
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaser illumination-induced dramatic catalytic activity change on Au nanospheres†
Zhongju
Ye
a,
Lin
Wei
b,
Lehui
Xiao
 *a and
Jianfang
Wang
c
*a and
Jianfang
Wang
c
aState Key Laboratory of Medicinal Chemical Biology, Tianjin Key Laboratory of Biosensing and Molecular Recognition, College of Chemistry, Nankai University, Tianjin, 300071, China. E-mail: lehuixiao@nankai.edu.cn; Web: http://www.xiaolhlab.cn
bKey Laboratory of Chemical Biology & Traditional Chinese Medicine Research, Key Laboratory of Phytochemical R&D of Hunan Province, College of Chemistry and Chemical Engineering, Hunan Normal University, Changsha, 410082, China
cDepartment of Physics, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, China
First published on 29th April 2019
Abstract
Understanding morphology dependent catalytic kinetics from a single nanoparticle plays a significant role in the development of robust nano-catalysts with high efficiency. Unfortunately, detailed knowledge of the morphology dependent catalytic properties of single nanoparticles after shape transitions is lacking. In this work, the distinct catalytic properties of a single gold nanoparticle (GNP) after symmetry breaking were disclosed at the single-particle level for the first time. The morphology of the spherical GNP was elongated into a rod shape (i.e., gold nanorod, GNR) with a tightly focused Gaussian laser beam based on the photothermal effect. By using the fluorogenic oxidation reaction (i.e., amplex red to resorufin) as a model reaction, noticeable variation in catalytic efficiency after the shape modulation process was found at the single-particle level. The GNP displays noticeably higher catalytic efficiency which might be ascribed to the heterogeneous lattice structure on the particle surface as confirmed by transmission electron microscopy (TEM) characterization. Rearrangement of surface atoms after shape modulation normally generates a more ordered crystal structure, resulting in a lower surface energy for catalytic reaction. However, both of these nanoparticles still exhibit dynamic activity fluctuation in a temporal dependent route, indicating a distinct spontaneous dynamic surface restructuring process. These kinetic evidences might facilitate the development nanoparticle-based heterogeneous catalysts, particularly based on the morphology effect.
Introduction
Nanoparticle-based heterogeneous catalysis has attracted considerable attention in many important chemical transformations, including organic synthesis, pollutant removal and energy production.1–3 Among those diverse nanostructures, metal nanoparticles are one of the most important heterogeneous catalyst with excellent or even new catalytic properties due to their high surface-to-volume ratio and chemical potentials.4–6 Due to this, many methods have been developed to fabricate metal nanoparticles with novel structures. Chemical synthesis is one of the most commonly used approaches in solution. However, the size- and shape-dependent catalytic properties of the nanoparticles made by most of these techniques are often obscured by broad size and shape distributions.7–10 For heterogeneous catalysis, different types of surface sites such as corner, edge and facet can lead to significant diversities even from the same type of nanoparticle.11–16 For instance, some works have proved that Au nanoparticles with smaller diameters were more susceptible to dynamic catalytic surface restructuring due to their higher surface energies.17–19 Atoms at the corner usually display the highest catalytic activity compared to other sites.11,20–22 As a consequence, exploration of the catalytic activity of nanostructures with different morphologies should be of great significance to better understand the catalytic mechanism. Unfortunately, a limited message has been deciphered so far on the aspect of the catalytic efficiency of the nanoparticle after morphology changes with single-particle resolution.According to earlier explorations, the catalytic properties of nanoparticles are mainly measured at ensemble level.23–26 However, because of the structural diversity, heterogeneous distribution of surface sites and surface restructuring dynamics under reaction conditions can further induce kinetic surface restructuring.14,27,28 The catalytic properties of nanoparticles are therefore fundamentally complicated. Single-particle measurement by removing ensemble averaging, that can uncover the heterogeneous and dynamic behaviors of individual nanoparticles, is highly desired to interrogate the catalytic properties of the particle with single-turnover resolution.29–32 Because of the difficult in morphology manipulation at single-particle level, it is still a grand challenge to disclose the catalytic properties of the same particle with different morphologies. Laser assisted shape modulation has been widely used for the controllable manipulation of metal nanoparticle morphology. Photothermal energy and optical force generated by a tightly focused laser beam can lead to optically-controlled nanolithography and nanofabrication.33–36 For example, a pulsed laser is often applied to fabricate and deform a rod structure to a spherical shape in solution.37 However, only rare examples have demonstrated the transformation of a symmetric structure into an anisotropic one.
In this work (Fig. 1), the catalytic dynamics of a gold nanostructure upon transformation from sphere to rod using controllable laser heating was explored for the first time. A tightly focused Gaussian laser beam with excitation wavelength (532 nm) comparable to the plasmonic resonance wavelength of GNPs was used to heat the nanoparticle via the photothermal effect. Radiation pressure induced elongation from a sphere to rod shape (GNR, with aspect ratio of 1.3 ± 0.06) was readily achieved according to the microscopic and spectroscopic characterizations, and is dependent on the heating time and laser power of the excitation light. By using the fluorogenic oxidation reaction (i.e., amplex red to resorufin) as a model reaction, the difference in catalytic efficiency between the GNP and GNR was dissected in detail at single-particle resolution. Interestingly, statistically analyzed results indicate that the GNR possesses a lower catalytic activity than that of the native spherical structure, indicating a slower spontaneous dynamic surface restructuring process which might be ascribed to the coordination saturation and reorganized surface lattice structure after morphology rearrangement. These new messages demonstrated herein provide deeper insight into the catalytic behaviors of single nanoparticles with different morphologies. Meanwhile, the single-particle shape modulation strategy as well as the single-molecule imaging approach introduced here should be applicable to study other nanoparticle catalysts and might facilitate the understanding of heterogeneous catalysis.
