
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous gold nanospheres via thiolate–Au(I) intermediates†
Hao
Lv‡
a,
Dongdong
Xu‡
 a,
Joel
Henzie
bc,
Ji
Feng
d,
Aaron
Lopes
e,
Yusuke
Yamauchi
*bfg and
Ben
Liu
*a
a,
Joel
Henzie
bc,
Ji
Feng
d,
Aaron
Lopes
e,
Yusuke
Yamauchi
*bfg and
Ben
Liu
*a
aJiangsu Key Laboratory of New Power Batteries, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing, Jiangsu 210023, China. E-mail: ben.liu@njnu.edu.cn
bKey Laboratory of Eco-chemical Engineering, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, China
cInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
dDepartment of Chemistry, University of California, Riverside, California 92521, USA
eDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
fSchool of Chemical Engineering and Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: y.yamauchi@uq.edu.au
gDepartment of Plant & Environmental New Resources, Kyung Hee University, 1732 Deogyeong-daero, Giheung-gu, Yongin-si, Gyeonggi-do 446-701, South Korea
First published on 28th May 2019
Abstract
Mesoporous gold (mesoAu) nanospheres support enhanced (electro)catalytic performance owing to their three-dimensional (3D) interior mesochannels that expose abundant active sites and facilitate electron/mass transfers. Although various porous Nanostructured Au has been fabricated by electrochemical reduction, alloying–dealloying and hard/soft templating methods, successful synthesis of mesoAu nanospheres with tailorable sizes and porosities remains a big challenge. Here we describe a novel surfactant-directed synthetic route to fabricate mesoAu nanospheres with 3D interconnected mesochannels by using the amphiphilic surfactant of C22H45N+(CH3)2–C3H6–SH (Cl−) (C22N–SH) as the mesopore directing agent. C22N–SH can not only self-reduce trivalent Au(III)Cl4− to monovalent Au(I), but also form polymeric C22N–S–Au(I) intermediates via covalent bonds. These C22N–S–Au(I) intermediates facilitate the self-assembly into spherical micelles and inhibit the mobility of Au precursors, enabling the crystallization nucleation and growth of the mesoAu nanospheres via in situ chemical reduction. The synthetic strategy can be further extended to tailor the sizes/porosities and surface optical properties of the mesoAu nanospheres. The mesoAu nanospheres exhibit remarkably enhanced mass/specific activity and improved stability in methanol electrooxidation, demonstrating far better performance than non-porous Au nanoparticles and previously reported Au nanocatalysts. The synthetic route differs markedly from other long-established soft-templating approaches, providing a new avenue to grow metal nanocrystals with desirable nanostructures and functions.
1. Introduction
Since the discovery of the high catalytic activity of gold (Au) in low-temperature CO oxidation reactions,1 Au nanostructures have received a great deal of attention for their wide range of applications in catalysis,2,3 electrocatalysis,4,5 surface enhanced Raman scattering (SERS),6–8 bioimaging and biotherapy.9,10 Among the Au nanostructures fabricated previously, mesoporous Au (i.e. mesoAu) is (electro)catalytically more active, due to large surface areas, highly accessible inner sites and abundance of low-coordination surface atoms.11–14 To date, various nanosynthesis methodologies and techniques, including alloying–dealloying,7,15,16 electrochemical reduction,12,17,18 soft-/hard-templating,12,13,19–21 and other methods,22–24 have been used to generate mesoAu. However, the accessible morphologies were generally limited to powders with irregular shapes or films on conductive substrates. This limitation restricts their potential in some applications. Irregular particles or mesoporous films have porous networks that are less accessible and have a higher tortuosity. By contrast, synthesizing mesoAu nanoparticles with uniform sizes and well-defined shapes ensures that all sides of the porous network are freely accessible for (electro)catalysis and also eliminates the average effects for distinguishing the intrinsic structure–performance relationship.11,13,25 Furthermore, some applications in biomedicines require nanosized spherical particles that can penetrate tumor cells for guided drug delivery.10,26Of the various nanosynthesis methodologies available, soft-templating approaches are the most powerful route to generate spherical mesoporous materials with three-dimensional (3D) interior mesochannels.27–29 In comparison with mesoporous carbon,30,31 oxides28 and even other noble metals,32–34 however, the synthesis of mesoAu nanospheres with precisely tailorable sizes and porosities has never been achieved with soft-templates, because of the challenge of controlling the reduction and crystallization of Au around surfactant templates. In addition, the high mobility of negatively charged AuCl4− (Au precursor) enables it to quickly migrate out of the soft templates, resulting in the formation of bulk nanoparticles and/or aggregates. Alkanethiolates (R–SH) are widely used as ligands to generate atomically precise Au nanostructures.35–39 These recipes use the self-reduction and binding interactions between Au precursors and R–SH to generate a stable thiolate–Au(I) polymer that serves as a starting material to generate Au nanostructures.36,40,41 Unfortunately, traditional R–SH is chemically hydrophobic and cannot serve as a template for the synthesis of the mesoAu nanospheres in an aqueous solution. We rationally hypothesize that a well-designed amphiphilic R–SH molecule with hydrophilic and hydrophobic moieties could simultaneously function as a thiolate–Au(I) polymer precursor and as a mesopore-directing agent to generate the mesoAu nanospheres.
In this work, we use a lab-synthesized amphiphilic surfactant of C22H45N+(CH3)2–C3H6–SH (Cl−) (denoted as C22N–SH hereafter) containing the hydrophobic long-chain alkyl R–SH covalently bonded to a hydrophilic quaternary ammonium group. C22N–SH is employed as the surfactant template to generate mesoAu nanospheres with 3D interconnected mesochannels in an aqueous solution. We propose that C22N–SH is able to chemically self-reduce the trivalent Au precursor (Au(III)Cl4−) into monovalent Au(I), generating polymeric C22N–S–Au(I) intermediates. Due to the amphiphilic nature of C22N–S–Au(I) and solvent conditions, it further self-assembles into spherical micelles that inhibit the mobility of Au precursors. Precisely controlling the reduction kinetics of these C22N–S–Au(I) hybrid micelles enables rational control over mesoAu nanospheres with tailorable sizes/porosities and optical properties. The resulting sub-100 nm mesoAu nanospheres have 3D interconnected mesochannels that expose abundant catalytic active sites, creating a material with an high surface area and fast mass/electron transfer rate. The mesoAu nanospheres thus demonstrate enhanced catalytic activity and stability in the electrochemical methanol oxidation reaction (MOR) compared to Au nanoparticle catalysts.
