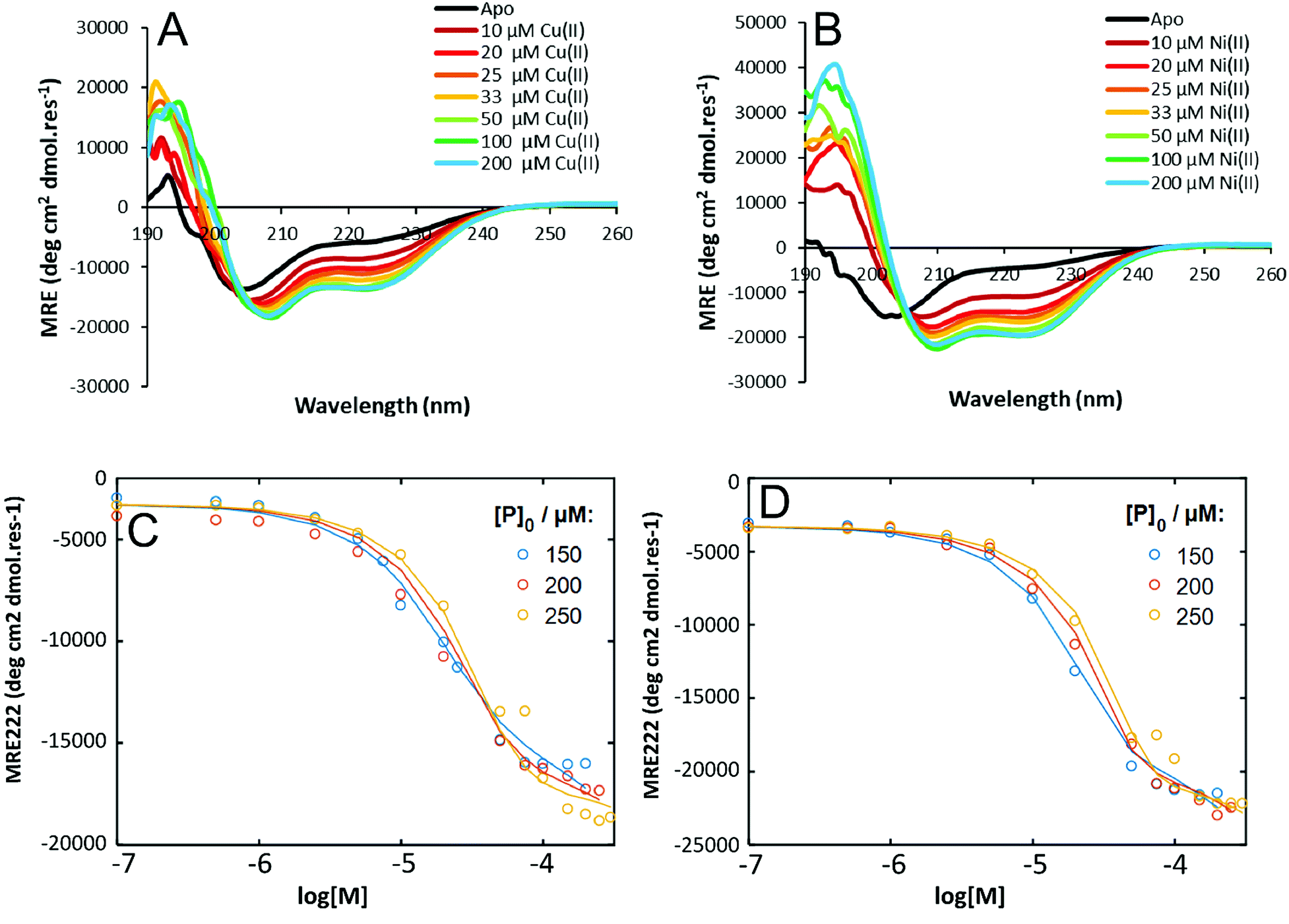 | ||
| Fig. 4 CD titration data for HisAD with Cu(II) and Ni(II). CD spectra of (A) HisAD-Cu(II), and; (B) HisAD-Ni(II) with increasing amounts of the respective metal. A transition from a largely unfolded to a helical state is observed. Experimental data (dots) and fitted data (lines) for a three-state model for (C) HisAD-Cu(II) and; (D) HisAD-Ni(II). Conditions for (A) and (B): [peptide] = 100 μM, 10 mM phosphate buffer, pH 7.4, 20 °C. | ||
Titrations were also performed with both Co(II) and Zn(II) to investigate whether folding of HisAD could be induced at elevated concentrations of these metals. Addition of Zn(II) at increasing concentrations failed to induce any helical structure of HisAD, even when Zn(II) was present in a three-fold molar excess, (Fig. S13A†). Interestingly, while a sub-stoichiometric ratio of Co(II) failed to induce helicity in HisAD; addition of an excess of the metal resulted in a helical structure being observed, (Fig. S13B†). This large excess of Co(II) which is required to induce a helical structure indicates that the affinity of HisAD for Co(II) is weak.
