
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUse of 15N NMR spectroscopy to probe covalency in a thorium nitride†
Selena L.
Staun
a,
Dumitru-Claudiu
Sergentu
 b,
Guang
Wu
a,
Jochen
Autschbach
*b and
Trevor W.
Hayton
*a
b,
Guang
Wu
a,
Jochen
Autschbach
*b and
Trevor W.
Hayton
*a
aDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, California 93106, USA. E-mail: hayton@chem.ucsb.edu
bDepartment of Chemistry, University at Buffalo, State University of New York, 312 Natural Sciences Complex, Buffalo, NY 14260-3000, USA. E-mail: jochena@buffalo.edu
First published on 4th June 2019
Abstract
Reaction of the thorium metallacycle, [Th{N(R)(SiMe2)CH2}(NR2)2] (R = SiMe3) with 1 equiv. of NaNH2 in THF, in the presence of 18-crown-6, results in formation of the bridged thorium nitride complex, [Na(18-crown-6)(Et2O)][(R2N)3Th(μ-N)(Th(NR2)3] ([Na][1]), which can be isolated in 66% yield after work-up. Complex [Na][1] is the first isolable molecular thorium nitride complex. Mechanistic studies suggest that the first step of the reaction is deprotonation of [Th{N(R)(SiMe2)CH2}(NR2)2] by NaNH2, which results in formation of the thorium bis(metallacycle) complex, [Na(THF)x][Th{N(R)(SiMe2CH2)}2(NR2)], and NH3. NH3 then reacts with unreacted [Th{N(R)(SiMe2)CH2}(NR2)2], forming [Th(NR2)3(NH2)] (2), which protonates [Na(THF)x][Th{N(R)(SiMe2CH2)}2(NR2)] to give [Na][1]. Consistent with hypothesis, addition of excess NH3 to a THF solution of [Th{N(R)(SiMe2)CH2}(NR2)2] results in formation of [Th(NR2)3(NH2)] (2), which can be isolated in 51% yield after work-up. Furthermore, reaction of [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)] with 2, in THF-d8, results in clean formation of [K][1], according to 1H NMR spectroscopy. The electronic structures of [1]− and 2 were investigated by 15N NMR spectroscopy and DFT calculations. This analysis reveals that the Th–Nnitride bond in [1]− features more covalency and a greater degree of bond multiplicity than the Th–NH2 bond in 2. Similarly, our analysis indicates a greater degree of covalency in [1]−vs. comparable thorium imido and oxo complexes.
Introduction
The past decade has seen a remarkable expansion of the chemistry of actinide-ligand multiple bonds.1–6 This is exemplified especially well by the chemistry of molecular uranium nitrides.6 Since the synthesis of the first molecular uranium nitride in 2002,7 many bridging and terminal uranium nitride complexes have been reported.2,8–21 The study of these complexes has allowed actinide chemists to reveal fundamentally important insights into 5f covalency, as well as uncover novel modes of reactivity.12,15–18,22–24In contrast, no isolable molecular thorium nitride complexes are known.25 A handful of thorium nitrides have been identified in matrix isolation studies, such as ThN, NThN, and NThO, but these are only stable at cryogenic temperatures.26–28 Recently, Liddle and co-workers reported the isolation of the bridged Th(IV) parent imido complex, [{Th(TrenDMBS)}2(μ-NH)] (TrenDMBS = {N(CH2CH2NSiMe2tBu)3}3−), which was thought to form via an unobserved Th nitride intermediate, [{Th(TrenDMBS)}2(μ-N)]−.25 It was postulated that the nitride ligand in this intermediate was exceptionally basic on account of its highly polarized Th–Nnitride bonds. As a result, it spontaneously deprotonated the solvent, forming the bridged parent imido. These results prompted the authors of ref. 25 to suggest that the Th![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NTh unit may be intrinsically more reactive than the more covalent UNU unit. Significantly, further work in this area would allow us to evaluate this hypothesis in more detail, as well as permit a better evaluation of the bonding within this functional group.
NTh unit may be intrinsically more reactive than the more covalent UNU unit. Significantly, further work in this area would allow us to evaluate this hypothesis in more detail, as well as permit a better evaluation of the bonding within this functional group.
Herein, we report the synthesis of the first isolable molecular thorium nitride complex, [Na(18-crown-6)(Et2O)][(R2N)3Th(μ-N)Th(NR2)3] (R = SiMe3). In addition, we report its characterization by 15N NMR spectroscopy and DFT calculations, which has allowed us to evaluate the degree of 5f covalency within the ThNTh unit. To provide context, we have synthesized the parent amide complex, [Th(NR2)3(NH2)]. This material was also characterized by 15N NMR spectroscopy and density functional theory (DFT) calculations.