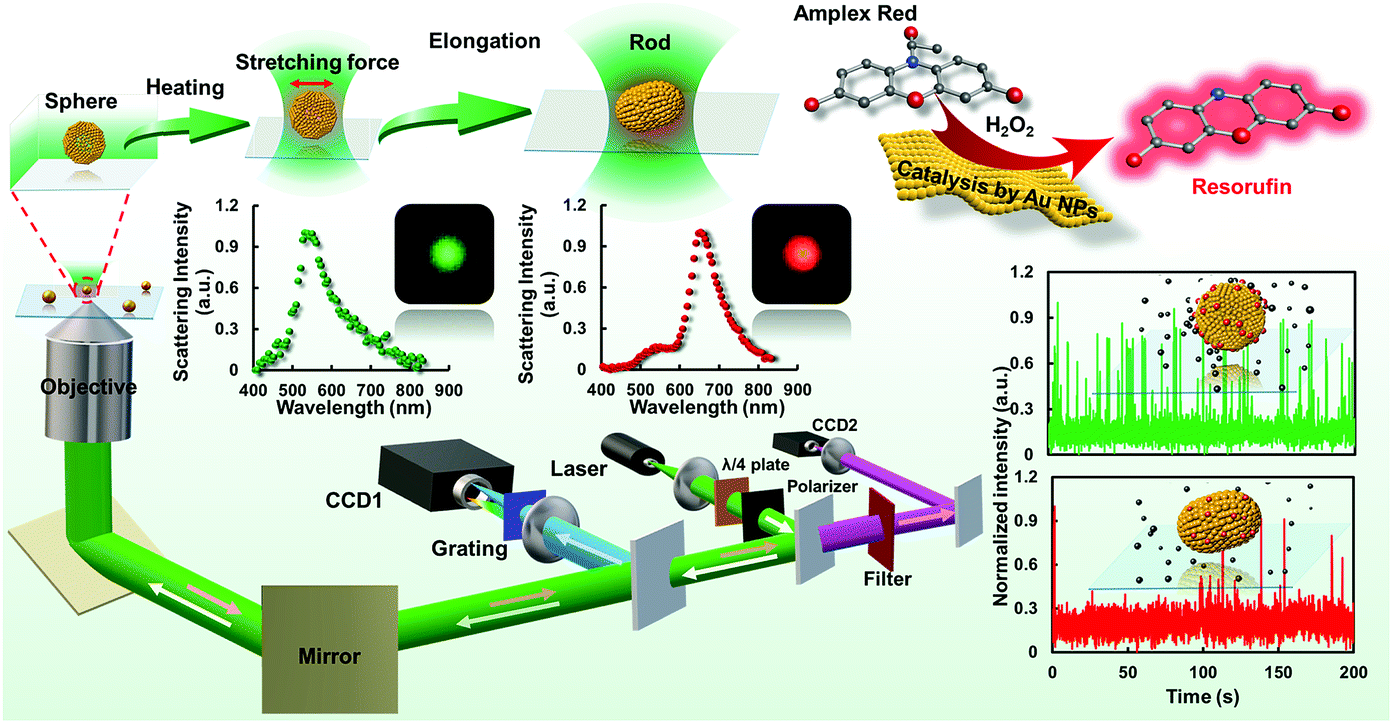 | ||
| Fig. 1 Schematic diagram of the procedures for laser-controlled shape modulation and single-molecule catalysis of gold nanostructures with different morphologies. | ||
Results and discussion
Elongation of GNPs with controllable laser melting
Even though many interesting works have been reported on the exploration of the catalytic efficiency of nanostructures with different morphologies in bulk solution, direct information on the catalytic properties of the same nanostructure in different morphologies, particularly at the single-particle level, is still lacking. Knowledge of this will contribute to a better understanding of the structure dependent catalytic activity across a variety of chemical transformations. In this regard, we modulated the morphology of a single GNP assisted by controllable laser melting. Firstly, a seed-mediated growth method was adopted to synthesize spherical GNPs. UV-vis absorption spectroscopy measurement illustrates that the GNPs show a characteristic absorption peak at 533 nm (Fig. 2a). Evidently, the GNPs exhibit a uniform size distribution with diameters around 60 nm as confirmed by the TEM image (Fig. 2b) which is further confirmed by the statistical result in Fig. 2c. The average value of the diameter is 59.0 ± 1.8 nm. The colloidal dispersibility of these GNPs was further confirmed by recording a color dark-field image on the cover-glass surface. An evenly distributed scattering signal and well-defined green color from individual GNPs were recorded as shown in Fig. 2d.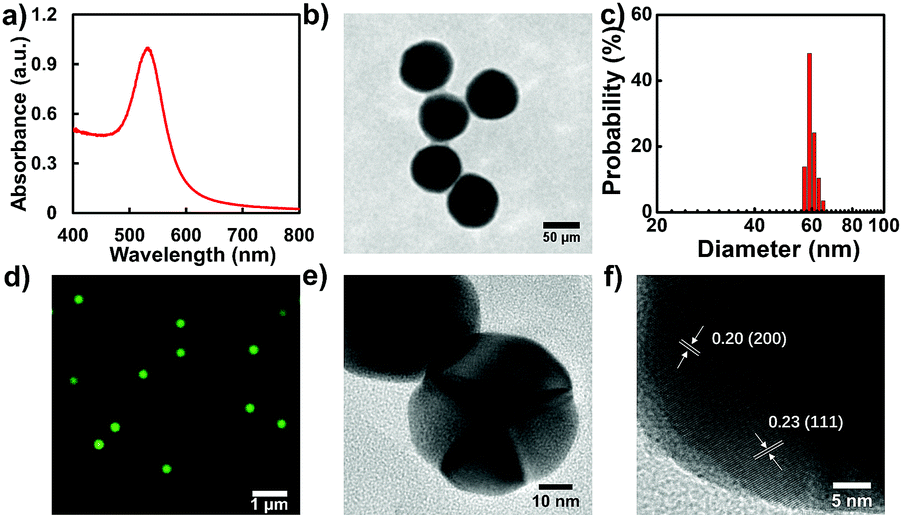 | ||
| Fig. 2 Spectroscopic and microscopic characterizations of GNPs. (a) UV-vis spectrum of GNPs. (b) TEM image of GNPs. (c) Size distribution of GNPs determined by TEM. (d) Representative dark-field scattering image of GNPs. (e) and (f) HRTEM images of GNPs. | ||
Since the photothermal effect is closely correlated with the strength of illumination power, the laser power-dependent elongation process was studied (Fig. 3a–d). Before shape elongation, the scattering spectrum of the particle was determined with a peak wavelength close to 537.5 nm as expected for a spherical structure with a diameter of approximately 60 nm. Herein, the laser power-dependent shape modulation kinetics were explored under the strengths of 10, 12.5, 15 and 20 mW. As expected, the scattering spectrum changed noticeably under higher laser power illumination. The rate of spectrum shift is positively correlated with the intensity of the laser beam as noted from the scattering spectrum. About a 106.5 nm red-shift in the scattering spectrum was obtained within 100 s under 20 mW compared to 3500 s under 10 mW. Detailed time-dependent elongation kinetics of the nanoparticle are shown in Fig. 3e–h. The scattering spectrum of the individual particle was gradually red-shifted with increasing heating time. A steady state was finally achieved which is correlated with the strength of the laser power. A higher laser power normally results in a faster saturation speed. It is worth noting that under these illumination conditions the scattering spectrum of the particle is basically within the range of 600–650 nm, which is essentially modulated by the wavelength of the excitation laser beam.