To quantify the thermodynamic properties of the Cu(II) and Ni(II)-containing complexes, titrations at different HisAD concentrations were performed, (Fig. S14†). As the X-ray crystal structure of the HisAD:Cu(II) complex revealed a 1:1 binding stoichiometry, a two-state model of P3M3 (three peptide chains and three metal ions) dissociation was fitted to the data, (Fig. S15†). However, this model failed to describe the observed ellipticity at intermediate metal concentrations (<50 μM) with reasonable physical parameters. Thus, it was concluded that an intermediate state with a HisAD:metal stoichiometric ratio >1 must form first and a three-state model was therefore devised, eqn (1):
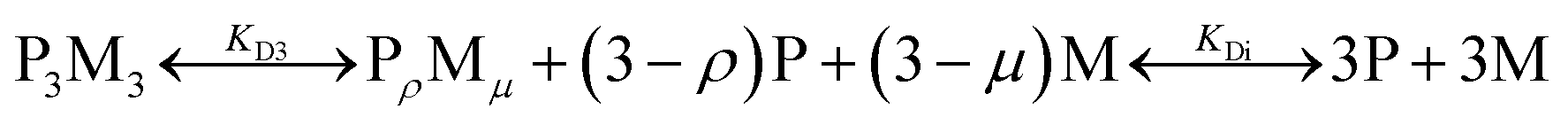 | (1) |
The intermediate state is represented by the notation, PρMμ, where ρ represents the number of peptide chains and μ the number of metal ions.
Models where (μ < ρ, μ ≤ 3, ρ ≤ 2) were tested and compared, (Fig. 4C and D, S16 and S17†). The three-state model that best fitted the titration data for both Cu(II) and Ni(II) complexes was with a P3M1 intermediate, (Fig. 4C and D), and from these data thermodynamic properties could be determined, (Table 3). The lower KD's of the HisAD-Ni(II) complexes, coupled with the more favourable free energies, indicated their increased stability with respect to the HisAD-Cu(II) assemblies, which is consistent with the thermal unfolding data.
| Peptide Complex | ΔGf (kJ mol−1)a | K d (μM) |
|---|---|---|
| a Both dissociation enthalpies and dissociation constants are obtained from fitting the titration data to the three-state model described above; a detailed explanation of the fitting procedure is provided is the ESI, along with the associated equations. | ||
| HisAD:Cu(II) 3:1 |
−69.8 ± 2.9 | 71.8 |
| HisAD:Cu(II) 3:3 |
−120.0 ± 8.0 | 0.075 |
| HisAD:Ni(II) 3:1 |
−75.9 ± 6.3 | 30.9 |
| HisAD:Ni(II) 3:3 |
−131.7 ± 17.8 | 0.015 |
The notion that these complexes form via a P3M1 intermediate state is intriguing, and suggests a subtle interplay between the overall stability of the complex, the binding preferences of metal ions, and the peptide folding. The P3M1 intermediates are reasonably stable, as evidenced by the μM dissociation constants, however the overall KD of the P3M3 assembly is reduced by several orders of magnitude; reflected in the increased dissociation enthalpies. This indicates that incorporation of two additional Cu(II) or Ni(II) ions results in a substantial energy gain of ∼50–55 kJ mol−1. What remains unclear is how the metal coordination changes from the P3M1 intermediate to the final P3M3 complex. It is hypothesised that the large energy gain is not only due to the complexation of two additional metal ions, but also to an alteration of the coordination geometry. No crystal structure of the P3M1 complex is available, but it is reasonable to assume that the metal ion is at the centre of the coiled coil and is complexed by some, or all, of the His residues. There is a literature precedent for such a coordination mode.31,35,36,47 In this arrangement the central metal would be unavailable for complexation by Glu16. It is therefore assumed that the additional complexation by the Glu residues contributes significantly to the increased stability of the trinuclear complex with respect to the mononuclear assembly. Indeed, Kuhlman et al. discovered that, when computationally designing binding sites for transition metal ions comprising multiple histidine residues, complexation by glutamic acid residues was preferred in one of the positions.46 The patterning of the histidine and glutamic acid residues in HisAD therefore favours this unusual trinuclear coordination over preserving the coiled-coil packing. This is evident from the thermodynamic data, which shows binding of three metal ions is more stabilising than binding one ion, and this increased binding energy compensates for the loss of knobs-into-holes packing.