Synthesis and characterization
Addition of 1 equiv. of NaNH2 to a cold (−25 °C) solution of the thorium metallacycle,29 [Th{N(R)(SiMe2)CH2}(NR2)2] (R = SiMe3), in tetrahydrofuran (THF), followed by addition of 1 equiv. of 18-crown-6, afforded the bridged nitride complex, [Na(18-crown-6)(Et2O)][(R2N)3Th(μ-N)(Th(NR2)3] ([Na][1]), after stirring for 24 h. This material could be isolated as colorless plates in 66% yield after work-up (eqn (1)). The reaction of [Th{N(R)(SiMe2)CH2}(NR2)2] with 0.5 equiv. of both NaNH2 and 18-crown-6 also generates [Na][1], but with reduced yields (30%). The isolation of [Na][1] contrasts with the recent results of Liddle and co-workers, who attempted to isolate a bridged thorium nitride complex by reduction of a Th azide precursor, but isolated the bridged parent imido complex, [{Th(TrenDMBS)}2(μ-NH)], instead.25 | (1) |
The connectivity of complex [Na][1] was verified by X-ray crystallography (Fig. 1; see ESI† for complete structural details). Complex [Na][1] crystallizes in the monoclinic space group P21/c. In the solid-state, each Th center features a pseudo-tetrahedral coordination geometry. In addition, the Th–Nnitride–Th linkage is linear (179(1)°), while its Th–Nnitride bond lengths (Th1–N1 = 2.14(2), Th2–N1 = 2.11(2) Å) are much shorter than the Th–Nsilylamido bond lengths (av. 2.41 Å), suggesting multiple-bond character in the former. A [Na(18-crown-6)(Et2O)]+ counterion is also present in the unit cell. The potassium analog, [K(18-crown-6)(THF)2][(R2N)3Th(μ-N)Th(NR2)3] ([K][1]), has also been structurally characterized. It features nearly identical metrical parameters to those of [Na][1] (See ESI† for full details).
 | ||
| Fig. 1 Solid-state molecular structure of [Na][1], shown with 50% probability ellipsoids. Hydrogen atoms and [Na(18-crown-6)(Et2O)]+ counterion removed for clarity. Selected bond lengths (Å) and angles (°): Th1–N1 = 2.14(2), Th2–N1 = 2.11(2), Th1–N1–Th2 = 179(1), av. Th1–Namido = 2.41, av. Th2–Namido = 2.41, av. Namido–Th1–Namido = 108.3, av. Namido–Th2–Namido = 108.7. | ||
Complex [Na][1] is the first nitrido complex reported for thorium. However, several thorium imido complexes have been structurally characterized.30–36 Generally, these complexes feature shorter Th–N bonds than those of [Na][1]. For example, [Cp2*Th(N-2,6-Me2C6H3)(THF)] features a Th–N distance of 2.045(8) Å,32 and [K(18-crown-6)][Th(NDipp)(NR2)3] (Dipp = 2,6-iPr2C6H3) features a Th–N distance of 2.072(3) Å.30 For further comparison, the bridged U(IV) nitride, [Na][(μ-N)(U(N[t-Bu]Ar)3)2] (Ar = 3,5-Me2C6H3), also features a linear UNU linkage (175.1(2)°), but shorter An–N bond distances (2.080(4) Å and 2.077(4) Å),11 consistent with the smaller ionic radius of uranium. Similar metric parameters are observed in [Cs][{U(OSi(OtBu)3)3}2(μ-N)] (U–N–U = 170.2(3)°; U1–N1 = 2.058(5) Å; U2–N1 = 2.079(5) Å).12
The 1H NMR spectrum of [Na][1] in THF-d8 features a sharp singlet at 0.36 ppm, assignable to the SiMe3 groups, along with a broad resonance at 3.62 ppm, assignable to the 18-crown-6 moiety. Complex [Na][1] is insoluble in pentane and benzene, but is quite soluble in Et2O and THF. It is stable as a THF-d8 solution at room temperature for at least 24 h, showing minimal signs of decomposition over this time. Finally, the IR spectrum of [Na][1] features a mode at 742 cm−1, which corresponds to the principal Th–N–Th asymmetric stretch (Fig. S15†). For comparison, this mode was calculated to occur at 758 cm−1 (Fig. S30, see ESI† for calculation details).