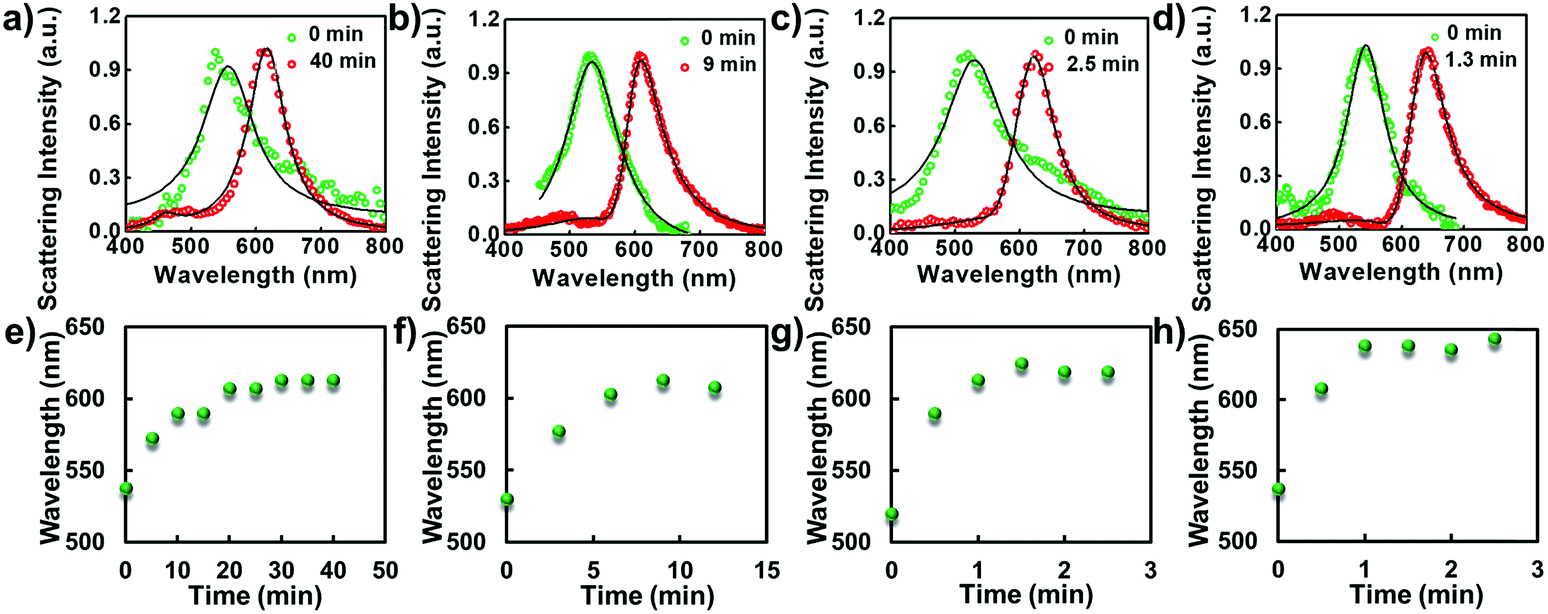 | ||
| Fig. 3 Scattering spectral characterizations of the laser induced shape modulation process under different laser powers and interval times. From (a to d) are 10, 12.5, 15 and 20 mW, respectively. (e–h) The corresponding time-dependent scattering spectral changes at the peak value as a function of time. | ||
Polarization measurements of the elongated nanoparticle
To disclose the morphology of the nanoparticle after the laser heating process, in situ microscopic characterizations were performed. Fig. 4a and b show the corresponding dark-field images of the nanoparticles captured by color CCD before and after laser heating. Prior to the laser heating process, all of the particles display a uniform green color. Given that the spherical particle was elongated to a rod shape after the laser heating process, a noticeable color change should be observed. For a GNR, the dark-field image normally displays red color, which is dependent on the corresponding aspect ratio. As confirmed in Fig. 4b, all of the particles are changed into a uniform red color after the laser heating process which is in good agreement with the scattering spectrum characterizations. Besides the dark-field microscopic imaging results, another robust strategy to characterize the anisotropic morphology of the particle beyond the optical diffraction limit is polarization measurement. Fig. 4c and d illustrate the polarization dependent scattering intensity measurements from a single particle before and after laser melting. Apparently, the intensity distribution pattern of the spherical GNP is noticeably different from that of the particle after laser heating.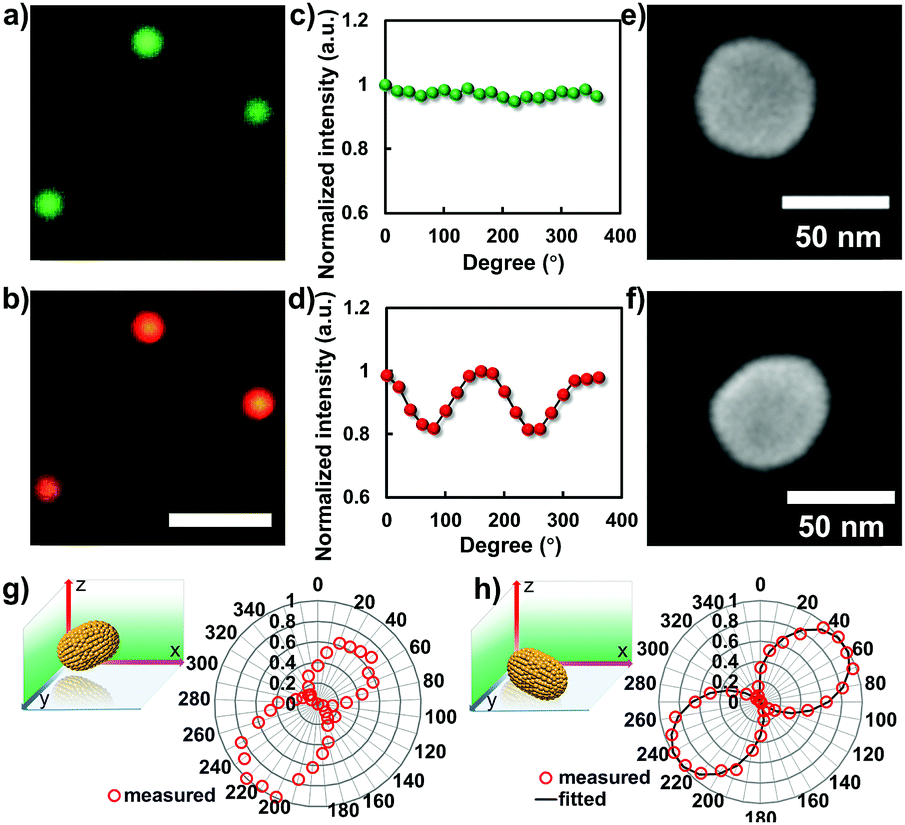 | ||
| Fig. 4 Microscopic characterizations and polarization measurements of gold nanostructures before and after laser-induced melting. (a and b) Representative dark-field scattering images of the gold nanostructures before and after laser melting, scale bar is 5 μm. (c and d) The measured polarization dependent scattering responses from a single particle before and after laser melting. (e and f) The SEM results of the particle before and after laser melting. Polarization dependent scattering responses from a single nanorod with (g) and without (h) apparent tilting angle in 3D. | ||
For spherical particles, the direction of the scattering signal in the far-field image plane is uniform, resulting in an orientation independent signal response in the polarization measurements. However, for a rod-shape nanoparticle laying down on the surface, the scattering response is anisotropic, and exhibits longitudinal and transverse surface plasmon modes with oscillation direction parallel to the long and short axes, respectively. In this case, given that the particle is laying down on the glass slide surface, the period of polarization dependent signal response should be π due to the symmetric effect, see Fig. 4d. When the particle is partially laying down with an apparent tilting angle, the symmetric effect is broken. The period of the polarization measurement degenerated into 2π. This argument is confirmed by embedding chemically synthesized GNRs in agarose gel where the particles are randomly distributed with an apparent tilting angle in 3D. As confirmed in the polarization measurement, two lobes with different strengths were observed in the scattering intensity plot (Fig. 4g), which is distinct from the result determined from the particle after laser heating on the glass slide surface (Fig. 4h). A more direct method to ascertain the laser assisted shape modulation process is SEM characterization. As shown in Fig. 4e, f and S1,† the apparent elongation process was observed after the laser heating process. The aspect ratio of the resulting GNR is ∼1.3.