Conclusions
Here we have presented the first example of an α-helical peptide, based on a coiled-coil scaffold, which binds three metal ions in identical coordination environments. Moreover, this peptide exhibits an unprecedented selectivity between different transition metal ions. We hypothesise that the reason for this selectivity is not due to ligand (i.e. hard versus soft) or coordination geometry preferences but is instead due to the relative stabilities of the resulting peptide-metal complexes. The Irving-Williams series indicates that the most stable assembly should be formed with Cu(II), whereas we observe most stability with Ni(II), however the differences in binding energies for the two complexes is relatively small. Further fundamental studies will focus on probing the basis for this selectivity further, by generating peptide scaffolds with differing stabilities and investigating the effects on metal binding. The metal-binding selectivity displayed by HisAD will be exploited to generate metal-ion sensing devices. Additionally, the HisAD-Cu(II) assembly will be used as a starting point for the generation of mimics of complex multi-copper containing enzymes; the fact that this assembly already coordinates multiple metal ions provides a distinct advantage over other metallopeptide structures in the realisation of this goal. These enzyme mimics will subsequently be evaluated for their electron transfer properties; the presence of multiple metal ions may allow for the simultaneous transfer of multiple electrons or for an enhanced transfer rate.Experimental
General experimental information
Dimethylformamide (DMF), piperidine, acetic anhydride, pyridine, trifluoroacetic acid (TFA) and acetonitrile (MeCN) were purchased from Biosolve. N,N-diisopropylethylamine (DIPEA), and Oxyma were obtained from Carl Roth. Dichloromethane (DCM) and diethyl ether were supplied by Honeywell. Tentagel HL-RAM was obtained from Rapp Polymere. All amino acids were supplied by NovaBioChem. All other chemicals were purchased from Sigma Aldrich. Ultrapure water was obtained from a MILLI-Q™ purification system. All buffers were made at pH 7.4 unless otherwise stated.Peptide synthesis and purification
Peptides were synthesized using a Liberty Blue, microwave-assisted, automated peptide synthesizer. Synthesis was performed on the solid-phase, at a 0.1 mmol scale, using Tentagel HL-RAM resin. Fmoc-deprotection was achieved using 20% piperidine in DMF, and coupling was facilitated with DIC as activator and Oxyma as base. Upon completion of synthesis, peptides were manually acetylated using acetic anhydride and pyridine, before cleavage from the resin was performed using a mixture of TFA:TIPS : water, 95:2.5:2.5. The peptide was precipitated using ice-cold diethyl ether; the precipitate was subsequently collected by centrifugation before being resuspended in a water and MeCN mixture and freeze-dried.
Purification was performed by reversed-phase HPLC on a Kinetic Evo C18 column with a Shimadzu system comprising two LC-8A pumps and an SPD-10AVP UV-vis detector. All peptides were purified using a gradient of 20–80% B, (where B is MeCN containing 0.1% TFA, and A is water with 0.1% TFA) over 20 minutes with a flow rate of 12 ml min−1. The collected fractions were analysed using LCMS, Fig. S18–22,† and those deemed to be >95% pure were pooled and freeze-dried.
Circular dichroism spectroscopy
CD spectra were measured using a JASCO-J815 spectrometer fitted with a Peltier temperature controller. Quartz cuvettes with a 1 mm path length were used for all measurements. Spectra were recorded at 20 °C from 260–190 nm, at 1 nm intervals, with a scanning speed of 100 nm min−1, a response rate of 1 s, and a bandwidth of 1 nm. Spectra were averaged over 5 scans.Melt spectra were monitored at 222 nm, between 5 and 95 °C, with a heating rate of 40 °C h−1, and a data point was recorded every 1 °C. Melting temperatures were obtained by taking the second derivative of the melting curve.