To better understand the mechanism of formation of [Na][1] we monitored the reaction of [Th{N(R)(SiMe2)CH2}(NR2)2] with NaNH2 and 18-crown-6, in THF-d8, by 1H NMR spectroscopy (Fig. S12†). A 1H NMR spectrum of this mixture after 7 h revealed an intense new resonance at 0.36 ppm, which is assignable to [Na][1]. Interestingly, this spectrum also features minor resonances at 0.30 and 0.18 ppm, which are assignable to the terminal parent amide complex, [Th(NR2)3(NH2)] (2), and the bis(metallacycle) complex, [Na(THF)x][Th{N(R)(SiMe2CH2)}2(NR2)],30 respectively. The assignment for the latter species was made by comparison with the 1H NMR spectrum of the known bis(metallacycle) complex, [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)].30 After 32 h, the resonance assignable to [Na][1] has grown in intensity, while the resonances assignable to [Th{N(R)(SiMe2)CH2}(NR2)2] and 2 have completely disappeared, and only trace amounts of [Na(THF)x][Th{N(R)(SiMe2CH2)}2(NR2)] are still present in solution. To explain these observations, we suggest that the first step of the reaction is deprotonation of [Th{N(R)(SiMe2)CH2}(NR2)2] by NaNH2, forming [Na(THF)x][Th{N(R)(SiMe2CH2)}2(NR2)] and NH3 (Scheme 1). NH3 then reacts with unreacted [Th{N(R)(SiMe2)CH2}(NR2)2], forming 2, which then protonates [Na(THF)x][Th{N(R)(SiMe2CH2)}2(NR2)] to give [Na][1].
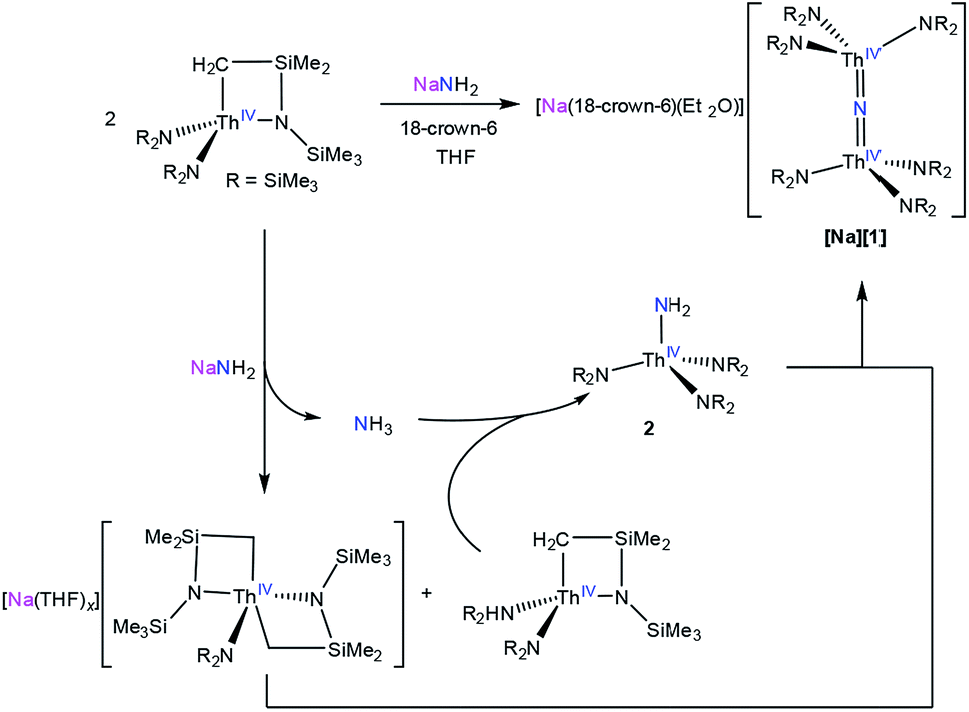 | ||
| Scheme 1 Proposed mechanism of formation of [Na][1]. | ||
To test this hypothesis we explored the reaction of [Th{N(R)(SiMe2)CH2}(NR2)2] with NH3. Thus, addition of 3 equiv. of NH3, as a 0.4 M solution in THF, to a THF solution of [Th{N(R)(SiMe2)CH2}(NR2)2] results in rapid formation of 2, which can be isolated in 51% yield after work-up (eqn (2)).