Distinct catalytic activity from gold nanostructures with different morphologies
Several interesting works have proved that GNPs possess the ability to catalyze the fluorogenic reduction of non-fluorescent amplex red to highly fluorescent resorufin by H2O2.17,18 According to this, by using single-molecule fluorescence microscopy, we dissected the catalytic effect of the gold nanostructure change from a sphere- to rod-shape at single-particle resolution. It is worth pointing out that, so far, no message has been elucidated on the difference in catalytic effects of a nanostructure after a shape transition process, particularly from a sphere to rod shape. In this work, the single-particle catalytic experiments were performed within a flow channel loaded with various concentrations of amplex and H2O2 (0.25 to 2 μM amplex red and 50 mM H2O2). An EMCCD mounted on the left port of the microscope was used to capture signals from the catalytic products with a time resolution of 20 Hz. The fluorescence from the oxidative product resorufin was excited by a 532 nm laser. Each fluorescent burst indicates the generation of a catalytic product (resorufin) from a single gold nanoparticle. To ascertain the observed catalytic process generated from the target particle beyond optical diffraction limit, the dark-field imaging modality was further coupled with the single-molecule fluorescence imaging system. The position of the gold nanoparticle was firstly recorded in the dark-field image (Fig. 5). The fluorogenic oxidation reaction from the desired nanoparticle was then recorded by changing the light path into fluorescence imaging mode. Representative dark-field images and the corresponding catalytic fluorescence image are shown in Fig. 5.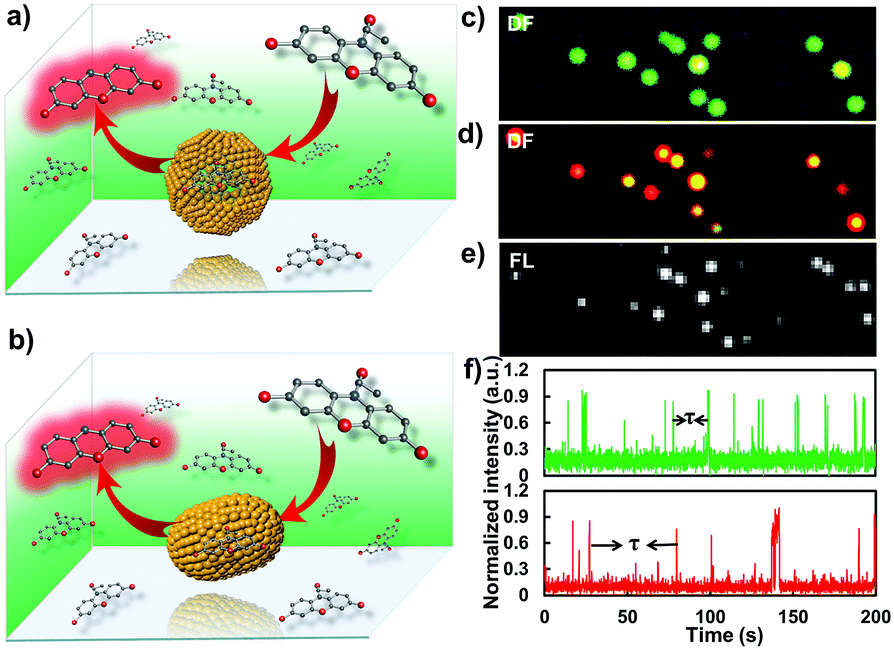 | ||
| Fig. 5 Fluorescence characterizations of the catalysis process from a single nanoparticle with single-turnover resolution. (a and b) illustrate the single-molecule catalysis from the gold nanoparticle before and after laser melting, respectively. (c and d) Dark-field images of the gold nanoparticles before and after the laser melting process, respectively. (e) The corresponding fluorescence microscopic characterization of the catalytic process from individual GNRs. (f) Representative time-dependent fluorescence intensity trajectories of the catalysis process from single gold nanoparticles before (green) and after (red) the laser melting process. | ||
Fig. 5f shows time dependent catalytic trajectories from gold nanostructures before and after laser melting, respectively. These two trajectories all contain many fluorescent bursts. We attributed the sudden intensity increase and sharp decline in the fluorescence trajectory to single catalytic turnover from a single nanoparticle. Each fluorescent burst indicates the formation of one fluorescent molecule and each intensity decrease represents a product dissociation event. The dissociated molecules in solution are undetectable at our imaging speed due to their fast diffusion coefficient. For a single nanoparticle, the interval time τ between two adjacent bursts is the time needed for the formation of a single product molecule. The individual value of time τ is stochastic, but the statistical properties, for example distributions and mean values, can well reveal the diversity of underlying catalytic reaction kinetics. Because the “on” time of the burst is far less than the “off” time that can be neglected in one turnover, the single-particle turnover rate can be derived directly by counting the number of bursts per unit time, and the value is equal to 〈τ〉−1, where 〈 〉 denotes ensemble averaging.