All data was converted to mean residue ellipticity, [θ], using eqn (2):
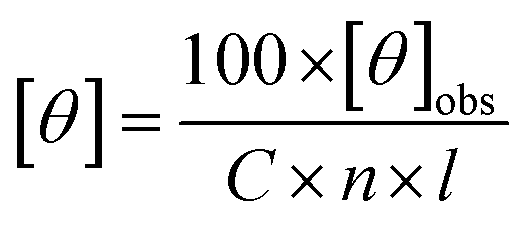 | (2) |
For fitting of the 3-state model the dissociation constants of the partial reactions KDi and KD3 from eqn (1) can be expressed as eqn (3) and (4) (cf. ESI eqn. (1)–(11)†):
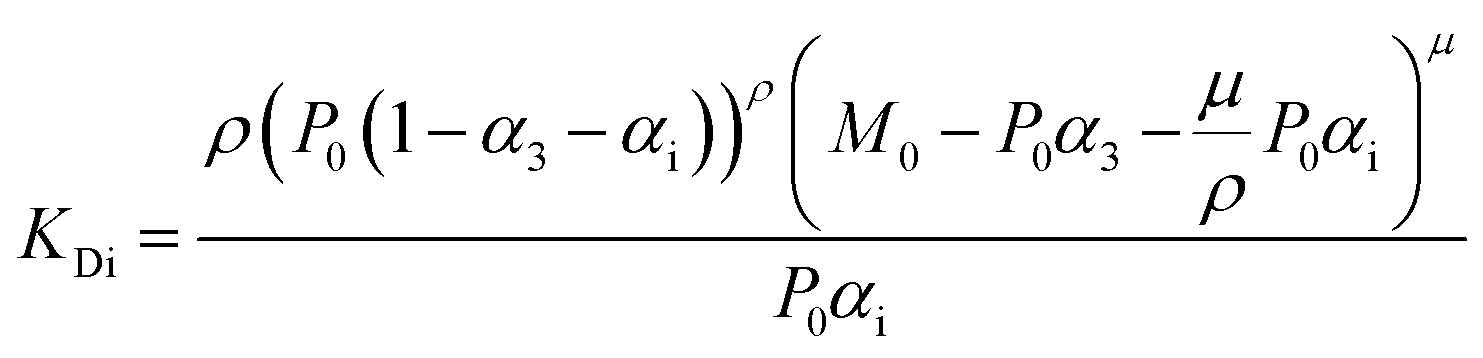 | (3) |
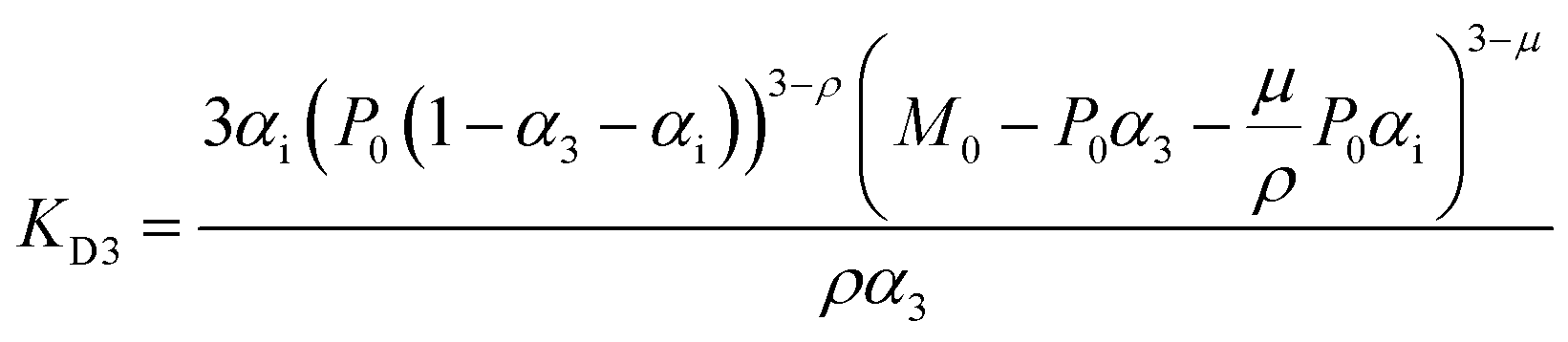 | (4) |
Fit parameters were pKD3, pKDi, (pKDi,D3 = −log10KDi,D3) and the mean residual ellipticities at 222 nm for 100% folded as P3M3 ([θ]s3) and P(3−ρ)M(3−μ) ([θ]si), (cf. ESI eqn (4)†) respectively. From the fit parameters the free energy of folding per peptide chain was determined by eqn (5):
| ΔGf = (−pKD) RTln10 | (5) |
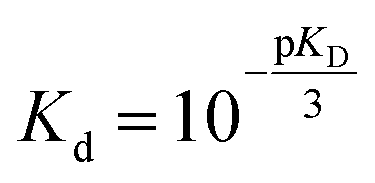 | (6) |
Analytical ultracentrifugation
AUC sedimentation equilibrium experiments were conducted at 20 °C in a Beckman ProteomeLab XL-I or XL-A analytical ultracentrifuge using an An-50 or An-60 Ti rotor (Beckmann Coulter). Solutions were made at 200 μM peptide concentration, in PBS, pH 7.4, with 100 mM NaCl to provide sufficient charge-paring ions. The experiments were run in triplicate in six-channel centrepieces. The samples were centrifuged at speeds in the range 30–48k rpm in 3k rpm intervals and scans at each recorded speed were duplicated. Data were fitted to single ideal-species models using Ultrascan II, (http:/www.ultrascan.uthscsa.edu), comprising a minimum of four speeds. 95% confidence limits were obtained via Monte Carlo analysis of the obtained fits.Sedimentation-velocity experiments were conducted at 20 °C in a Beckman ProteomeLab XL-A or Beckman ProteomeLab XL-I analytical ultracentrifuge using an An-60 Ti rotor. Solutions of 310 μl volume were made at 200 μM peptide concentration and placed in a sedimentation-velocity cell with an epon two-channel centrepiece and quartz windows. The reference channel was loaded with 325 μl of buffer. The samples were centrifuged at 60k rpm, with absorbance scans taken across a radial range of 5.8 to 7.3 cm at 5 min intervals to a total of 120 scans. Data from a single run were fitted to a continuous c(s) distribution model using Sedfit,48 at 95% confidence level. The partial specific volume (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) ) for the peptide and the buffer densities and viscosities were calculated using Ultrascan II (http:/www.ultrascan.uthscsa.edu).
) for the peptide and the buffer densities and viscosities were calculated using Ultrascan II (http:/www.ultrascan.uthscsa.edu).
NMR spectroscopy