2. Results and discussion
2.1 Synthesis and structural characterization
Metallic mesoAu nanospheres were synthesized by an aqueous soft-templating method where C22N–SH, HAuCl4 and N2H4 are used as the micelle template, Au precursor and reducing agent, respectively. The mesoAu nanospheres were then collected by consecutively centrifuging and washing them with H2SO4/H2O2 and H2O. The morphology and mesoporous network of the mesoAu nanospheres were initially observed with electron microscopy. The low-magnification transmission electron microscope (TEM) image shows that the morphology of mesoAu is nearly spherical with high uniformity, dispersity, and purity (Fig. 1a and S1†). The average diameter of the nanospheres is 74 nm (defined as mesoAu-74 hereafter). The mesotructured pores were interconnected in 3D and homogeneously distributed throughout the entire nanosphere, rather than solely on the surface (Fig. 1b). High-angle annular dark-field scanning TEM (HAADF-STEM) offers higher contrast of the mesopores, showing the 3D interconnected nature of the mesopores (Fig. 1c and S2†). The size of the mesopores is ∼3 to 5 nm, and the average framework thickness is ca. 4 nm (Fig. 1d). STEM is also used to map the concentrations of elements in Fig. 1e. The mesoporous frameworks appear to be crystallographically continuous which should further facilitate mass/electron transfer during the electrocatalysis and improve electrocatalytic performance accordingly.32,42 The selected area electron diffraction (SAED) micrograph of an individual nanosphere exhibited a series of spots within concentric rings, indicating mesoAu is polycrystalline with a face-centered cubic (fcc) crystalline structure (Fig. 1f). Furthermore, high-resolution TEM observation displayed the high crystallinity of the mesoporous framework, with a 0.238 nm lattice fringe that assigned to the (111) plane of fcc Au (Fig. 1g). Interestingly, some uneven atomic steps and hierarchical kink sites are observable on the surfaces of the mesoporous frameworks (indicated by arrows). The presence of unsaturated surface active sites typically leads to enhanced (electro)catalytic performance.11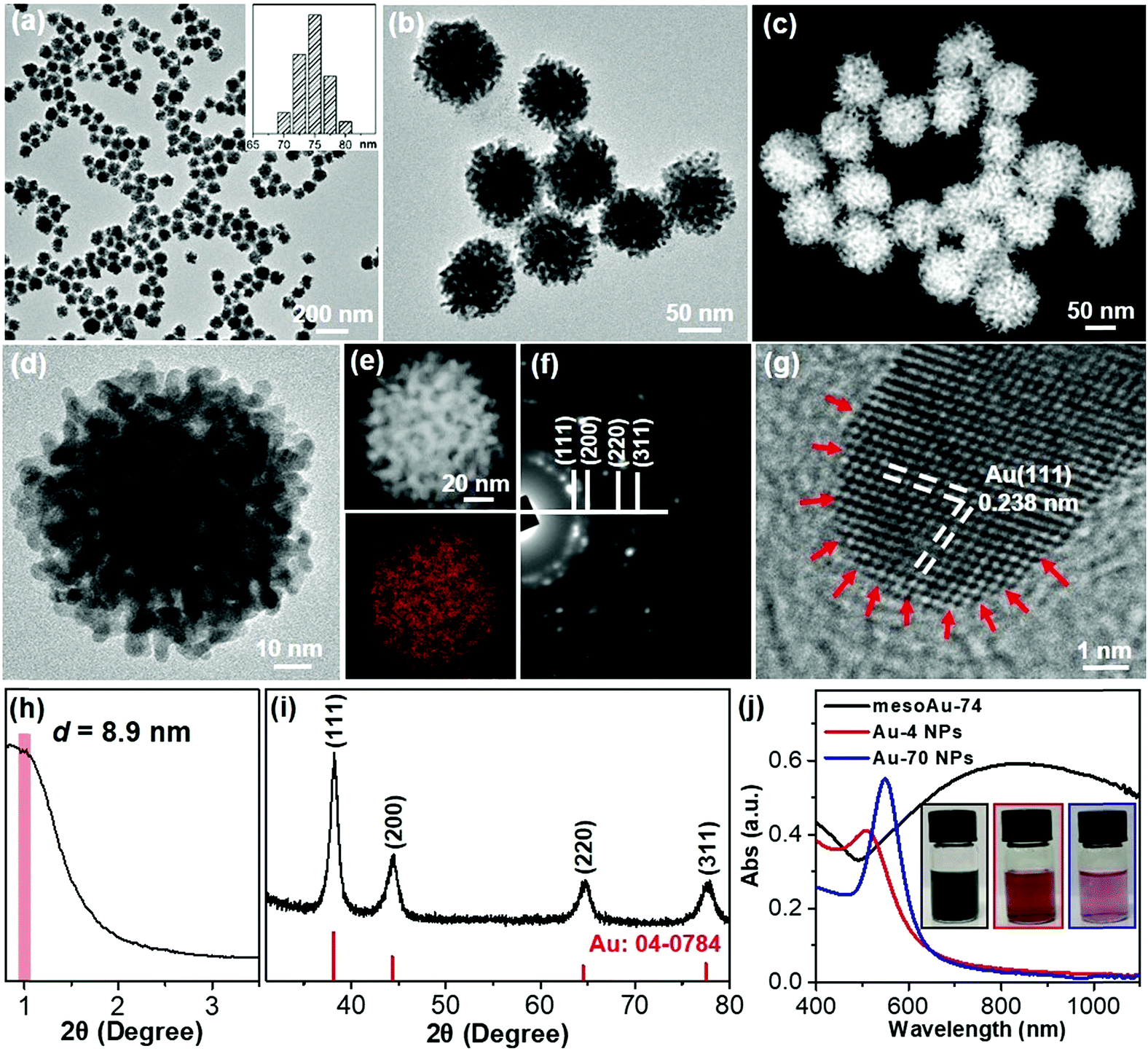 | ||
| Fig. 1 Structural characterization methods. (a and b) Low-magnification TEM, (c) HAADF-STEM and (d) high-magnification TEM images, (e) STEM elemental mapping, (f) SAED pattern, (g) high-resolution TEM image, and (h) low-angle and (i) wide-angle XRD patterns of mesoAu-74 nanospheres. (j) UV-vis spectra and (inset) corresponding digital pictures of mesoAu-74 nanospheres, Au-4 and Au-70 nanoparticles. | ||
The mesoporous periodicity of mesoAu nanospheres was also recorded by low-angle X-ray diffraction (XRD). The peak located at 2θ = ∼0.99° indicates that the average d-spacing (pore-to-pore distance) of the mesoAu nanospheres is 8.9 nm (Fig. 1h). The result closely matches the TEM observation. Wide-angle XRD further displayed four characteristic peaks, assigned to the (111), (200), (220) and (311) planes, respectively, of the fcc Au lattice (Fig. 1i). According to the Scherrer equation, the grain size of the mesoAu nanoparticles is ∼4.6 nm, roughly equivalent to the thickness of the mesoporous frameworks. Ultraviolet-visible (UV-vis) absorption spectroscopy was further used to reveal localized surface plasmon resonance (LSPR) of the mesoAu nanospheres (Fig. 1j), since the LSPR peak of Au nanocrystals is strongly sensitive to their nanostructures.7,8 The MesoAu nanosphere solution is dark-blue in color (see picture inset in Fig. 1j) and has a strong LSPR with a main extinction band at 828 nm. For the purpose of comparison, the solutions containing nanoparticles with diameters of 4.2 nm and 70 nm (defined as Au-4 and Au-70, Fig. S3†) were examined in UV-vis, to mimic the thickness of the mesoporous framework and the diameter of mesoAu nanospheres. The solutions have a bright red color and sharp LSPR peaks at 510 and 545 nm, respectively. Interestingly, the extinction band of mesoAu nanospheres was broad and significantly red-shifted. This is likely due to the collective plasmonic coupling between adjacent Au domains of the mesopores.7,12 These results further indicate the successful formation of mesoAu nanospheres with 3D interconnected mesochannels via a solution-phase surfactant-directed synthesis.