To disclose the difference in the catalytic kinetics of these gold nanostructures, we firstly examined how the turnover rate 〈τ〉−1 depends on the reactant concentrations by conducting the catalytic reactions with different reactant concentrations. Here, the concentration of amplex red was changed while H2O2 was kept constant at a saturated concentration (50 mM). Apparently, the turnover rate of these two types of particle all increase linearly with the increase of substrate concentration. Steady states were gradually achieved under different amplex red concentrations. It is evident that the turnover rate of the GNP was much higher than that of the elongated GNR, indicating a higher catalytic activity of the GNP for this oxidation reaction (Fig. 6a).
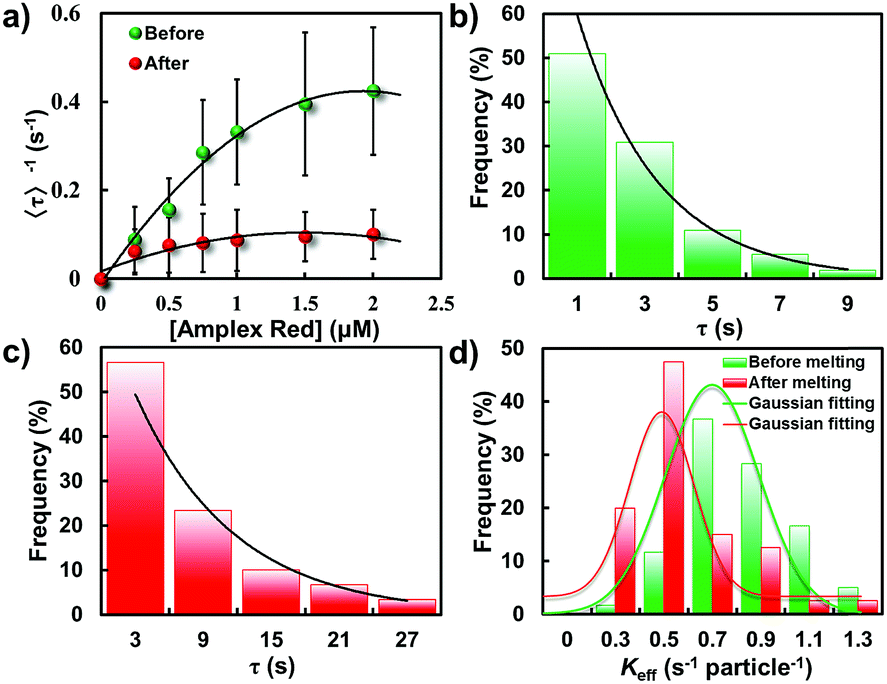 | ||
| Fig. 6 Statistically analyzed catalytic activity of GNPs and GNRs. (a) Amplex red concentration dependence of 〈τ〉−1 from gold nanoparticles before (green) and after (red) laser melting. Distributions of τ from a single nanoparticle before (b) and after (c) laser melting, respectively. Solid lines are the exponential fits with a decay constant of Keff = 0.84 and 0.69 s per particle for GNPs and GNRs, respectively. (d) Distributions of Keff from individual gold nanostructures for the oxidative N-deacetylation reaction. | ||
According to previous work, the kinetics of this nanoparticle-based catalytic reaction can be interpreted by the Langmuir–Hinshelwood mechanism, which has been widely used for heterogeneous catalysis. Based on this mechanism, the turnover rate from a single-particle was deduced according to the following equation:
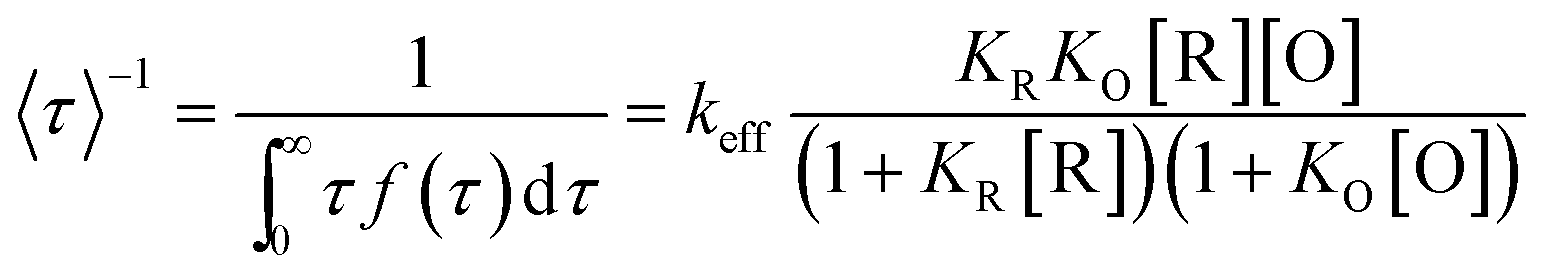 | (1) |
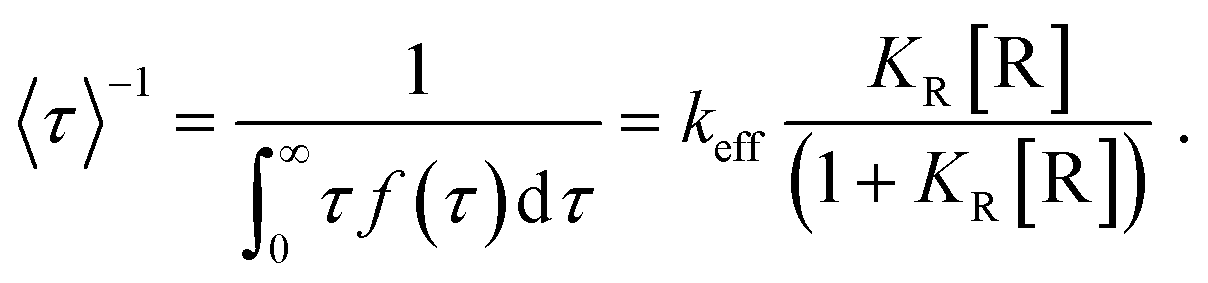 | (2) |
Fitting the nanoparticle-averaged data of 〈τ〉−1 at different [R] in eqn (2) gives keff = 0.73 ± 0.23 and 0.54 ± 0.31 s per particle, and KR = 1.25 ± 1.09 and 0.12 ± 0.09 μM, respectively. As the data in Fig. 6a are averaged over many particles, all the values illustrated here reflect the statistical properties of gold nanoparticles.