Samples were prepared in water at 500 μM peptide concentration, with 0 or 1 equivalent of Cu(II) or Ni(II). The samples were freeze dried and redissolved in 10 mM phosphate buffer, prepared in D2O. NMR spectra were recorded at ambient temperature (298 K) using a Bruker Avance 500 MHz spectrometer equipped with a Bruker BBFO probe. Spectra were recorded using a standard pulse sequence with HDO presaturation for 512, or 1024 scans, when the sample contained higher metal concentrations. 1H chemical shifts (δ) were reported relative to the residual solvent peak (δ 4.79 ppm). Data processing and analysis was performed using MestReNova with manual peak assignment and integration.X-ray crystallography: crystallization
The peptide solution was prepared at a concentration of 15 mg mL−1, with 1 equivalent of Cu(II) in ultrapure water. Crystallization was achieved using the sitting-drop vapour-diffusion method at 20 °C. Initial screens were performed using the commercially available PACT premier and JCSG+ screens from Molecular Dimensions. Initial crystallization plates were prepared using an NT8 robot (Formulatrix) for pipetting the reservoir solution (70 μL) and making the drops (500 nL). Crystals were seen in many conditions, but the largest were in JCSG+, condition D7 (0.2 M LiSO4, 0.1 M TRIS 8.5 pH, 40% v/v PEG 400). This condition was further optimized by varying the concentration of the buffer, precipitant and salt as well as employing an additive screen. Crystals giving the best diffraction grew in ∼1 month in 0.09 M TRIS pH 8.5, 0.18 M LiSO4, 36% v/v PEG 400 and 5% v/v Jeffamine M-600. The crystals were mounted on cryo-loops and, before flash-cooling in liquid nitrogen, were cryo-protected in a solution consisting of mother liquor and 15% v/v glycerol.X-ray crystallography: data collection and refinement
X-ray data collection was performed at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) on beamline ID30B,49 at a wavelength tuned to 1.34765 Angstroms to optimize the anomalous signal from the intrinsic copper atoms. The data sets were auto-processed by the EDNA auto-processing package that used XDS50 to integrate the intensities and AIMLESS51 to scale and merge the intensities. The diffraction patterns indicated severe anisotropy that was evident in the merging statistics that showed weaker diffraction along the h–k plane. Although the anomalous signal was large (as judged by the anomalous correlation coefficient), all attempts to solve the structure from the anomalous signal alone failed. A molecular replacement solution was found using ARCIMOBOLDO,52–55 and this model was further built with CRANK2.56 The model was further refined with iterative manual model building using REFMAC,57 and COOT,58 from the CCP4 suite.59 Given the very small size of the data set, all the data was used in the refinement and to prevent overfitting, ‘jelly-body’ refinement within REFMAC was used. In order to validate the final model, the R-complete60 was used. The final R-complete and R-work were 25.5%, and 26.7% respectively. The data collection and refinement statistics are presented in Table 2 and the final coordinates have been deposited in the protein data bank, PDB code: 6I1J.