2.2 Formation mechanism
The utilization of lab-synthesized amphiphilic C22N–SH (see the Method in ESI†) as a mesopore-forming agent is critical in the formation of mesoAu nanospheres. The conformational states of the metal precursors in solution with the associated nanostructures is strongly sensitive to the interactions between metal ions and surfactants.35,37,41,43 UV-vis spectroscopy was first used to reveal the conformational states of the Au ions and the structural states of C22N–SH as it complexes with Au (Fig. 2a). The spectrum of pure HAuCl4 has an intense absorption band at 217 nm and a weak shoulder at 286 nm, matching the typical ligand-to-metal charge transfer (LMCT) transitions of AuCl4− between Au(III) and its chloro ligands.41,43 After mixing the Au(III)Cl4− with C22N–SH for 5 seconds (s), the absorption spectrum became featureless, similar to pure C22N–SH. This indicates that the Au(III)Cl4− was totally reduced by the thiol group of C22N–SH to Au(I). Furthermore, it forms a polymer containing C22N–SH/Au(I) complex intermediates (named as C22N–S–Au(I) hereafter).35,37,44 No new absorption band characteristic of an LSPR appears at this time, also indicating that Au(I) was not further reduced to Au(0). In other words, these results imply that the functional thiol group in C22N–SH assists the self-reduction of Au(III)Cl4− to Au(I) and further forms chemically stable C22N–S–Au(I) intermediates by covalent bonds, both of which may be beneficial the formation of mesoAu nanospheres. | ||
| Fig. 2 Formation of polymeric C22N–S–Au(I) hybrid micelles. (a) UV-vis spectra and (inset) corresponding digital pictures of C22N–SH, Au(III)Cl4− and C22N–S–Au(I) hybrid micelles. (b) High-resolution XPS Au 4f and (c) S 2p spectra of C22N–S–Au(I) hybrid micelles. Low-angle XRD patterns of C22N–S–Au(I) hybrid micelles in (d) aqueous and (e) solid states. (f) FT-IR spectra of C22N–SH and C22N–S–Au(I) hybrid micelles. | ||
High-resolution X-ray photoelectron spectroscopy (XPS) was employed to further investigate the chemical states of Au and S in the C22N–S–Au(I) precursor by freeze-drying it to remove the solvent (Fig. 2b). It had two characteristic peaks at 84.0 and 87.6 eV that match with Au 4f7/2 and Au 4f5/2, respectively. The Au 4f peak was further deconvoluted to separate the signal from Au(I) and Au(0). Au(I) has a binding energy of 84.4 eV and contributes ∼74% of the overall signal. Previously reported thiolate–Au(I) complexes also had a high amount of Au(I),40,45 offering further proof of the formation of polymeric C22N–S–Au(I) intermediates in our synthetic system. Fig. 2c shows the S 2p spectrum with two signals at 163.1 and 161.6 eV, which are slightly higher in energy than the values for Au nanoparticles with adsorbed thiols.45 Fitting the S 2p peak revealed three pairs of components: thiol groups in the alkanethiolate (R–SH) at 163.1 and 164.2 eV, thiolate–Au (S–Au) at 162.9 and 163.9 eV, and sulfide at 161.5 and 162.5 eV. Obviously, the presence of covalent S–Au bonds further indicates that C22N–SH could reduce AuCl4− to Au(I) and facilitates the formation of polymeric C22N–S–Au(I) intermediates by covalent bonds.40,46
Considering the amphiphilic nature of C22N–SH with the hydrophobic alkyl chain (C22) and hydrophilic quaternary ammonium group, polymeric C22N–S–Au(I) intermediates could further self-assemble into ordered mesophases. The mesopores of mesoAu are nearly spherical, so we deduced that the polymeric C22N–S–Au(I) intermediates originally form clusters of micelles in an aqueous solution. Such behavior has been previously reported in the synthesis of other kinds of mesoporous materials with amphiphilic surfactants.47 To confirm the existence of spherical C22N–S–Au(I) hybrid micelles, a solution containing C22N–S–Au(I) hybrids was directly investigated by XRD. A low-angle XRD peak at 1.51° (d = 5.8 nm) indicated the formation of micelles (Fig. 2d). Meanwhile, the solution exhibited the Tyndall effect, also showing the presence of micelles (inset in Fig. 2d). The C22N–S–Au(I) hybrid micelles are slightly larger compared to the size of the mesopores in mesoAu as observed in TEM (3 to 5 nm), indicating the spherical micelles may shrink during the crystallization of mesoAu.12 Low-angle XRD was further performed on C22N–S–Au(I) micelles after freeze-drying (Fig. 2e). Well-defined 1st and 2nd diffraction peaks in between 2 and 5° indicate that the spherical C22N–S–Au(I) hybrid micelles self-assembled into an ordered mesophase during the drying step.35,41 The d-spacing is ∼4.0 nm, which is almost similar to the size of the mesopores in mesoAu. Furthermore, Fourier transform infrared (FT-IR) spectroscopy of polymeric C22N–S–Au(I) hybrid micelles exhibited two strong signals at 2926 and 2858 nm (Fig. 2f). These originated from symmetric (d+) and antisymmetric (d−) CH2 stretching band, respectively.41,48 Obviously, the two peaks positively shifted compared to pure C22N–SH (2919 and 2850 nm), indicating that the formation of C22N–S–Au(I) increased the population of the gauche conformation and thus enlarged/stabilized the spherical hybrid micelles.