 | (2) |
The 1H NMR spectrum of 2 in C6D6 features a sharp resonance at 0.37 ppm (54H), assigned to the SiMe3 groups. In addition, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet (2H, JNH = 45.8 Hz) at 3.67 ppm is assigned to the –NH2 resonance (Fig. S5†). For comparison, the JNH values for the isostructural group(IV) complexes, [M(NR2)3(NH2)] (M = Zr, Hf), were found to be 45.6 Hz (Zr) and 46.0 Hz (Hf).37 Interestingly, the only other known thorium NH2 complex, [K(DME)4][(DME)Th(NH2)(diphenolate)2], featured a broad singlet at 2.0 ppm in its 1H NMR spectrum, which was assignable to the –NH2 group.38
:1:1 triplet (2H, JNH = 45.8 Hz) at 3.67 ppm is assigned to the –NH2 resonance (Fig. S5†). For comparison, the JNH values for the isostructural group(IV) complexes, [M(NR2)3(NH2)] (M = Zr, Hf), were found to be 45.6 Hz (Zr) and 46.0 Hz (Hf).37 Interestingly, the only other known thorium NH2 complex, [K(DME)4][(DME)Th(NH2)(diphenolate)2], featured a broad singlet at 2.0 ppm in its 1H NMR spectrum, which was assignable to the –NH2 group.38
The connectivity of complex 2 was verified by X-ray crystallography (Fig. 2, see ESI† for complete structural details). Complex 2 crystallizes in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c. In the solid-state, complex 2 is disordered over two positions in a 50:50 ratio, which somewhat lowers the precision of the resulting metrical parameters. It features a pseudo-tetrahedral geometry about the thorium center. Due to the large ESDs, the Th–NH2 distance (2.24(6) Å) in 2 is statistically identical to its Th–Nsilylamido distances (2.36(2) Å).30,39 The other Th–NH2 complex has a similar Th–NH2 bond length (2.2431(6) Å).38 For further comparison, the uranium(IV) terminal amide complexes, [U(NH2)(TrenTIPS)] (TrenTIPS = {N(CH2CH2NSiiPr3)3}3−) and [{η5-1,2,4-C5H2tBu3}2U(NH2)2], feature U–NH2 bond lengths of 2.228(4) and 2.19 Å (av.), respectively.40,41
c. In the solid-state, complex 2 is disordered over two positions in a 50:50 ratio, which somewhat lowers the precision of the resulting metrical parameters. It features a pseudo-tetrahedral geometry about the thorium center. Due to the large ESDs, the Th–NH2 distance (2.24(6) Å) in 2 is statistically identical to its Th–Nsilylamido distances (2.36(2) Å).30,39 The other Th–NH2 complex has a similar Th–NH2 bond length (2.2431(6) Å).38 For further comparison, the uranium(IV) terminal amide complexes, [U(NH2)(TrenTIPS)] (TrenTIPS = {N(CH2CH2NSiiPr3)3}3−) and [{η5-1,2,4-C5H2tBu3}2U(NH2)2], feature U–NH2 bond lengths of 2.228(4) and 2.19 Å (av.), respectively.40,41
 | ||
| Fig. 2 Solid-state molecular structure of 2, shown with 50% probability ellipsoids. Hydrogen atoms removed for clarity. Selected bond lengths (Å) and angles (°): Th1–N1 = 2.24(6), Th1–N2 = 2.36(2), N1–Th1–N2 = 100.7(3), N2–Th1–N2* = 116.7(2). | ||
Complex 2 is highly soluble in pentane, benzene, Et2O and THF. Furthermore, 2 is stable as a C6D6 solution for over 36 h with minimal signs of decomposition. In addition, the IR spectrum of 2 features a prominent N–H stretching mode at 3321 cm−1 (Fig. S21, ESI†), providing further support for our formulation. For comparison, this mode is observed at 3342 cm−1 (Zr) and 3364 cm−1 (Hf) for [M(NR2)3(NH2)] (M = Zr, Hf).37
To further test our proposed mechanistic hypothesis, we monitored the reaction of [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)]30 with 2 in THF-d8, in the presence of 1 equiv. of 18-crown-6, by 1H NMR spectroscopy (eqn (3)). A 1H NMR spectrum of this solution, after standing at room temperature for 4 h, reveals a new resonance at 0.36 ppm, which is assignable to [K][1] (Fig. S13†). After 3d, the peak assignable to the nitride has increased in intensity, while resonances assignable to complex 2 and [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)] have decreased in intensity. These three complexes are present in a ratio of 1:3:6.7 in the 3d spectrum. Overall, this result supports the proposed mechanism (Scheme 1), but it is important to note that formation of [1]− under these conditions is much slower than its rate of formation under the conditions described in eqn (1), suggesting that this experiment does not perfectly duplicate the original reaction conditions.