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The X-ray data collection was performed on beamline ID30-B at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We are grateful to Alexander Popov at the ESRF for providing assistance using beamline ID30-B. The work was supported by the NWO via a VENI grant (722.015.006) to ALB. MR acknowledges funding from the European Union’s Horizon 2020 research and innovation programme through a Marie Skłodowska-Curie Grant (Agreement No. 705857). A VICI grant (724.014.001) awarded to AK funded NSAC. GGR is supported by a European Research Council Advanced Grant (340764). NSP acknowledges the support of an NWO ZonMW grant (91116025). NS acknowledges grant number BIO2015-64216-P (MINECO/AEI/FEDER/UE).References
- F. H. C. Crick, Acta Crystallogr., 1953, 6, 689–697 CrossRef CAS.
- A. N. Lupas and M. Gruber, Fibrous Proteins: Coiled-Coils, Collagen and Elastomers, 2005, vol. 70, pp. 37–78 Search PubMed.
- D. N. Woolfson, Fibrous Proteins: Coiled-Coils, Collagen and Elastomers, 2005, vol. 70, pp. 79–112 Search PubMed.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. C. Bromley, P. J. Booth, R. L. Brady, A. R. Thomson and D. N. Woolfson, ACS Synth. Biol., 2012, 1, 240–250 CrossRef CAS PubMed.
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, Science, 1993, 262, 1401–1407 CrossRef CAS PubMed.
- G. R. Dieckmann, D. K. McRorie, D. L. Tierney, L. M. Utschig, C. P. Singer, T. V. Ohalloran, J. E. PennerHahn, W. F. DeGrado and V. L. Pecoraro, J. Am. Chem. Soc., 1997, 119, 6195–6196 CrossRef CAS.
- D. Ghosh and V. L. Pecoraro, Inorg. Chem., 2004, 43, 7902–7915 CrossRef CAS PubMed.
- D. Ghosh and V. L. Pecoraro, Curr. Opin. Chem. Biol., 2005, 9, 97–103 CrossRef CAS PubMed.
- S. Chakraborty, J. Y. Kravitz, P. W. Thulstrup, L. Hemmingsen, W. F. DeGrado and V. L. Pecoraro, Angew. Chem., Int. Ed., 2011, 50, 2049–2053 CrossRef CAS PubMed.
- J. S. Plegaria, S. P. Dzul, E. R. P. Zuiderweg, T. L. Stemmler and V. L. Pecoraro, Biochemistry, 2015, 54, 2858–2873 CrossRef CAS PubMed.
- K. J. Koebke, L. Ruckthong, J. L. Meagher, E. Mathieu, J. Harland, A. Deb, N. Lehnert, C. Policar, C. Tard, J. E. Penner-Hahn, J. A. Stuckey and V. L. Pecoraro, Inorg. Chem., 2018, 57, 12291–12302 CrossRef CAS PubMed.
- J. S. Plegaria, M. Duca, C. Tard, T. J. Friedlander, A. Deb, J. E. Penner-Hahn and V. L. Pecoraro, Inorg. Chem., 2015, 54, 9470–9482 CrossRef CAS PubMed.