The combined observations described above reveal that the synthesis of mesoAu nanospheres is based on the in situ chemical reduction of the Au precursor (intermediate C22N–S–Au(I) hybrid micelles) by the C22N–SH surfactant (Fig. 3). Using C22N–SH as the surfactant template has three key features: (i) the hydrophobic C22 tail helps drive and stabilize the assembly of the C22N–SH into spherical micelles in an aqueous solution. This feature is essential for the formation of mesopores. (ii) The hydrophilic quaternary ammonium maintains the solubility of the surfactant in an aqueous solution and electrostatically coordinates with negatively charged Au precursors (Au(III)Cl4−). These features enable the templates to confine and direct the crystalline growth. And (iii) the functional thiol group self-reduces Au(III)Cl4− to Au(I) and covalently drives the formation of polymeric C22N–S–Au(I) micelles. This feature inhibits the mobility of Au precursors and thus optimizes the crystallization growth of Au by localizing it on the surface of the micelles. Subsequently, polymeric C22N–S–Au(I) micelles behave as the mesopore-forming sacrificial surfactant template, and in situ reduction by N2H4 with optimal reduction kinetics results in the formation of monodispersed mesoAu nanospheres. Lastly, the surfactant can be removed from the mesoporous channels of mesoAu by H2SO4/H2O2.
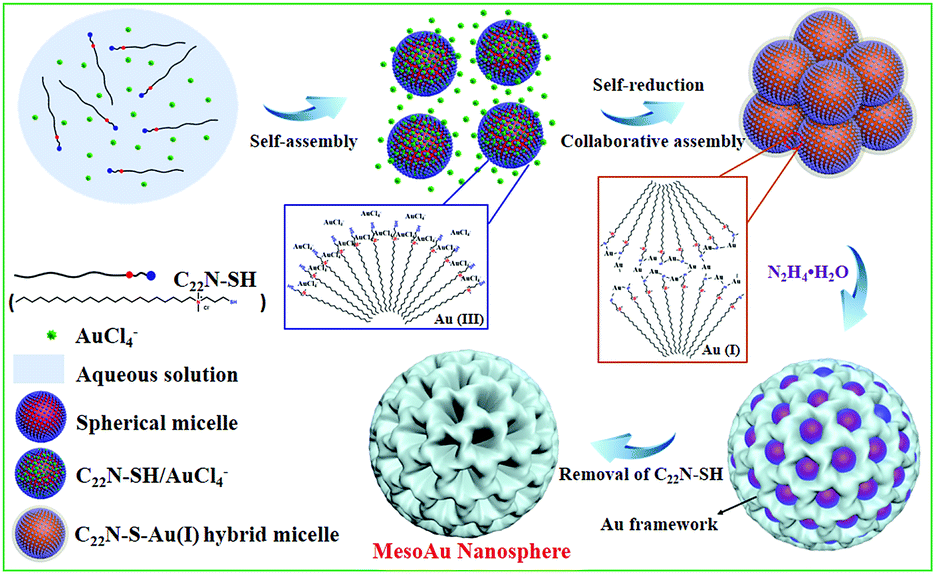 | ||
| Fig. 3 Scheme illustrating the soft-templating synthesis route of mesoAu nanospheres via C22N–S–Au(I) intermediate micelles. | ||
To further highlight the critical effect of amphiphilic C22N–SH as the surfactant template for the formation of mesoAu nanospheres, a series of control experiments were carried out in detail. First, in the absence of the surfactant, non-porous large nanoparticles (NPs) and their aggregates formed because nanoparticles with lower surface areas are more thermodynamically stable (Fig. S4†). Second, traditional amphiphilic quaternary ammonium-functionalized surfactants (C16N, C22N and (C18)2N) were examined. All three surfactants generated large disordered NPs and/or attached as aggregates with UV-vis absorption in the range of 500–600 nm, regardless of the hydrophobicity of the surfactants (Fig. S5–S7†). Meanwhile, the absorbance of the AuCl4− was retained in solutions containing these surfactants and AuCl4−, further indicating the importance of the SH functional group in the formation of R–S–Au(I) intermediates. Similar results were also achieved using the amphiphilic bifunctional surfactant of C22N–COOH49 (Fig. S8†). Third, we used C22–SH without any hydrophilic quaternary ammonium group. It could not function as a pore directing agent because it is insoluble in an aqueous solution. In addition, the water-soluble thiol-containing molecule of thiourea and mixed surfactants of C22N/thiourea were also investigated (Fig. S9 and S10†). Obviously, the introduction of a thiol group could result in the formation of R–S–Au(I) intermediates, as indicated by the disappearance of UV-vis signals for AuCl4−. However, the final Au products were also spherical NP aggregates rather than mesoAu. These results further confirmed the critical effect of C22N–SH as the template for the synthesis of mesoAu nanospheres via covalently bonded C22N–S–Au(I) intermediates.