To differentiate the catalytic efficiencies of the sphere- and rod-shaped particles, we then quantified the catalytic activity from individual nanoparticles and determined their activity distributions. With the above kinetic messages, the probability density functions f(τ) of turnover time τ was derived:
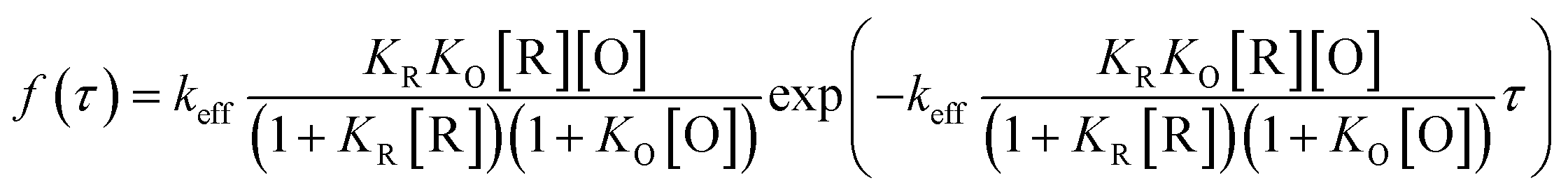 | (3) |
f(τ) = keff![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−keffτ) exp(−keffτ) | (4) |
Fig. 6b and c illustrate the distributions of τ from the fluorescence turnover trajectories of the GNP and GNR at a saturating [R] (1.5 μM). It is noticeable that τ of the GNR is larger than that of the GNP. The distributions all follow an exponential decay with a rate constant of keff. Statistically analyzing keff for GNPs and GNRs, broad distributions from both of them are observed (Fig. 6d), indicating a heterogeneously distributed catalytic efficiency among individual nanoparticles. This direct quantification of heterogeneous activity among individual gold nanostructures is uniquely available from single-particle measurements, and is difficult to realize from ensemble-averaged studies.
Importantly, knowledge of the difference of the catalytic efficiency between these two types of particles can be easily distinguished from the statistical analysis. The value of keff for GNPs essentially distributes with a peak value of 0.80 ± 0.03 s per particle which is much larger than that of GNRs (0.49 ± 0.02 s per particle), indicating a faster turnover rate of the GNPs than that of the physically deformed one. Furthermore, the heterogeneity of keff between these two kinds of particle can be quantified by the parameter of heterogeneity index h, which is defined as the value of the full width at half-maximum (fwhm) of the Gaussian distribution divided by the center of the Gaussian distribution.12,29,38 This parameter can be used as a reference to evaluate the spread of values from the average of keff. A larger heterogeneity index h indicates greater heterogeneity of the catalytic activity. In this work, the heterogeneity index h of the GNP is 71% while for the GNR is 57%, manifesting reduced heterogeneity after shape modulation.
Facet-dependent catalytic dynamics
Besides the direct quantification of the activity differences from one nanoparticle to another, the underlying dynamic activity fluctuations of a single nanoparticle can be examined by the temporal variation frequency of their catalytic turnovers. Temporal activity fluctuations of a single nanoparticle can be demonstrated from the burst kinetics of single-molecule fluorescence signals. On this basis, the rate of turnovers (the number of turnovers per unit time) is determined as shown in Fig. 7a and e. Broad temporal variations were observed in both these two types of particle, indicating dynamic activity fluctuation on a single nanoparticle from time to time. The dynamic activity fluctuation is essentially ascribed to the reaction changes in each single-turnover trajectory. Evaluation of the reaction changes on the activity fluctuation can be done by extracting the sequences of individual τ from their turnover trajectories and analyzing their autocorrelation functions Cτ(m) = 〈Δτ(0)Δτ(m)〉/〈Δτ2〉. Here, m is the turnover index number in the sequences, and Δτ(m) = τ(m) − 〈τ〉. If activity fluctuations occurred in the catalytic reaction, Cτ(m) ≥ 0 and shows a decay behavior with a decay time constant. The decay time constant is defined as the fluctuation correlation time.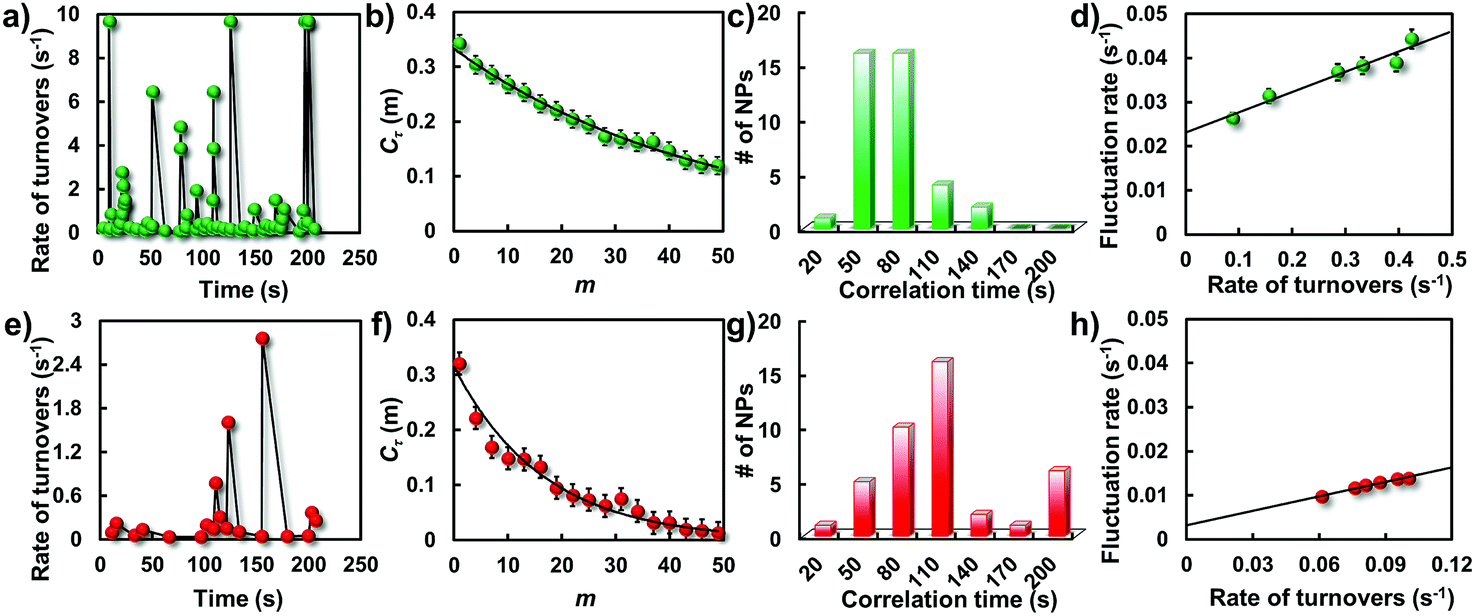 | ||
| Fig. 7 Single-particle catalytic dynamics. Trajectories of rate of turnovers from a single gold nanostructure before (a) and after (e) laser melting at an amplex red concentration of 1.5 μM. Autocorrelation functions of τ derived from the single-turnover trajectories before (b) and after (f) laser melting. Solid lines are exponential fits with decay constants of m = 47.6 and 16.7 turnovers, respectively. Histograms of fluctuation correlation times for the catalytic reaction before (c) and after (g) laser melting. Dependence of the activity fluctuation rate (the inverse of fluctuation correlation time) on the rate of turnover before (d) and after (h) laser melting. | ||
Fig. 7b and f show two such autocorrelation functions (Cτ(m)) from two representative particles at a saturating [R]. Both autocorrelation functions exhibit an exponential decay behavior, indicating that the two reaction rates all fluctuated from time to time. For the GNP, the exponential decay constant of Cτ(m) is 47.6 turnovers with an average turnover time of 2.4 s. The determined activity fluctuation correlation time for the corresponding GNP is 114.2 s which is evidently faster than that for the GNR (175.4 s). Since the correlation time reflects the time scales of the dynamic surface restructuring among those active surface sites on the nanoparticle, distributions of the correlation times from these two particles were explored to further determine the difference of the catalytic dynamics as shown in Fig. 7c and g. Consistent with the above observations, broad distributions were again observed from these two types of particle, indicating different surface restructuring time scales from different nanoparticles even with the same type of morphology. It is worth pointing out that the correlation time of the GNRs is evidently larger than the value from the GNPs, indicating a longer restructuring time for most deformed particles.