- S. T. R. Walsh, H. Cheng, J. W. Bryson, H. Roder and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5486–5491 CrossRef CAS PubMed.
- K. Suzuki, H. Hiroaki, D. Kohda, H. Nakamura and T. Tanaka, J. Am. Chem. Soc., 1998, 120, 13008–13015 CrossRef CAS.
- D. Shiga, D. Nakane, T. Inomata, H. Masuda, M. Oda, M. Noda, S. Uchiyama, K. Fukui, Y. Takano, H. Nakamura, T. Mizuno and T. Tanaka, Biopolymers, 2009, 91, 907–916 CrossRef CAS PubMed.
- O. A. Kharenko and M. Y. Ogawa, J. Inorg. Biochem., 2004, 98, 1971–1974 CrossRef CAS PubMed.
- J. Aupic, F. Lapenta and R. Jerala, ChemBioChem, 2018, 19, 2453–2457 CrossRef CAS PubMed.
- S. Studer, D. A. Hansen, Z. L. Pianowski, P. R. E. Mittl, A. Debon, S. L. Guffy, B. S. Der, B. Kuhlman and D. Hilvert, Science, 2018, 362, 1285–1288 CrossRef CAS PubMed.
- M. R. Berwick, D. J. Lewis, A. W. Jones, R. A. Parslow, T. R. Dafforn, H. J. Cooper, J. Wilkie, Z. Pikramenou, M. M. Britton and A. F. A. Peacock, J. Am. Chem. Soc., 2014, 136, 1166–1169 CrossRef CAS PubMed.
- M. Chino, L. Leone, O. Maglio, D. D'Alonzo, F. Pirro, V. Pavone, F. Nastri and A. Lombardi, Angew. Chem., Int. Ed., 2017, 56, 15580–15583 CrossRef CAS PubMed.
- M. Faiella, C. Andreozzi, R. T. M. de Rosales, V. Pavone, O. Maglio, F. Nastri, W. F. DeGrado and A. Lombardi, Nat. Chem. Biol., 2009, 5, 882–884 CrossRef CAS PubMed.
- A. Lombardi, C. M. Summa, S. Geremia, L. Randaccio, V. Pavone and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6298–6305 CrossRef CAS PubMed.
- A. Pasternak, S. Kaplan, J. D. Lear and W. F. DeGrado, Protein Sci., 2001, 10, 958–969 CrossRef CAS PubMed.
- S. Q. Zhang, M. Chino, L. J. Liu, Y. Z. Tang, X. Z. Hu, W. F. DeGrado and A. Lombardi, J. Am. Chem. Soc., 2018, 140, 1294–1304 CrossRef CAS PubMed.
- T. L. Olson, E. Espiritu, S. Edwardraja, E. Canarie, M. Flores, J. C. Williams, G. Ghirlanda and J. P. Allen, Biochim. Biophys. Acta, Bioenerg., 2017, 1858, 945–954 CrossRef CAS PubMed.
- M. Flores, T. L. Olson, D. Wang, S. Edwardraja, S. Shinde, J. C. Williams, G. Ghirlanda and J. P. Allen, J. Phys. Chem. B, 2015, 119, 13825–13833 CrossRef CAS PubMed.
- D. Shiga, Y. Funahashi, H. Masuda, A. Kikuchi, M. Noda, S. Uchiyama, K. Fukui, K. Kanaori, K. Tajima, Y. Takano, H. Nakamura, M. Kamei and T. Tanaka, Biochemistry, 2012, 51, 7901–7907 CrossRef CAS PubMed.
- O. A. Kharenko, D. C. Kennedy, B. Demeler, M. J. Maroney and M. Y. Ogawa, J. Am. Chem. Soc., 2005, 127, 7678–7679 CrossRef CAS PubMed.
- D. V. Zaytsev, V. A. Morozov, J. F. Fan, X. C. Zhu, M. Mukherjee, S. S. Ni, M. A. Kennedy and M. Y. Ogawa, J. Inorg. Biochem., 2013, 119, 1–9 CrossRef CAS PubMed.
- A. Roy, I. Sarrou, M. D. Vaughn, A. V. Astashkin and G. Ghirlanda, Biochemistry, 2013, 52, 7586–7594 CrossRef CAS PubMed.