 | (3) |
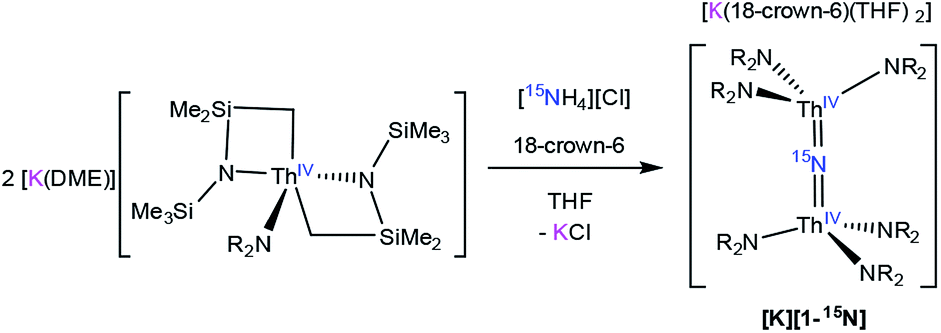
To facilitate our covalency analysis we endeavoured to synthesize [1-15N]−. Given the proposed intermediacy of [Na(THF)x][Th{N(R)(SiMe2CH2)}2(NR2)] in the formation of [Na][1], we rationalized that reaction of NH4Cl with 2 equiv. of [Th{N(R)(SiMe2CH2)}2(NR2)]− would generate the nitride complex. Thus, addition of 1 equiv. of finely ground 15NH4Cl to a pale yellow THF solution containing 2 equiv. of [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)],30 followed by addition of 1 equiv. of 18-crown-6, results in formation of [K(18-crown-6)(THF)2][(R2N)3Th(μ-15N)(Th(NR2)3] ([K][1-15N]), which can be isolated as a white powder in 13% yield after work-up (eqn (4)).
 | (4) |
The low yield of [K][1-15N] under these conditions can be partly ascribed to unselective protonation of [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)] by 15NH4Cl, which results in the formation of copious amounts of HNR2 (observed in the in situ1H NMR spectrum), and results in the presence of unreacted [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)] in the final reaction mixture. The unreacted [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)] can be conveniently removed from the nitride product by rinsing with toluene. The 1H NMR spectrum of [K][1-15N] in THF-d8 matches those recorded for both [Na][1] and [K][1] (see ESI† for full details), where a singlet at 0.36 ppm can be assigned to the SiMe3 groups and a resonance at 3.63 ppm can be assigned to 18-crown-6. Furthermore, the 15N{1H} NMR spectrum of [K][1-15N] (referenced to CH3NO2) reveals a sharp singlet at 298.8 ppm. No other resonances are observed in this spectrum. This is the first observation of an 15N chemical shift for an actinide nitride. For comparison, the group 4 nitride complexes [{(η5-C5H2-1,2,4-Me3)2Hf}2(μ-N)(NCO)(DMAP)] and [{Cp*TiCl2}(μ-N){Cp*TiCl(NH3)}] feature 15N resonances at 567.19 ppm and 431.6 ppm, respectively, for their bridging nitride ligands.42,43 Finally, the IR spectrum of [K][1-15N] features a stretch at 735 cm−1, which corresponds to the principal Th–N–Th asymmetric stretch (Fig. S17†), and is redshifted by 7 cm−1 from that observed for [Na][1].
Finally, access to 2-15N was achieved by reaction of [Th{N(R)(SiMe2)CH2}(NR2)2] with 1 equiv. of 15NH3 gas in THF. Synthesized in this manner, colorless crystals of 2-15N could be isolated in 85% yield after work-up. Similar to 2, the 1H NMR spectrum in benzene-d6 shows a singlet at 0.36 ppm (54H), assignable to the SiMe3 groups. In addition, a 1:1 doublet (2H, JNH = 62.3 Hz) at 3.67 ppm is assignable to the –NH2 resonance (Fig. S9, ESI†). The 15N NMR spectrum of 2-15N (referenced to CH3NO2) reveals a sharp resonance at −198.4 ppm. For comparison, the previously reported thorium amide 15N NMR spectrum featured a sharp triplet centered at 155.01 ppm (J = 57.2 Hz).38 It is not readily apparent why this chemical shift is so different from that recorded for complex 2-15N. Finally, the IR spectrum of 2-15N features an N–H stretch at 3317 cm−1, and a Th–NH2 stretch at 482 cm−1 (Fig. S22, ESI†). The identity of the latter stretch was confirmed by comparison with the calculated IR spectrum (Fig. S31†), where it is predicted to occur at 508 cm−1.