2.3 Size control
Our approach for the formation of mesoAu nanospheres with 3D interior mesopores is based on in situ chemical reduction of the intermediate C22N–S–Au(I) hybrid micelles which inhibit the mobility of the Au precursor and nanoconfine the crystallization growth of Au. Therefore, various means to control the kinetics of the reduction rate of stable C22N–S–Au(I) intermediates should enable us to precisely tailor the sizes and porosities of the mesoAu nanospheres and in turn modify their LSPR resonances (Fig. 4). As shown in Fig. 4a, the higher reaction temperature and pH result in larger mesoAu nanospheres. Representative TEM images show that, at the evaluated reduction temperatures, the diameter of mesoAu nanospheres gradually increases from 17 nm at 0 °C, to 48 nm at 25 °C, 74 nm at 35 °C, 99 nm at 50 °C, and finally to 175 nm at 75 °C and 182 nm to 95 °C, respectively (defined as mesoAu-17, mesoAu-48, mesoAu-99, mesoAu-175 and mesoAu-182, respectively) (Fig. 4c–f and S11†). At the same time, the mesopores became larger, and the size distribution of the particles broadened as the C22N–S–Au(I) micelles became disordered at higher temperature.48 The diameters of the nanospheres increased almost linearly with reaction temperature, indicating that the size of the particles and mesoporosity are well controlled. The size of the mesoAu nanospheres was also controlled by changing the pH of the reduction solution, although the largest size was limited to 92 nm (see the TEM images in Fig. S12†).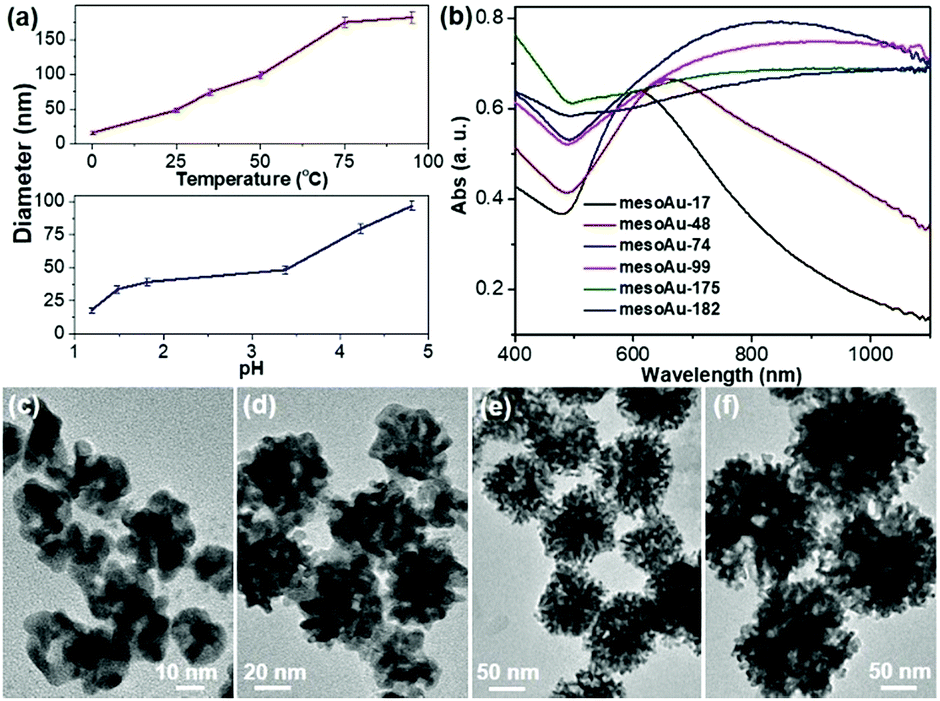 | ||
| Fig. 4 Size-controlled synthesis. (a) Size-controlled synthesis diagrams of mesoAu nanospheres by reaction temperature (pH = 3.4) and pH (25 °C). (b) UV-vis spectra and (c–f) representative TEM images of mesoAu nanospheres with different sizes synthesized at different temperatures of (c) 0 °C, (d) 25 °C, (e) 50 °C, and (f) 75 °C. | ||
MesoAu nanospheres with tailorable sizes and porosities also result in different LSPR responses. Fig. 4b shows that the peaks of the LSPR gradually red-shifted in the range of 600–1100 nm with different sizes (porosities), although a broader adsorption band was also obtained for a larger size. Specifically, a well-resolved peak appeared at 610 nm for mesoAu-17, which progressively red-shifted to 658 nm for mesoAu-48, 828 nm for mesoAu-74, 907 nm for mesoAu-99, 1012 nm for mesoAu-175, and eventually to 1098 nm for mesoAu-182. Precisely controlling the diameters of the particles and sizes of the mesopores should enable broad tunability of LSPRs for plasmon-based applications.7,50
2.4 Catalytic performance
The spherical morphology and presence of interconnected mesoporous channels in mesoAu should help to accelerate the electron/mass transfers in (electro)catalysis. Meanwhile, the rough mesoporous framework with uneven atomic steps and unsaturated hierarchical kink sites will expose more (electro)catalytically active sites. Therefore, the mesoAu nanospheres are expected to perform well in the (electro)catalysis and far beyond the capability of bulk Au nanoparticles. The MOR was employed as a model reaction to evaluate the electrocatalytic performance of mesoAu-74 nanospheres. As a comparison, non-porous Au-4 and Au-70 NPs (Fig. S3†) were also studied, to highlight the advantages created by the mesostructuring of Au. Oxide stripping was first performed for all three samples in 0.5 M H2SO4. Fig. 5a shows two characteristic peaks with a very broad peak at 0.2–0.5 V (vs. the saturated calomel electrode (SCE)) in the forward scan that matches the formation of Au oxides by adsorption of OH on Au (Au-OHads) and a sharp peak at 0.8–1.1 V in the backward scan that corresponds to the reduction of Au oxides.11,51 These are the potentials of oxide reactions on Au nanocatalysts in acidic electrolyte. The electrochemically active surface area (ECSA) was estimated to be 9.61 m2 g−1 for mesoAu-74, 13.64 m2 g−1 for Au-4 and 2.22 m2 g−1 for Au-70, respectively (inset in Fig. 5a), based on the area of Au oxide reduction peaks. These figures match the structural features of the samples.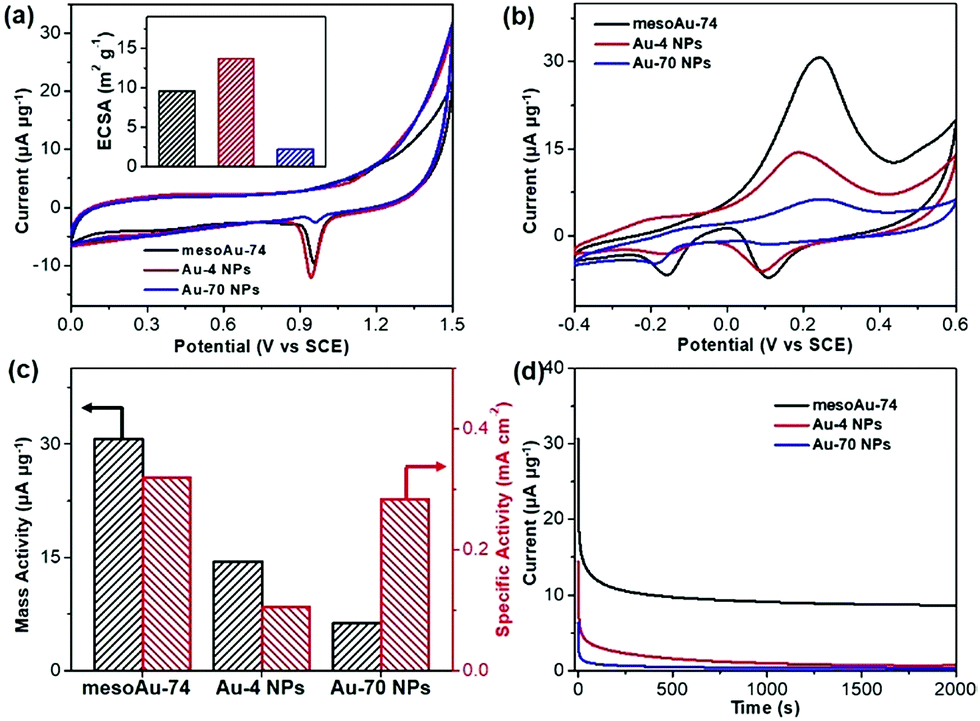 | ||
| Fig. 5 Electrocatalytic performance. (a) Oxide stripping curves and (inset) corresponding ECSAs, (b) CV curves of methanol electrooxidation, (c) summarized mass and specific activities, and (d) i–t chronoamperometric stability curves of mesoAu-74 nanospheres, and Au-4 and Au-70 NPs (at peak potentials). CVs and stability curves were collected in 0.5 M KOH and 2.0 M methanol at a scan rate of 20 mV s−1, while oxide striping was performed in 0.5 M H2SO4 at a scan rate of 10 mV s−1. | ||
Electrocatalytic MOR activities of the three Au nanocatalysts were determined in 0.5 M KOH and 2.0 M methanol (Fig. 5b). Two sets of peaks associated with the MOR can be seen in the cyclic voltammogram (CV) curves.52–54 The methanol electrooxidation peak for the mesoAu-74 catalyst appeared at 0.231 V in the forward scan, while the peaks associated with the reduction of Au-OHads to Au and further removal of incompletely oxidized carbonaceous intermediates were located at 0.089 and −0.156 V, respectively, in the backward scan.11,13 The mass activity of mesoAu-74 in the MOR is 30.7 μA μg−1, whereas both Au-4 and Au-70 had lower mass activities at 14.5 and 6.3 μA μg−1, respectively. Thus in this comparison under equivalent conditions, the mesoAu-74 nanospheres exhibited 2.1- and 4.9-times higher mass activity compared to its non-porous NP counterpart catalysts. A high forward current to backward current (If/Ib) ratio of 4.43 for the mesoAu nanospheres was also seen, as compared with 2.77 and 2.46 of Au-4 and Au-70 nanoparticles, respectively. This indicated the better tolerance to the poisoning intermediates (Fig. S13†). Meanwhile, a more negative onset potential of −70 mV indicated that it is easier for mesoAu-74 to oxidize methanol (lower activation energy).55,56 Considering both the ECSA and mass, mesoAu-74 nanospheres also had a higher specific activity of 0.320 mA cm−2, which was 3.05- and 1.13-times higher than that of Au-4 (0.105 mA cm−2) and Au-70 nanoparticles (0.284 mA cm−2), respectively (Fig. 5c). Table S1† summarizes the mass and specific activities of previously reported state-of-the-art Au catalysts for the MOR in alkaline media. Obviously, the mesoAu nanospheres exhibit far better performance in the electrocatalytic MOR than previously reported Au nanocatalysts. The 3D mesochannels enable better charge/mass transfers and expose more unsaturated surface active sites, while simultaneously increasing electrocatalytic stability by facilitating the removal of carbonaceous intermediates that tend to poison the surfaces of noble metal catalysts.11,13,51Fig. 5d shows current–time (i–t) chronoamperometric measurements for the three Au nanocatalysts examined in this study. After 2000 s, mesoAu-74 still retained a steady current of 8.61 μA μg−1 (∼27.9%). By contrast, that of Au-4 and Au-70 nanoparticles decreased to 0.7 and 0.3 μA μg−1 (only ∼4.9% and 4.4%, respectively).