- T. Tanaka, T. Mizuno, S. Fukui, H. Hiroaki, J. Oku, K. Kanaori, K. Tajima and M. Shirakawa, J. Am. Chem. Soc., 2004, 126, 14023–14028 CrossRef CAS PubMed.
- M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Nat. Chem., 2012, 4, 118–123 CrossRef CAS PubMed.
- M. L. Zastrow and V. L. Pecoraro, J. Am. Chem. Soc., 2013, 135, 5895–5903 CrossRef CAS PubMed.
- P. Teare, C. F. Smith, S. J. Adams, S. Anbu, B. Ciani, L. J. C. Jeuken and A. F. A. Peacock, Dalton Trans., 2018, 47, 10784–10790 RSC.
- S. N. Dublin and V. P. Conticello, J. Am. Chem. Soc., 2008, 130, 49–51 CrossRef CAS PubMed.
- T. Kiyokawa, K. Kanaori, K. Tajima, M. Koike, T. Mizuno, J. I. Oku and T. Tanaka, J. Pept. Res., 2004, 63, 347–353 CrossRef CAS PubMed.
- M. Matzapetakis and V. L. Pecoraro, J. Am. Chem. Soc., 2005, 127, 18229–18233 CrossRef CAS PubMed.
- J. M. Fletcher, G. J. Bartlett, A. L. Boyle, J. J. Danon, L. E. Rush, A. N. Lupas and D. N. Woolfson, ACS Chem. Biol., 2017, 12, 528–538 CrossRef CAS PubMed.
- S. Barber-Zucker, B. Shaanan and R. Zarivach, Sci. Rep., 2017, 7, 16381 CrossRef PubMed.
- L. Rulisek and J. Vondrasek, J. Inorg. Biochem., 1998, 71, 115–127 CrossRef CAS PubMed.
- H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192–3210, 10.1039/jr9530003192.
- I. Bertini, P. Turano and A. J. Vila, Chem. Rev., 1993, 93, 2833–2932 CrossRef CAS.
- G. R. Dieckmann, D. K. McRorie, J. D. Lear, K. A. Sharp, W. F. DeGrado and V. L. Pecoraro, J. Mol. Biol., 1998, 280, 897–912 CrossRef CAS PubMed.
- M. V. Tsurkan and M. Y. Ogawa, Inorg. Chem., 2007, 46, 6849–6851 CrossRef CAS PubMed.
- J. Walshaw and D. N. Woolfson, J. Mol. Biol., 2001, 307, 1427–1450 CrossRef CAS PubMed.
- B. S. Der, M. Machius, M. J. Miley, J. L. Mills, T. Szyperski and B. Kuhlman, J. Am. Chem. Soc., 2012, 134, 375–385 CrossRef CAS PubMed.
- M. Tegoni, F. T. Yu, M. Bersellini, J. E. Penner-Hahn and V. L. Pecoraro, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21234–21239 CrossRef CAS PubMed.
- P. Schuck, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS PubMed.
- A. A. McCarthy, R. Barrett, A. Beteva, H. Caserotto, F. Dobias, F. Felisaz, T. Giraud, M. Guijarro, R. Janocha, A. Khadrouche, M. Lentini, G. A. Leonard, M. L. Marrero, S. Malbet-Monaco, S. McSweeney, D. Nurizzo, G. Papp, C. Rossi, J. Sinoir, C. Sorez, J. Surr, O. Svensson, U. Zander, F. Cipriani, P. Theveneau and C. Mueller-Dieckmann, J. Synchrotron Radiat., 2018, 25, 1249–1260 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS PubMed.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204–1214 CrossRef CAS PubMed.
- I. Caballero, M. Sammito, C. Millan, A. Lebedev, N. Soler and I. Uson, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 194–204 CrossRef CAS PubMed.
- A. J. Mccoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- D. D. Rodriguez, C. Grosse, S. Himmel, C. Gonzalez, I. M. de Ilarduya, S. Becker, G. M. Sheldrick and I. Uson, Nat. Methods, 2009, 6, 651–U639 CrossRef CAS PubMed.
- I. Uson and G. M. Sheldrick, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 106–116 CrossRef CAS PubMed.
- P. Skubak and N. S. Pannu, Nat. Commun., 2013, 4, 2777 CrossRef PubMed.
- G. N. Murshudov, P. Skubak, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- J. Luebben and T. Gruene, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8999–9003 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01165j |
| This journal is © The Royal Society of Chemistry 2019 |