Electronic structure analysis
We analyzed the electronic structures of [1]− and 2 with DFT. Using the B3LYP functional, we observe excellent agreement between the calculated and experimentally determined structural parameters for both complexes. For example, the calculated Th–Nnitride and Th–Nsilylamide bond lengths for [1]− are within 0.02 Å of the distances found in the solid state. Similarly, the calculated Th–Namide and Th–Nsilylamide bond lengths in 2 are within 0.02 Å of those found in its crystal structure.An NBO/NLMO analysis of [1]− reveals that the Th–N–Th interaction consists of two orthogonal 3c–2e π bonds and two predominantly 2c–2e σ bonds that feature some three-center character (Fig. 3), suggestive of overall ThN double bond character. The covalency in the Th–Nnitride bonds in [1]− is greater than that observed for the related Th imido, [Th(NAr)(NR2)3]− (Ar = 2,6-iPr2C6H3),30 and Th oxo, [Th(O)(NR2)3]−,44 with a greater magnitude of 5f orbital involvement. For example, the Th–N π interaction in [1]− features 16% Th character (58% 6d, 42% 5f) (Table 1), whereas [Th(NAr)(NR2)3]− and [Th(O)(NR2)3]− feature 0% and 12% Th character (65% 6d, 35% 5f), respectively, in their Th–E π bonds. For further comparison, the degree of covalency in [1]− is comparable to that observed for the thorium sulfide, [Th(S)(NR2)3]−, which features 17% Th character (61% 6d, 38% 5f) in its Th–S π interaction.44 The Wiberg bond index of the Th–Nnitride bond is 0.94, which is greater than that calculated for [Th(NAr)(NR2)3]− (0.88).30 Overall, these combined computational metrics indicate a greater degree of covalency in [1]−vs. the comparable imido and oxo complexes. Similar observations have been made for uranium(V) nitride and oxo complexes.21,40,45
 | ||
| Fig. 3 Th–N (2σ + 2π) bonding NLMOs in [(NR2)3Th(μ-N)Th(NR2)3]− (isosurface plots ±0.03 au; hydrogen atoms are omitted for clarity). Color code for atoms: Th, light blue; N, blue; Si, beige; C, gray. | ||
For complex 2, an NBO/NLMO analysis reveals that the Th–N interaction consists of 2c–2e π bond and a 2c–2e σ bond (Fig. S28†). Not surprisingly, the degree of covalency within the Th–Namide bond in 2 is less than that observed for the ThNTh bonds of [1]−. Specifically, the σ bond in 2 features 7% Th character (63% 6d, 21% 5f, 5% 7p, 11% 7s) and the π bond features 10% Th character (59% 6d, 41% 5f). Accordingly, the Wiberg bond index of the Th–Namide bond (0.65) is substantially less than that observed for the Th–Nnitride bonds in [1]−.
Chemical shift analysis
To assess the accuracy of our computational approach, we calculated the 15N chemical shift of the nitride ligand in the known group(IV) nitride complex, [{Cp*TiCl2}(μ-N){Cp*TiCl(NH3)}].42 The 15N chemical shift of the nitride ligand for the B3LYP-optimized structure was calculated to be 406.8/421.8 ppm using the PBE0/B3LYP functionals and the scalar relativistic (SR) all-electron ZORA Hamiltonian. For comparison, the experimentally determined chemical shift is 431.6 ppm.42 Even better agreement was achieved by performing the calculation with two-component ZORA, i.e., including the spin–orbit (SO) coupling variationally. At this level, the calculated chemical shift (414.5/430.5 ppm) is in very good agreement with experiment.With these results in hand, the 15N NMR chemical shifts for the nitride and NH2 ligands in [1]− and 2 were calculated using the PBE0 functional. We, and others, have found that this functional typically works better than B3LYP for NMR shift calculations in actinide-containing molecules.46 For [1]−, the calculated 15N chemical shift without spin orbit coupling (ZORA-SR) is 226 ppm – substantially upfield from the experimental result (298.8 ppm). Considerably better agreement is obtained when SO coupling is taken into account, with a calculated 15N shift of 305 ppm. The 79 ppm downfield shift induced by SO coupling is evidence of 5f (and 6d) character in the Th–Nnitride bonds. For 2, the calculated 15N chemical shift without SO coupling is −254 ppm. Upon inclusion of SO coupling, the shift changes to −210 ppm, which is much closer to the measured value (−198.4 ppm). The smaller downfield shift induced by SO coupling in 2 (Δδ = 44 ppm) is consistent with the reduced covalency, and reduced bond multiplicity, of the Th–Namide bond. Perhaps most importantly, the good agreement between the experimental and calculated shifts for both [1]− and 2 gives credence to the NBO analysis presented above.
Conclusions
We have synthesized and characterized the first isolable molecular thorium nitride complex, [(NR2)3Th(μ-N)Th(NR2)3]−. This complex is thermally stable, in contrast to the bridged thorium nitride recently proposed by Liddle and co-workers.25 The origin of this stability difference is not known, but it may be related to the lack of a trans donor ligand in [(NR2)3Th(μ-N)Th(NR2)3]−vs. [{Th(TrenDMBS)}2(μ-N)]−. Alternatively, it could relate to the different method of synthesis. 15N NMR spectroscopic characterization of [(NR2)3Th(μ-N)Th(NR2)3]−, in combination with a DFT analysis, reveals the presence of 5f orbital participation within the ThNTh unit. In line with the reduced electronegativity of nitrogen vs. oxygen, our data suggests greater levels of covalency in [(NR2)3Th(μ-N)Th(NR2)3]− than in the closely related oxo, [Th(O)(NR2)3]−. However, we find comparable covalency in [(NR2)3Th(μ-N)Th(NR2)3]− to that found in the thorium sulfide, [Th(S)(NR2)3]−, likely on account of the greater “energy-driven” overlap in the latter.47 To better contextualize our results, we also synthesized and characterized the thorium parent amide complex, [Th(NR2)3(NH2)]. According to 15N NMR spectroscopy and DFT calculations, this complex features a lesser degree of 5f covalency in its Th–NH2 bond than that found for the bridging nitride complex, which is not surprising given its reduced bond order.
This work further solidifies the use of NMR spectroscopy as an important tool for probing the electronic structure of the actinides. Previously, 13C, 77Se, and 125Te NMR spectroscopies had been used to evaluate covalency in An–E bonds.39,46,48–51 In the case of An–C bonding, large downfield 13C shifts have been consistently observed for the 13C nuclei bonded directly to an actinide center. More significantly, the degree of deshielding was found to correlate with the amount of 5f covalency within the An–C bond. For example, the 13C NMR shift of acetylide carbon in the U(VI) acetylide complexes, UVI(O)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC6H4-p-R)(NR2)3 (R = NMe2, OMe, Me, Ph, H, Cl), correlated well with two measures of covalency, the QTAIM delocalization index and the Wiberg bond order of the U–C bond.48 Highly deshielded 13C resonances are also observed for the carbene resonance in [Th(CHPPh3)(NR2)3] and the methylene resonances in [UO2(CH2SiMe3)4]2− and [U(CH2SiMe3)6]−.46,52 Our results demonstrate that 15N NMR spectroscopy can also be used to evaluate covalency in actinide-ligand bonding, and like 13C NMR spectroscopy, the magnitude of the downfield shift correlates with the degree of 5f character in the An–N bond. Going forward, we propose to characterize other actinide nitrides by 15N NMR spectroscopy. Of particular interest is the measurement of the 15N chemical shift of a U(VI) nitride complex, which, on account of the high anticipated covalency, should exhibit an extreme downfield shift of its nitride resonance.
CC6H4-p-R)(NR2)3 (R = NMe2, OMe, Me, Ph, H, Cl), correlated well with two measures of covalency, the QTAIM delocalization index and the Wiberg bond order of the U–C bond.48 Highly deshielded 13C resonances are also observed for the carbene resonance in [Th(CHPPh3)(NR2)3] and the methylene resonances in [UO2(CH2SiMe3)4]2− and [U(CH2SiMe3)6]−.46,52 Our results demonstrate that 15N NMR spectroscopy can also be used to evaluate covalency in actinide-ligand bonding, and like 13C NMR spectroscopy, the magnitude of the downfield shift correlates with the degree of 5f character in the An–N bond. Going forward, we propose to characterize other actinide nitrides by 15N NMR spectroscopy. Of particular interest is the measurement of the 15N chemical shift of a U(VI) nitride complex, which, on account of the high anticipated covalency, should exhibit an extreme downfield shift of its nitride resonance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Biosciences, and Geosciences Division, under Contract DE-SC0001861. D. C. S. and J. A. acknowledge support from the U.S. Department of Energy, Office of Basic Energy Sciences, Heavy Element Chemistry program under grant DE-SC0001136 and the UB Center for Computational Research (CCR) for the computational part of this study. The authors thank the Ménard group (UCSB) for the use of their 15NH3.Notes and references
- T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC
.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed
- D. Patel and S. T. Liddle, Prog. Inorg. Chem., 2012, 32, 1 CAS
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed
- D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266–267, 2–15 CrossRef CAS
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2002, 41, 3433–3436 CrossRef CAS PubMed
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Science, 2005, 309, 1835 CrossRef CAS PubMed
- M. Falcone, L. Chatelain, R. Scopelliti, I. Živković and M. Mazzanti, Nature, 2017, 547, 332 CrossRef CAS PubMed
- S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed
- A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed
- L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed
- P. A. Cleaves, C. E. Kefalidis, B. M. Gardner, F. Tuna, E. J. L. McInnes, W. Lewis, L. Maron and S. T. Liddle, Chem.–Eur. J., 2017, 23, 2950–2959 CrossRef CAS PubMed
- P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 10412–10415 CrossRef CAS PubMed
- M. Falcone, L. Chatelain and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 4074–4078 CrossRef CAS PubMed
- M. Falcone, L. Chatelain, R. Scopelliti and M. Mazzanti, Chimia, 2017, 71, 209–212 CrossRef CAS PubMed
- M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 12290–12294 CrossRef CAS PubMed
- M. Falcone, L. N. Poon, F. Fadaei Tirani and M. Mazzanti, Angew. Chem., Int. Ed., 2018, 57, 3697–3700 CrossRef CAS PubMed
- T. W. Hayton, Nat. Chem., 2013, 5, 451 CrossRef CAS PubMed
- D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS PubMed
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717 CrossRef CAS PubMed
- K. C. Mullane, H. Ryu, T. Cheisson, L. N. Grant, J. Y. Park, B. C. Manor, P. J. Carroll, M.-H. Baik, D. J. Mindiola and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 11335–11340 CrossRef CAS PubMed
- R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723 CrossRef CAS PubMed
- M. Falcone, L. Barluzzi, J. Andrez, F. Fadaei Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed
- J. Du, D. M. King, L. Chatelain, E. Lu, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC
- D. W. Green and G. T. Reedy, J. Mol. Spectrosc., 1979, 74, 423–434 CrossRef CAS
- G. P. Kushto, P. F. Souter and L. Andrews, J. Chem. Phys., 1998, 108, 7121–7130 CrossRef CAS
- B. Vlaisavljevich, L. Andrews, X. Wang, Y. Gong, G. P. Kushto and B. E. Bursten, J. Am. Chem. Soc., 2016, 138, 893–905 CrossRef CAS PubMed
- T. Cantat, B. L. Scott and J. L. Kiplinger, Chem. Commun., 2010, 46, 919–921 RSC
- N. L. Bell, L. Maron and P. L. Arnold, J. Am. Chem. Soc., 2015, 137, 10492–10495 CrossRef CAS PubMed
- M. E. Garner, S. Hohloch, L. Maron and J. Arnold, Organometallics, 2016, 35, 2915–2922 CrossRef CAS
- A. Haskel, T. Straub and M. S. Eisen, Organometallics, 1996, 15, 3773–3775 CrossRef CAS
- W. Ren, G. Zi and M. D. Walter, Organometallics, 2012, 31, 672–679 CrossRef CAS
- T. Straub, A. Haskel, T. G. Neyroud, M. Kapon, M. Botoshansky and M. S. Eisen, Organometallics, 2001, 20, 5017–5035 CrossRef CAS
- P. Yang, E. Zhou, G. Hou, G. Zi, W. Ding and M. D. Walter, Chem.–Eur. J., 2016, 22, 13845–13849 CrossRef CAS PubMed
- C. Zhang, P. Yang, E. Zhou, X. Deng, G. Zi and M. D. Walter, Organometallics, 2017, 36, 4525–4538 CrossRef CAS
- X. Yu and Z.-L. Xue, Inorg. Chem., 2005, 44, 1505–1510 CrossRef CAS PubMed
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2003, 42, 4958–4961 CrossRef CAS PubMed
- D. E. Smiles, G. Wu, P. Hrobárik and T. W. Hayton, J. Am. Chem. Soc., 2016, 138, 814–825 CrossRef CAS PubMed
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482 CrossRef CAS PubMed
- G. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS
- A. Abarca, P. Gómez-Sal, A. Martín, M. Mena, J. M. Poblet and C. Yélamos, Inorg. Chem., 2000, 39, 642–651 CrossRef CAS PubMed
- S. P. Semproni, C. Milsmann and P. J. Chirik, Angew. Chem., 2012, 51, 5213–5216 CrossRef CAS PubMed
- D. E. Smiles, G. Wu, N. Kaltsoyannis and T. W. Hayton, Chem. Sci., 2015, 6, 3891–3899 RSC
- D. M. King, F. Tuna, J. McMaster, W. Lewis, A. J. Blake, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 4921–4924 CrossRef CAS PubMed
- D. E. Smiles, G. Wu, P. Hrobárik and T. W. Hayton, Organometallics, 2017, 36, 4519–4524 CrossRef CAS
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS
- K. C. Mullane, P. Hrobárik, T. Cheisson, B. C. Manor, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2019, 58, 4152–4163 CrossRef CAS PubMed
- E. A. Pedrick, P. Hrobárik, L. A. Seaman, G. Wu and T. W. Hayton, Chem. Commun., 2016, 52, 689–692 RSC
- L. A. Seaman, J. R. Walensky, G. Wu and T. W. Hayton, Inorg. Chem., 2013, 52, 3556–3564 CrossRef CAS PubMed
- W. Wu, D. Rehe, P. Hrobárik, A. Y. Kornienko, T. J. Emge and J. G. Brennan, Inorg. Chem., 2018, 57, 14821–14833 CrossRef CAS PubMed
- L. A. Seaman, P. Hrobárik, M. F. Schettini, S. Fortier, M. Kaupp and T. W. Hayton, Angew. Chem., 2013, 52, 3259–3263 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, crystallographic details (as CIF files), computational results, and spectral data for complexes [Na][1], [K][1], 2, [K][1-15N], and 2-15N. CCDC 1911146–1911148. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01960j |
| This journal is © The Royal Society of Chemistry 2019 |