3. Conclusion
In summary, we have successfully developed a novel soft-templating strategy to generate mesoporous Au nanospheres containing an interior space of 3D interconnected mesochannels. To our knowledge, this is the first demonstration of the use of a soft-templating method for synthesizing highly uniform mesoAu nanoparticles. Through systematically studying the formation mechanism, we found that the amphiphilic C22N–SH pore directing agent served initially as a reducing agent to form polymeric C22N–S–Au(I) intermediates. This step was important in generating mesoAu because it limits the ionic mobility of Au and localizes it to the surfaces of the self-assembled spherical C22N–S–Au(I) micelles and likely triggers the formation of a mesoporous network. This synthetic strategy can be adapted to tune the size and porosity of the nanoparticles by changing the reduction kinetics of C22N–S–Au(I) via reaction temperature and pH. Modifying the size and porosity of mesoAu allows us to tune the LSPR of the particles from visible to near infrared frequencies. The mesoAu nanospheres are electrocatalytically active because the morphology of mesoAu exposes abundant active sites via its high surface area and accessibility, which accelerates both electron and mass transfers in addition to assisting in the removal of carbonaceous species. As a model catalysis, the mesoAu nanospheres exhibited remarkably enhanced catalytic activity and stability in the electrochemical MOR as compared to bulk Au nanoparticles and previously reported Au nanocatalysts. We believe that the synthetic protocol, which differed from the long-established soft-templating approaches, would open a new avenue to explore nanostructured metal nanocrystals for a wide range of applications.4. Methods
4.1 Synthesis of mesoAu nanospheres
In a typical synthesis of mesoAu-74, 1.0 mL of HAuCl4 aqueous solution (10 mM) was added to 5 mL of C22N–SH aqueous solution (19 mM). The color of the solution changed from yellow to colorless in 5 s under gentle shaking. After homogeneously mixing the solution at 35 °C for 0.5 h, 6.4 mL of N2H4 was injected into the solution. Its color immediately changed from clear to dark-blue. It was incubated at 35 °C for another 0.5 h, then the product was collected by suspending it in a solution of H2SO4 and H2O2 (H2SO4/H2O2 = 3/1) for 0.5 h, and then washed with H2O several times. The clean procedures were repeated one more time to further remove the surfactant. Similarly, mesoAu nanospheres with different sizes were synthesized at different reaction temperatures from 0 to 95 °C or in the pH range of 1.2 to 4.8. Meanwhile, Au nanostructures were also investigated by other surfactants with the same procedures for mesoAu-74.4.2 Electrocatalytic measurements
Electrocatalytic tests were performed on the CHI 660E electrochemical analyzer at 25 °C. A three-electrodes system was used in all electrochemical tests, which consisted of a carbon rod as the counter electrode, a silver/silver chloride electrode (SCE) as the reference electrode, and the mesoAu nanospheres coated onto a glassy carbon electrode (GCE, 0.07065 cm2) as the working electrode. The potentials used in this work were reported with respect to the saturated calomel electrode (SCE). The preparation of the ink was as follows: 1 mg of catalysts and 4 mg of Vulcan XC-72 were dissolved in 1 mL co-solvent of ethanol and water (ethanol/water = 4/1); after sonication for 30 min, 50 μL of Nafion was injected in the ink with another 30 min sonication. Then, 6.0 μL of the ink was dropped on the GCE electrode and dried at room temperature for electrocatalytic investigations. All the electrochemical tests were performed in N2-saturated 0.5 M H2SO4 or 0.5 M KOH and 2 M methanol solution.4.3 Structural and compositional characterization
TEM observations were performed with a JEOL JEM-2100 microscope operated at 200 kV (Cs 0.5 mm, point resolution 1.9 Å). High-angle annular dark-field scanning STEM was performed on a JEOL JEM-2100F microscope equipped with STEM and EDS detectors for elemental mapping analysis. TEM and STEM samples were prepared by casting a suspension of the samples on a carbon coated copper grid (300 mesh). XRD patterns were recorded on powder samples using a D/max 2500 VL/PC diffractometer (Japan) equipped with graphite-monochromatized Cu Kα radiation. XPS was performed on a scanning X-ray microprobe (Thermo ESCALAB 250Xi) that uses Al Kα radiation. The binding energy of the C 1s peak (284.8 eV) was employed as a standard to calibrate the binding energies of other elements. UV-vis absorption spectroscopy was performed on a Shimadzu UV-2450 ultraviolet-visible spectrophotometer. FT-IR spectroscopy was performed on a Bruker Hyperion 2000. 1H NMR spectra were recorded on an Avance III HD 400 spectrometer (400 MHz), and the chemical shifts were reported in ppm relative to the residual deuterated solvent and the internal standard tetramethylsilane.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Jiangsu Specially Appointed Professor Plan, National Natural Science Foundation of China (No. 21501095), Natural Science Foundation of Jiangsu Province (No. BK20180723), research fund from the Priority Academic Program Development of Jiangsu Higher Education Institutions, and National and Local Joint Engineering Research Center of Biomedical Functional Materials. This work was performed in part at the Queensland node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano- and micro-fabrication facilities for Australia's researchers.Notes and references
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- G. J. Hutchings, ACS Cent. Sci., 2018, 4, 1095–1101 CrossRef CAS.
- L. Prati and A. Villa, Acc. Chem. Res., 2013, 47, 855–863 CrossRef PubMed.
- P. Rodriguez and M. T. M. Koper, Phys. Chem. Chem. Phys., 2014, 16, 13583–13594 RSC.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan and C. Cao, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- M. R. Jones, K. D. Osberg, R. J. Macfarlane, M. R. Langille and C. A. Mirkin, Chem. Rev., 2011, 111, 3736–3827 CrossRef CAS PubMed.
- K. Liu, Y. Bai, L. Zhang, Z. Yang, Q. Fan, H. Zheng, Y. Yin and C. Gao, Nano Lett., 2016, 16, 3675–3681 CrossRef CAS PubMed.
- H.-E. Lee, H.-Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. T. Nam, Nature, 2018, 556, 360–365 CrossRef CAS PubMed.
- C. Yi, S. Zhang, K. T. Webb and Z. Nie, Acc. Chem. Res., 2016, 50, 12–21 CrossRef PubMed.
- J. He, Y. Liu, T. Babu, Z. Wei and Z. Nei, J. Am. Chem. Soc., 2012, 134, 11342–11345 CrossRef CAS PubMed.
- S. Pedireddy, H. K. Lee, W. W. Tjiu, I. Y. Phang, H. R. Tan, S. Q. Chua, C. Troadec and X. Y. Ling, Nat. Commun., 2014, 5, 4947 CrossRef CAS PubMed.
- C. Li, Ö. Dag, T. D. Dao, T. Nagao, Y. Sakamoto, T. Kimura, O. Terasaki and Y. Yamauchi, Nat. Commun., 2015, 6, 6608 CrossRef CAS PubMed.
- J. Fang, L. Zhang, J. Li, L. Lu, C. Ma, S. Cheng, Z. Li, Q. Xiong and H. You, Nat. Commun., 2018, 9, 521 CrossRef PubMed.
- C. Zhu, D. Du, A. Eychmüller and Y. Lin, Chem. Rev., 2015, 115, 8896–8943 CrossRef CAS PubMed.
- J. Biener, A. Wittstock, L. Zepeda-Ruiz, M. Biener, V. Zielasek, D. Kramer, R. Viswanath, J. Weissmüller, M. Bäumer and A. Hamza, Nat. Mater., 2009, 8, 47–51 CrossRef CAS PubMed.
- A. Wittstock, V. Zielasek, J. Biener, C. Friend and M. Bäumer, Science, 2010, 327, 319–322 CrossRef CAS PubMed.
- K. Nishio and H. Masuda, Angew. Chem., Int. Ed., 2011, 50, 1603–1607 CrossRef CAS PubMed.
- C. Li, M. Iqbal, J. Lin, X. Luo, B. Jiang, V. Malgras, K. C.-W. Wu, J. Kim and Y. Yamauchi, Acc. Chem. Res., 2018, 51, 1764–1773 CrossRef CAS PubMed.