Because of the nanometer size effect, the surface of nanoparticles is unstable and can reconstruct dynamically, especially under catalysis, where the constantly changing adsorbate–surface interactions can further induce dynamic surface restructuring.28,39,40 These structural dynamics can cause temporal dynamics of catalytic activity from individual nanoparticles. We attribute the dynamics of catalytic activity to the underlying surface restructuring dynamics, that is, the activity fluctuation (the inverse of the correlation time). To support this point, we determined the dependence of the activity fluctuation rates on the rate of turnover at various concentrations. As shown in Fig. 7d and h, the activity fluctuation rates for all particles increase linearly with increasing turnover rate, reflecting that the underlying dynamic surface restructuring caused activity fluctuation is dependent on the turnover rate. In addition, an evident difference in the fluctuation rates for these two kinds of particle can be seen. The spontaneous surface restructuring rate of the GNPs is slightly faster than that of the GNRs at any rate of turnover, showing the distinct surface restructuring dynamics they experienced. The intercept of the GNPs is higher than that of the GNRs, further indicating the faster spontaneous dynamic surface restructuring they possess.
Elongation of the nanoparticle during the laser melting process significantly affects the photophysical properties of the nanoparticle, for instance, the structure of surface atoms and the surface energy.16,18,22,41 As confirmed by previous work, the growth rate of GNPs during seed-mediated synthesis is not constant. Generally, the particle grew faster at the beginning and slowed down linearly with increasing volume.42,43 Therefore, a gradient of surface defect density should be developed, manifesting in diverse crystal faces of GNPs as indicated by high-resolution TEM (HRTEM) results (Fig. 2e and f). This observation confirms well the active catalytic efficiency of GNPs. The higher surface defect density on the GNPs might facilitate the oxidation reaction because the surface atoms of GNPs have greater coordination unsaturation. In the case of GNRs, photothermal effect induced surface atom reshaping should associate with rearrangement of the surface lattice structure which might largely restore the surface defects and drive the surface atoms from an unstable condition to a more inactive state. The energetics for adsorbate binding and breaking in the catalytic reaction are dependent on the surface free energy, which can effectively affect the spontaneous surface restructuring. Therefore, surface defects of the facets and stability of the surface atoms have a vital impact on the catalytic activity of the nanoparticle. In addition, as described in the Experimental section in the ESI,† the colloidal GNPs studied here have citrate ions on their surfaces as stabilization ligands. Although these nanoparticles were extensively purified under centrifugation and washed with DI water many times, residual citrate can still be present and may play roles in the catalysis and the dynamic surface restructuring. Laser assisted photothermal modulation might attenuate the ligand effect on the heterogeneity of the catalytic efficiency. Taken together, this detailed knowledge of the change in catalytic dynamics after sphere to rod transformation by laser melting affords a new message on the better understanding of single-particle catalysis.
Conclusion
In summary, the morphology-correlated catalytic dynamics and activities of single gold nanoparticles after laser-controlled shape manipulation were dissected at the single-particle level for the first time. Real-time single-molecule catalysis reactions disclosed a distinct catalytic efficiency of the gold nanoparticle before and after laser melting. GNPs made by a seed-mediated growth method displayed a higher catalytic turnover rate compared to the particle elongated by laser melting, which might be essentially ascribed to the reduced surface defect density after the melting process. Meanwhile, the surface restructuring-coupled catalytic dynamics were also strongly influenced after shape deformation due to the deactivation of surface atoms in the catalytic reaction. This work factors out ensemble averaged effects and provides direct insight into the catalytic behaviors of single nanoparticles after shape transformation which might provide a deeper understanding of nanoparticle based heterogeneous catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, Project no. 21522502), the Fundamental Research Funds for the Central Universities, the Excellent Youth Scholars of Hunan Provincial Education Department (17B155) and the Opening Fund of Key Laboratory of Chemical Biology and Tradition Chinese Medicine Research (Ministry of Education of China), Hunan Normal University.Notes and references
- A. T. Bell, Science, 2003, 299, 1688 CrossRef CAS PubMed.
- B. M. Weckhuysen, Angew. Chem., Int. Ed., 2009, 48, 4910 CrossRef CAS.
- B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4557 RSC.
- D. Y. Murzin, Catal. Sci. Technol., 2011, 1, 380 RSC.
- S. Zhang, L. Nguyen, Y. Zhu, S. Zhan, C. Tsung and F. F. Tao, Acc. Chem. Res., 2013, 46, 1731 CrossRef CAS PubMed.
- L. Kun, W. W. Qin, X. Yan, T. H. Peng and L. Di, Front. Optoelectron., 2015, 8, 379 CrossRef.
- S. E. Habas, H. Lee, V. Radmilovic, G. A. Somorjai and P. Yang, Nat. Mater., 2007, 6, 692 CrossRef CAS PubMed.
- J. E. Millstone, S. J. Hurst, G. S. Métraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646 CrossRef CAS PubMed.
- S. Wu, A. W. Schell, M. Lublow, J. Kaiser, T. Aichele, S. Schietinger, F. Polzer, S. Kühn, X. Guo, O. Benson, M. Ballauff and Y. Lu, Colloid Polym. Sci., 2013, 291, 585 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed.
- I. L. C. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873 CrossRef CAS PubMed.
- T. Chen, S. Chen, P. Song, Y. Zhang, H. Su, W. Xu and J. Zeng, ACS Catal., 2017, 7, 2967 CrossRef CAS.
- T. Chen, S. Chen, Y. Zhang, Y. Qi, Y. Zhao, W. Xu and J. Zeng, Angew. Chem., Int. Ed., 2016, 55, 1839 CrossRef CAS PubMed.
- M. A. Newton, C. Belver-Coldeira, A. Martínez-Arias and M. Fernández-García, Nat. Mater., 2007, 6, 528 CrossRef CAS PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. Elsayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS PubMed.
- X. Zhou, N. M. Andoy, G. Liu, E. Choudhary, K. Han, H. Shen and P. Chen, Nat. Nanotechnol., 2012, 7, 237 CrossRef CAS PubMed.
- W. Xu, J. S. Kong, Y. E. Yeh and P. Chen, Nat. Mater., 2008, 7, 992 CrossRef CAS PubMed.
- X. Zhou, W. Xu, G. Liu, D. Panda and P. Chen, J. Am. Chem. Soc., 2010, 132, 138 CrossRef CAS PubMed.
- T. Chen, Y. Zhang and W. Xu, Phys. Chem. Chem. Phys., 2016, 18, 22494 RSC.
- T. Tachikawa, S. Yamashita and T. Majima, J. Am. Chem. Soc., 2011, 133, 7197 CrossRef CAS PubMed.
- Y. Zhang, J. M. Lucas, P. Song, B. J. Beberwyck, Q. Fu, W. Xu and A. P. Alivisatos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8959 CrossRef CAS PubMed.
- N. M. Andoy, X. Zhou, E. Choudhary, H. Shen, G. Liu and P. Chen, J. Am. Chem. Soc., 2013, 135, 1845 CrossRef CAS PubMed.
- H. Lee, S. E. Habas, S. J. Kweskin, D. R. Butcher, G. A. Somorjai and P. Yang, Angew. Chem., Int. Ed., 2006, 45, 7824 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed.
- M. D. Hughes, Y. Xu, P. Jenkins, P. Mcmorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings and F. King, Nature, 2005, 437, 1132 CrossRef CAS PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS PubMed.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932 CrossRef CAS PubMed.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrupnielsen, B. S. Clausen and H. Topsoe, Science, 2002, 295, 2053 CrossRef CAS PubMed.
- K. S. Han, G. Liu, X. Zhou, R. E. Medina and P. Chen, Nano Lett., 2012, 12, 1253 CrossRef CAS PubMed.
- P. Chen, X. Zhou, N. M. Andoy, K. Han, E. Choudhary, N. Zou, G. Chen and H. Shen, Chem. Soc. Rev., 2014, 43, 1107 RSC.
- P. Chen, X. Zhou, H. Shen, N. M. Andoy, E. Choudhary, K. Han, G. Liu and W. Meng, Chem. Soc. Rev., 2010, 39, 4560 RSC.
- T. Chen, B. Dong, K. Chen, F. Zhao, X. Cheng, C. Ma, S. Lee, P. Zhang, S. H. Kang and J. W. Ha, Chem. Rev., 2017, 117, 7510 CrossRef CAS PubMed.
- M. Fedoruk, M. Meixner, S. Carreteropalacios, T. Lohmuller and J. Feldmann, ACS Nano, 2013, 7, 7648 CrossRef CAS PubMed.
- M. Fedoruk, A. A. Lutich and J. Feldmann, ACS Nano, 2011, 5, 7377 CrossRef CAS PubMed.
- M. Enders, S. Mukai, T. Uwada and S. Hashimoto, J. Phys. Chem. C, 2016, 120, 6723 CrossRef CAS.
- S. Wang and T. Ding, ACS Nano, 2018, 13, 32 CrossRef PubMed.
- A. Plech, V. Kotaidis, M. Lorenc and J. Boneberg, Nat. Phys., 2006, 2, 44 Search PubMed.
- W. Xu, J. S. Kong and P. Chen, Phys. Chem. Chem. Phys., 2009, 11, 2767 RSC.
- R. Imbihl and G. Ertl, Chem. Rev., 1995, 95, 697 CrossRef CAS.
- D. A. King, Surf. Rev. Lett., 1994, 01, 435 CrossRef CAS.
- R. A. Van Santen, Acc. Chem. Res., 2009, 42, 57 CrossRef CAS PubMed.
- J. J. Martin and A. F. Armington, J. Cryst. Growth, 1983, 62, 203 CrossRef CAS.
- A. Gulati, H. Liao and J. H. Hafner, J. Phys. Chem. B, 2006, 110, 22323 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01666j |
| This journal is © The Royal Society of Chemistry 2019 |