- S. Salvatore, A. Demetriadou, S. Vignolini, S. S. Oh, S. Wuestner, N. A. Yufa, M. Stefik, U. Wiesner, J. J. Baumberg and O. Hess, Adv. Mater., 2013, 25, 2713–2716 CrossRef CAS PubMed.
- H. Y. Hsueh, H. Y. Chen, Y. C. Hung, Y. C. Ling, S. Gwo and R. M. Ho, Adv. Mater., 2013, 25, 1780–1786 CrossRef CAS PubMed.
- Y. Kuroda and K. Kuroda, Angew. Chem., Int. Ed., 2010, 49, 6993–6997 CrossRef CAS PubMed.
- C.-W. Liao, Y.-S. Lin, K. Chanda, Y.-F. Song and M. H. Huang, J. Am. Chem. Soc., 2013, 135, 2684–2693 CrossRef CAS PubMed.
- Z. Li, K. Hur, H. Sai, T. Higuchi, A. Takahara, H. Jinnai, S. M. Gruner and U. Wiesner, Nat. Commun., 2014, 5, 3247 CrossRef PubMed.
- E. Auyeung, W. Morris, J. E. Mondloch, J. T. Hupp, O. K. Farha and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 1658–1662 CrossRef CAS PubMed.
- Q. Ruan, L. Shao, Y. Shu, J. Wang and H. Wu, Adv. Opt. Mater., 2014, 2, 65–73 CrossRef.
- G. Song, C. Liang, X. Yi, Q. Zhao, L. Cheng, K. Yang and Z. Liu, Adv. Mater., 2016, 28, 2716–2723 CrossRef CAS PubMed.
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- D. Gu and F. Schüth, Chem. Soc. Rev., 2014, 43, 313–344 RSC.
- V. Malgras, H. Ataee-Esfahani, H. Wang, B. Jiang, C. Li, K. C. W. Wu, J. H. Kim and Y. Yamauchi, Adv. Mater., 2016, 28, 993–1010 CrossRef CAS PubMed.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- B. Liu, L. Jin, H. Zheng, H. Yao, Y. Wu, A. Lopes and J. He, ACS Appl. Mater. Interfaces, 2017, 9, 1746–1758 CrossRef CAS PubMed.
- H. Lv, L. Sun, L. Zou, D. Xu, H. Yao and B. Liu, Chem. Sci., 2019, 10, 1986–1993 RSC.
- B. Jiang, C. Li, V. Malgras, M. Imura, S. Tominaka and Y. Yamauchi, Chem. Sci., 2016, 7, 1575–1581 RSC.
- B. Jiang, C. Li, Ö. Dag, H. Abe, T. Takei, T. Imai, M. S. A. Hossain, M. T. Islam, K. Wood and J. Henzie, Nat. Commun., 2017, 8, 15581 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Z. Luo, V. Nachammai, B. Zhang, N. Yan, D. T. Leong, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 10577–10580 CrossRef CAS PubMed.
- P. J. Goulet and R. B. Lennox, J. Am. Chem. Soc., 2010, 132, 9582–9584 CrossRef CAS PubMed.
- D. M. Chevrier, L. Raich, C. Rovira, A. Das, Z. Luo, Q. Yao, A. Chatt, J. Xie, R. Jin and J. Akola, J. Am. Chem. Soc., 2018, 140, 15430–15436 CrossRef CAS PubMed.
- R. Takahata, S. Yamazoe, K. Koyasu, K. Imura and T. Tsukuda, J. Am. Chem. Soc., 2018, 140, 6640–6647 CrossRef CAS PubMed.
- G. Corthey, L. J. Giovanetti, J. M. Ramallo-López, E. Zelaya, A. A. Rubert, G. A. Benitez, F. G. Requejo, M. H. Fonticelli and R. C. Salvarezza, ACS Nano, 2010, 4, 3413–3421 CrossRef CAS PubMed.
- S.-H. Cha, J.-U. Kim, K.-H. Kim and J.-C. Lee, Chem. Mater., 2007, 19, 6297–6303 CrossRef CAS.
- H. Lv, A. Lopes, D. Xu and B. Liu, ACS Cent. Sci., 2018, 4, 1412–1419 CrossRef CAS PubMed.
- H. Schmidbaur, Gold Bull., 1990, 23, 11–21 CrossRef CAS.
- N. K. Chaki, Y. Negishi, H. Tsunoyama, Y. Shichibu and T. Tsukuda, J. Am. Chem. Soc., 2008, 130, 8608–8610 CrossRef CAS PubMed.
- M.-C. Bourg, A. Badia and R. B. Lennox, J. Phys. Chem. B, 2000, 104, 6562–6567 CrossRef CAS.
- M. H. Schoenfisch and J. E. Pemberton, J. Am. Chem. Soc., 1998, 120, 4502–4513 CrossRef CAS.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- S.-H. Cha, K.-H. Kim, J.-U. Kim, W.-K. Lee and J.-C. Lee, J. Phys. Chem. C, 2008, 112, 13862–13868 CrossRef CAS.
- D. Xu, X. Liu, H. Lv, Y. Liu, S. Zhao, M. Han, J. Bao, J. He and B. Liu, Chem. Sci., 2018, 9, 4451–4455 RSC.
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- J. Zhang, P. Liu, H. Ma and Y. Ding, J. Phys. Chem. C, 2007, 111, 10382–10388 CrossRef CAS.
- B. Y. Xia, H. B. Wu, N. Li, Y. Yan, X. W. Lou and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 3797–3801 CrossRef CAS PubMed.
- Y. Lou, M. M. Maye, L. Han, J. Luo and C.-J. Zhong, Chem. Commun., 2001, 473–474 RSC.
- J. Kim, S. W. Lee, P. T. Hammond and Y. Shao-Horn, Chem. Mater., 2009, 21, 2993–3001 CrossRef CAS.
- J. Sun, F. Wang, Y. Liu, Y. Ni, H. Zhou, C. F. Guo and S. Chen, RSC Adv., 2017, 7, 22479–22484 RSC.
- L. Lu, Y. Nie, Y. Wang, G. Wu, L. Li, J. Li, X. Qi and Z. Wei, J. Mater. Chem. A, 2018, 6, 104–109 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details of the surfactant, additional TEM images and electrocatalytic MOR performances. See DOI: 10.1039/c9sc01728c |
| ‡ H. Lv and D. Xu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |