
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual confinement properties of a water insoluble small peptide hydrogel†
Nilotpal
Singha
a,
Arpita
Srivastava
b,
Bapan
Pramanik
a,
Sahnawaz
Ahmed
a,
Payel
Dowari
a,
Sumit
Chowdhuri
a,
Basab Kanti
Das
a,
Ananya
Debnath
 *b and
Debapratim
Das
*a
*b and
Debapratim
Das
*a
aDepartment of Chemistry, Indian Institute of Technology Guwahati, Assam 781039, India. E-mail: ddas@iitg.ac.in
bDepartment of Chemistry, Indian Institute of Technology Jodhpur, Rajasthan 342037, India. E-mail: ananya@iitj.ac.in
First published on 6th May 2019
Abstract
Unlike polymeric hydrogels, in the case of supramolecular hydrogels, the cross-linked network formation is governed by non-covalent forces. Hence, in these cases, the gelator molecules inside the network retain their characteristic physicochemical properties as no covalent modification is involved. Supramolecular hydrogels thus get dissolved easily in aqueous medium as the dissolution leads to a gain in entropy. Thus, any supramolecular hydrogel, insoluble in bulk water, is beyond the present understanding and hitherto not reported as well. Herein, we present a peptide-based (PyKC) hydrogel which remained insoluble in water for more than a year. Moreover, in the gel state, any movement of solvent or solute to and from the hydrogel is highly restricted resulting in a high degree of compartmentalization. The hydrogel could be re-dissolved in the presence of some biomolecules which makes it a prospective material for in vivo applications. Experimental studies and all atom molecular dynamics simulations revealed that a cysteine containing gelator forms dimers through disulfide linkage which self-assemble into PyKC layers with a distinct PyKC–water interface. The hydrogel is stabilized by intra-molecular hydrogen bonds within the peptide-conjugates and the π–π stacking of the pyrene rings. The unique confinement ability of the hydrogel is attributed to the slow dynamics of water which remains confined in the core region of PyKCvia hydrogen bonds. The hydrogen bonds present in the confined water need activation energies to move through the water depleted hydrophobic environment of pyrene rings which significantly reduces water transport across the hydrogel. The compartmentalizing ability is effectively used to protect enzymes for a long time from denaturing agents like urea, heat or methanol. Overall, the presented system shows unique insolubility and confinement properties that could be a milestone in the research of soft-materials.
Introduction
Hydrogels are an important class of soft materials owing to their widespread applications.1–4 Classically, in a hydrogel, cross-linked networks are formed by covalent linkages within natural or synthetic polymers where water molecules get trapped through cohesive forces.5–8 The entrapment leads to immobilization of the solvent molecules which eventually results in self-supporting hydrogels. The monomers used to construct cross-linked polymeric networks no longer retain their original physicochemical properties as they get covalently modified. Thus, though the monomers are water soluble, a cross-linked polymeric hydrogel, when dispersed in water, may not get dissolved in bulk water. Similarly, a xerogel (dried gel) of a polymeric hydrogel swells back to its original shape and size in the presence of water. Moreover, solvent molecules of these hydrogels remain in dynamic motion with the solvent outside and both solvent and solute can pass through the gel.7Over the last few decades, supramolecular hydrogels of small molecules have emerged as an attractive alternative to polymeric hydrogels.6,8–10 In this case, hierarchical self-assembly of small molecules utilizing non-covalent forces results in fibrous structures. These fibres further crosslink through supramolecular interactions to form the gel-network where water molecules get entrapped to form the hydrogel. Unlike polymeric hydrogels, owing to the non-covalent nature of the interactions, each constituent molecule (monomer/gelator) in the network retains its physicochemical characteristics.6,8 Thus, in contrast to polymeric hydrogels, dissolution of supramolecular hydrogels in bulk water can easily be achieved. Dissolution of a supramolecular hydrogel results in a gain in the entropy of the overall system and thus is a thermodynamically favourable process.8
Supramolecular hydrogels especially the small peptide based ones have major advantages over polymeric hydrogels as these systems are in general non-toxic and biodegradable in nature.3,11–18 However, a drawback for bio-medical application comes from the poor stability of such systems. It is important to clarify the term “stability” for this particular report: by stability we mean that the gel remains in the gel state without getting dissolved and retains its rheological properties when immersed in bulk water, buffer, solvent or bio-fluid for a long time. Since dissolution in bulk aqueous medium is a thermodynamically favourable process for small molecule hydrogels, when used as a localized drug delivery system, there is a possibility of easy dissolution of the gel in the bloodstream. Prolonged and sustained use of these hydrogels is thus difficult inside the human body.14 Although the stability of such hydrogels in buffers and cell culture medium is reported in the literature, it is not clear whether they can fulfil the above-mentioned criteria since no quantitative analysis is available.19–21 An attractive alternative could be a water insoluble and highly stable supramolecular hydrogel which may get dissolved slowly by a biomolecule (stimuli to break the gel) present in the bio-fluid and thus result in sustained and prolonged application. A supramolecular hydrogel made of small molecules has not been reported so far, where the gel does not get dissolved in bulk solvent systems and exchange of solutes as well as solvents with an external bulk medium is highly restricted.
Herein, we report a small peptide based (PyKC,22Scheme 1A) water insoluble supramolecular hydrogel. When immerged in water and various other solutions, the hydrogel did not dissolve and remained insoluble for more than a year. The underlying mechanism behind this exceptional property is revealed with a combination of various analytical experiments and theoretical calculations. Moreover, the unique confinement property is efficiently used to trap and protect protein molecules from external denaturing agents for a long period.
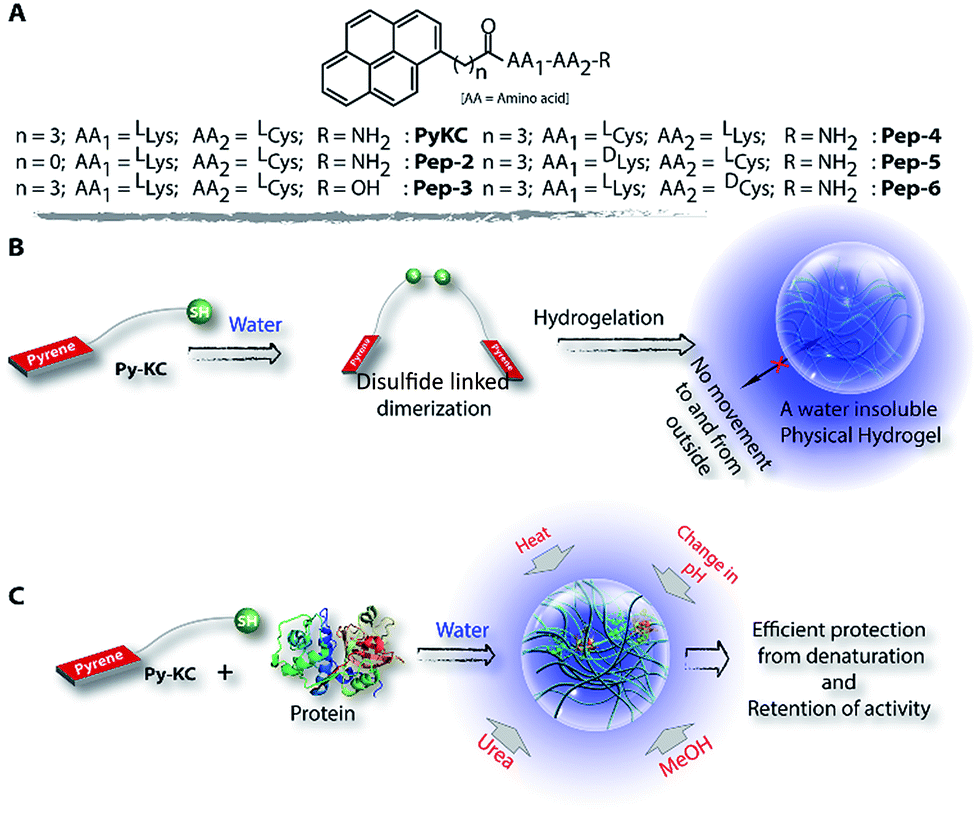 | ||
| Scheme 1 Chemical structures, aggregation pattern and enzyme encapsulation. (A) Chemical structures of peptides used in this study. (B) Pictorial presentation of hydrogelation of PyKC in water through dimerization and the insolubility of the formed hydrogel in water. (C) Schematic representation of the encapsulation of proteins and prevention from denaturation in the presence of various denaturing agents. | ||
Results and discussion
PyKC forms a self-supporting hydrogel in Tris buffer (20 mM, pH 8.0) with a minimum gelation concentration (MGC) of 1 wt%. The hydrogel was thermo-reversible and the gel to sol transition temperature (Tg) was measured to be 75 ± 0.5 °C. The rheological analyses (Fig. 1A) showed that the storage modulus (G′) of the gel was found to be considerably higher than the loss modulus (G′′) over a range of applied angular frequencies.23 ESI-MS analysis of the hydrogel disclosed the presence of only the disulfide linked dimer of PyKC. Time dependent analytical HPLC analyses of the solution of PyKC revealed that complete conversion to the dimer takes ∼12 h at room temperature (Fig. 1B). In the presence of a well-known disulfide bond breaker, dithiothreitol (DTT) (0.1 equivalent), no hydrogelation was observed confirming the critical role of the dimer in gelation. To further verify the actual time required for gelation, time dependent rheology was performed which shows that sol-to-gel transformation takes ∼8 h (Fig. S18†) while the equilibrium was obtained at around 12 h when no further change in the G′′ and G′ values was observed. The similar completion time for the dimerization process and the equilibration time suggest a critical role of the dimer formation in the gelation process.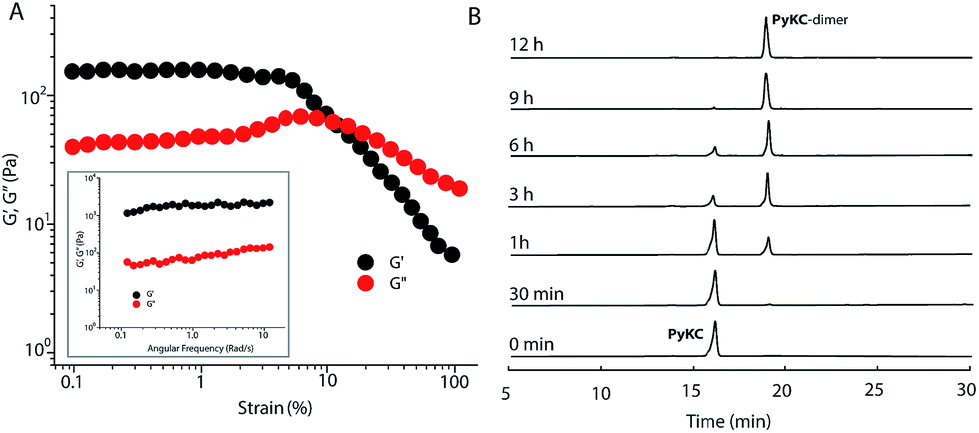 | ||
| Fig. 1 (A) Changes in the storage and loss modulus as a function of shear strain and (inset) angular frequency for a 1 wt% hydrogel of PyKC. (B) Time dependent chromatographic analysis of a 0.01 wt% aqueous solution of PyKC showing the formation of a disulfide linked dimer. | ||
Interestingly, when a small portion of the hydrogel was added to water and stirred vigorously, no dissolution of the hydrogel in that bulk water was observed (Fig. 2A and ESI Video S1†). The sample was found to be as it was for more than 18 months when kept at room temperature. This observation is unprecedented for any supramolecular hydrogel and prompted us to quantify the dis-solution/insolubility of the hydrogel. A 1 wt% hydrogel of PyKC was added to bulk water and incubated with slow shaking at room temperature (for details, follow the Experimental section). Analyses of aliquots from the bulk water taken at different time intervals showed up to 5% dissolution of the hydrogel within the first seven days and it remained constant thereafter for more than 1 year (Fig. S19†). The initial small dissolution of the PyKC dimer can be attributed to the residues which connect the hydrophobic layers to form the fibre as seen from the molecular dynamics simulations discussed later. These residues could easily get dissolved in water at the initial stage. However, no further dissolution of the hydrogel proved the insolubility of the hydrogel in water. The rheological properties of the hydrogel remained unchanged even after keeping the hydrogel in water for seven days.
 | ||
| Fig. 2 (A) Photographs of a portion of the 1 wt% hydrogel prepared from PyKC and immerged in bulk water. Photographs taken at different times to show the insolubility of the hydrogel even after 12 months. (B) Photographs of vials containing different aqueous solutions where small portions of the hydrogel (1 wt% PyKC) containing rhodamine B were immerged and stirred for 10 min. | ||
It is worth mentioning that, for a cross-linked polymeric hydrogel, though the network remained insoluble, the water molecules inside the network can move out and that is why their xerogels swell in water to form the hydrogel reproducibly.1,7,24 Interestingly, in the present case, the xerogel remained insoluble and unable to swell when water or buffers up to pH 12 were added. Six different dyes (two of each cationic, anionic and neutral category) were separately entrapped in the gels while preparing the hydrogel and the release profiles of the dyes showed very similar results as only ∼5% release (within the first hour) over seven days was observed for all these dyes (Fig. S20†). A reverse experiment of incubating the hydrogel (without any dye) in an aqueous solution of methylene blue for seven days showed no incorporation of dye into the hydrogel (Fig. S21†). Interestingly, a similar test with perylene (a fused π-system) in acetonitrile also resulted in similar observation (Fig. S22†). These results clearly demonstrate that passage of the tested solutes to and from the hydrogel is highly restricted.
The observations also raised the question about the exchangeability of the gel entrapped water molecules. To get an insight into that, we took the help of 1H NMR experiments. Hydrogels prepared in water were added to D2O, NaOD and DCl solutions and incubated while shaking. The amount of water exchanged from the hydrogels was estimated from 1H NMR spectra of the supernatant deuterated solvents after different time intervals (see the Experimental section for details). As can be seen from Fig. 3 and S22,† only ∼3% water molecules could come out of the hydrogel within the first day and no further noticeable change was observed after that. The initial increase can be attributed to the loosely bound water molecules at the surface of the hydrogel. To further confirm this fact, a reverse experiment was carried out where a portion of the hydrogel prepared in D2O was suspended in water and the amount of external water that entered the hydrogel was monitored. No noticeable increase in the H2O content was observed in that case as well (Fig. S23†). Similar studies were performed with two of our previously reported hydrogels.25,26 However, in both cases, the hydrogels get dissolved in bulk H2O/D2O and all the H2O molecules were found in the NMR samples. These results demonstrate that the self-assembly creates a system where both the gel-network and the water of gelation are inaccessible to the surrounding environment. Neither the solvent nor the solute could pass through the envelope created by the hydrogel. This unique confinement property is unprecedented in the literature.
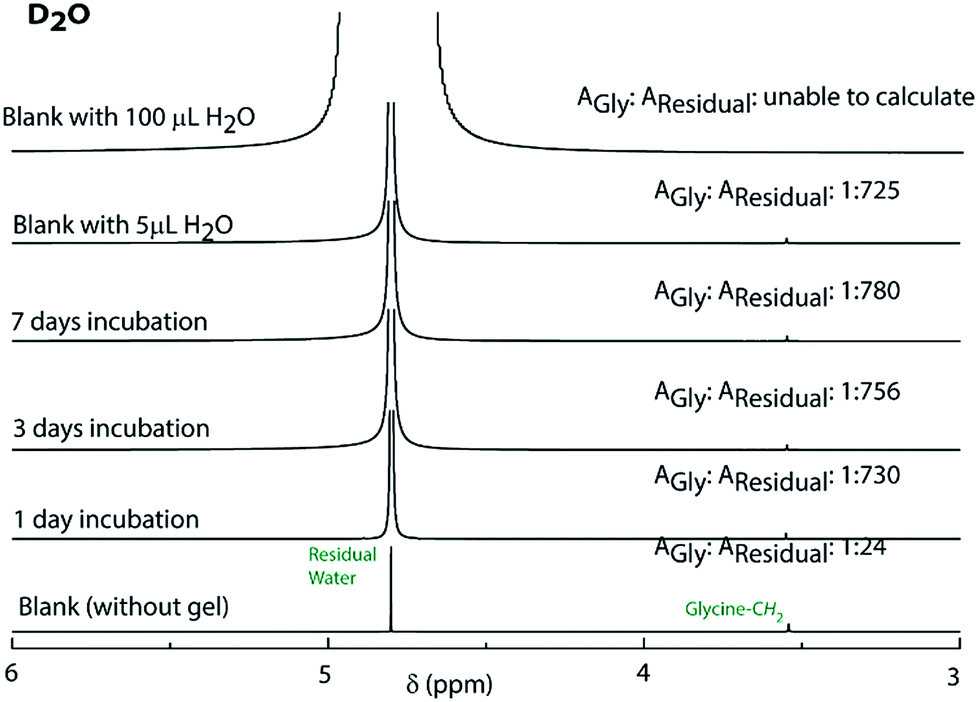 | ||
| Fig. 3 1H NMR spectra of D2O containing glycine before and after incubating the PyKC hydrogel inside the solvent for different time periods to determine the extent of exchange of water of gelation. | ||
Buffers (20 mM) of different pH also failed to solubilize the hydrogel (Fig. 2B, 4A and S24†). Several water-miscible organic solvents and aqueous solutions were tested to find the solubility of the hydrogel. The results from all these tests are shown in Fig. 4A (also Fig. S25†). Interestingly, though DMF and DMSO could dissolve the hydrogel within 5 min, other solvents showed 4–8% dissolution within the first 24 h with no further changes thereafter up to seven days. When exposed to some extreme conditions like 2 M NaOH, 12 N HCl, and 6 M urea (well-known chaotropic agent), only the acid could solubilize the gel (Fig. 4A and S26†). It is interesting to find that though 6 M urea or 2 M NaOH solutions were unable to dissolve the gel, DMSO, DMF and 12 N HCl dissolve the gel efficiently. At this point, the reason behind such observation is not clear to the authors. The gel also remained insoluble in biological fluids like human blood serum (HBS) (Fig. 4A and S27†). Next, the hydrogel was dispersed in bulk water and the sample was heated at a particular temperature for 1 h with constant shaking and then cooled to room temperature before analysing the bulk water. As can be seen from Fig. 4B, even at 80 °C, only ∼8% dissolution was recorded. Interestingly at 80 °C, which is above the Tg of the hydrogel, though the gel melts, the network structure and the insolubility remain intact (Fig. S28†). Bringing that sample to room temperature brings back the gel state.
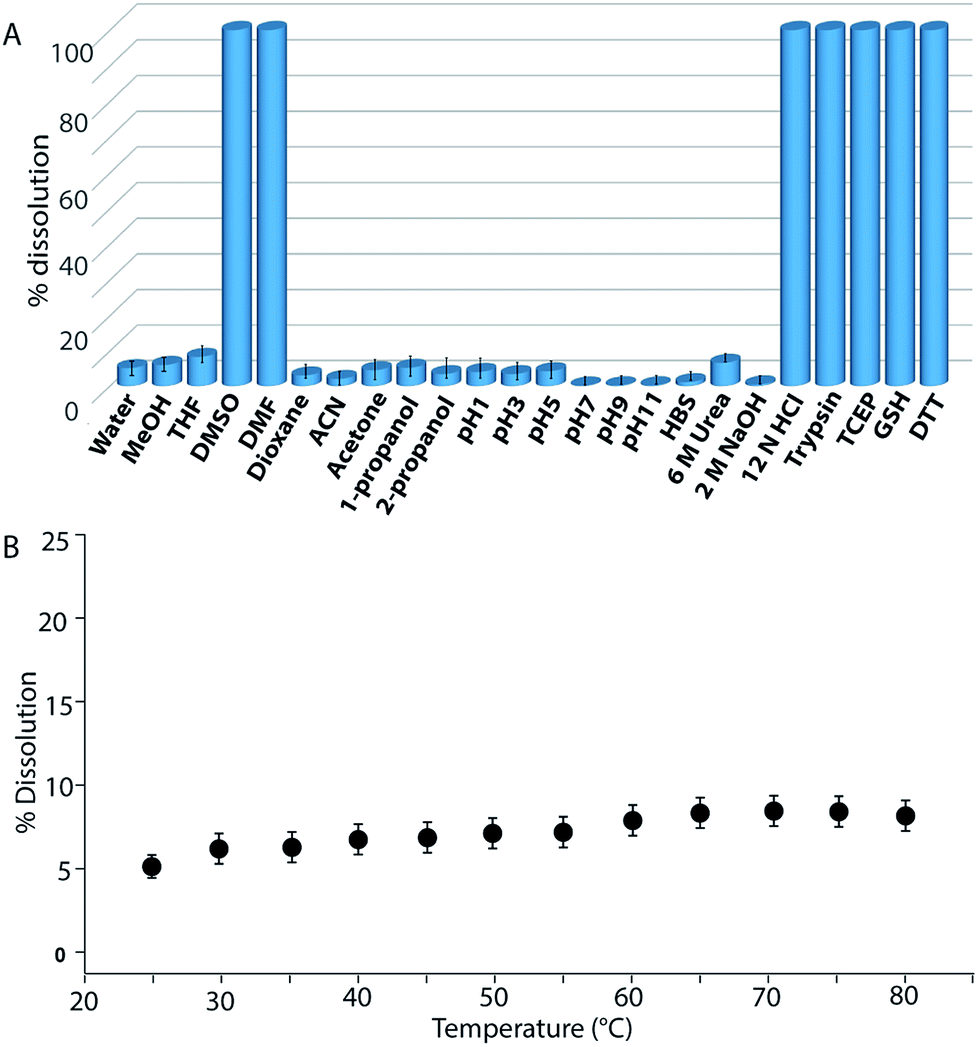 | ||
| Fig. 4 (A) Percentage dissolution of a 1 wt% PyKC hydrogel (20 mM Tris-buffer, pH 8) in different media after 168 h. (B) Percentage dissolution of a 1 wt% hydrogel prepared from PyKC in Tris-buffer (20 mM, pH 8) in bulk water at different temperatures. | ||
Though concentrated acid can dissolve the hydrogel, such extreme conditions are not a good choice for future applications. During the search for a suitable system which can break/dissolve the hydrogel, we found that aqueous solutions of disulfide breakers, tris(2-carboxyethyl)phosphine hydrochloride (TCEP), DTT or glutathione (GSH) can do so effectively (Fig. 4A and S29†). When subjected to dissolution with a proteolytic enzyme, trypsin, the gel dissolves and smaller fragments of the peptide were observed in the ESI-MS spectrum of the solution indicating the breakdown of the peptide (Fig. 4A and S30†). Being biomolecules, both trypsin and GSH could be interesting options to break the hydrogel during biomedical applications.
The insolubility of this hydrogel is fascinating and it is important to evaluate the gelation process. Densely packed thin fibres (diameter ∼ 9 nm) with an average length of ∼500 nm were observed from FETEM (Fig. 5A) of the hydrogel while no such network structure could be found from DTT treated solutions of PyKC. Notably, the fibrous network structure was retained by the hydrogel even after keeping it in water for more than 12 months (Fig. 5B). Interestingly, even at a very low concentration of 0.005 wt%, an intense pyrene excimer band (∼475 nm, Fig. 5C) was observed in the emission spectra of PyKC.27 The unusually high intensity of the pyrene excimer band can be attributed to possible strong π–π stacking. Further insight into the π–π stacking is obtained from 1H NMR and PXRD analyses. A PyKC solution in DMSO-d6 was titrated with H2O and the 1H NMR spectrum was monitored for the aromatic region. In DMSO, the molecule remains in a non-aggregated state and as the water content increased, the aggregation starts. With increasing amount of water, the protons corresponding to the pyrene group showed a clear up-field shift (Fig. 5D). The up-field shift is confirmatory evidence for π–π stacking of the pyrene rings. PXRD of a dried film of the hydrogel showed a π–π stacking distance of 3.4 Å (Fig. S31†) representing strong π–π stacking. The stacking distance is in agreement with the distance found from electronic structure calculations (2.97 Å) and from the MD simulations (3.9 Å) discussed later. Additionally, to quantify the pores formed within the gel network, gas-adsorption/desorption analysis of the lyophilized hydrogel sample was performed and an average pore diameter of 3.05 nm was observed. The extremely small pores could possibly justify the confinement properties of the hydrogel.
 | ||
| Fig. 5 (A) FETEM images of the 1 wt% PyKC-hydrogel in 20 mM Tris buffer pH 8 (inset: zoomed-in picture of the same sample showing the diameter of a fibre). (B) FETEM image of the hydrogel after incubating in bulk water under ambient conditions for 12 months. (C) Concentration (0.001–0.05 wt%) dependent emission spectra of 24 h matured PyKC solution (in 20 mM Tris buffer pH 8) with an increase in concentration (λex = 337 nm). (D) 1H NMR spectra of 24 h matured samples of PyKC in DMSO-d6 with varying amounts of water showing strong π–π interaction between the pyrene rings. | ||
Though the hydrogel forms via dimerization of PyKC, a separately synthesized PyKC-dimer failed to form the hydrogel. Buffers of pH 7–11 were tested but no gelation was observed under any condition. There are some literature reports which also demonstrate that both the aggregated structure and the gelation highly depend on the method used.19,28 The failure of the dimer to form a gel suggests that, here, dimerization and aggregation proceed simultaneously leading to a network structure. During this process, the water molecules get entrapped within the network. The entrapment is strong enough to prevent the water molecules from exchanging with the exterior. In order to obtain information regarding the importance of the peptide sequence, some control molecules (Pep-2–6, Scheme 1A) were tested for their gelation ability. Only Pep-3, 5 and 6 were able to form hydrogels but all these hydrogels were found to be spontaneously soluble in water. Hence, it can be concluded that the insolubility is an exclusive property of the PyKC-hydrogel and the molecular structure certainly plays a significant role in providing this property.
To understand the molecular origin of the insolubility and unique confinement ability of the PyKC-hydrogel, the molecular packing of the most stable building block of the hydrogel is analyzed. As a first step, to obtain the most stable conformer, the dispersion energies, total energies and binding energies of the geometry optimized PyKC molecules were evaluated using electronic structure calculations. The binding energies for the open and folded conformers were found to be −57.92 and −78.5 kcal mol−1 respectively (Table S1†). This signifies that the stacking of the folded dimer of PyKC is energetically more favorable than that of the open dimer. The coordinates of the most stable folded dimer and the charges on each atom are summarized in Table S2.† The snapshot of the geometry optimized folded PyKC dimer with atom labels is shown in Fig. S33.†Fig. 6A and B show the optimized geometry of the folded dimer and the stacked dimer respectively. The closest inter-planar distance in the stacked dimer was found to be 2.44 Å which is consistent with the PXRD results (Fig. S31†). The stability of the stacked folded dimer is attributed to both T-type and H-type π–π interactions of the pyrene rings as shown in Fig. 6B.
 | ||
| Fig. 6 (A) Optimized geometry of a folded PyKC dimer showing a parallel stacking of the pyrene rings. (B) Optimized geometry of two stacked folded PyKC dimers. Pyrene rings are shown in magenta color. (C) Snapshot of the PyKC layer in the presence of water for system (c) from the all-atom MD simulation after 42.5 ns run-length. PyKC is shown in licorice representation. Color code: pyrene – magenta, water – green. Distinct interfaces of PyKC–water are visible showing compartmentalization of water and the hydrogel. The inset shows that the intra-molecular hydrogen bonds in the peptide conjugate and π–π stacking across pyrene rings stabilize the self-assembly of PyKC in the hydrogel. | ||
The most stable folded conformer of PyKC was chosen as the initial structure for Molecular Dynamics (MD) simulations. A snapshot of the final configuration of the system (c) from the MD simulation (as mentioned in simulation details) is shown in Fig. 6C (the equilibration of the system is examined by calculating the potential energy and SASA shown in Fig. S34(a) and (b) in the ESI†).29 We find that PyKC-dimers self-assemble into a layer-like structure with two distinct interfaces of water and PyKC layers (shown in Fig. 6C). The inset in the figure demonstrates the molecular structure of the interior of the aggregated PyKC phase. The pyrene–pyrene distance falls within the π–π stacking distance where the nitrogens (N) and oxygens (O) of the peptide conjugates form intramolecular hydrogen bonds with each other stabilizing the conformer. To understand the molecular arrangements of water, pyrene rings and PyKC in the self-assembled structure, we determined density profiles across PyKC layers (shown in Fig. 7A) where the direction across the layers is considered as the direction of the normal vector. Distinct interfaces between water and PyKC layers are observed demonstrating compartmentalization of water molecules and PyKC. Interestingly, there is a strong overlap between the locations of the pyrene rings and the N/O atoms of the peptide. This clearly confirms that the significant part of the hydrophilic atoms of the peptide conjugates is buried in the core region of the hydrophobic aromatic pyrene rings unlike the arrangements of lipids/surfactants in biological membranes/vesicles.30–33 The hydrophilic peptide conjugates are stabilized by the intra-molecular hydrogen bonds between N–H and O as shown in the inset of Fig. 6C. The number of intramolecular hydrogen bonds of PyKC is calculated using the widely accepted geometric criteria where the distance between the donor and the acceptor is ∼0.35 nm and the angle between the donor–H–acceptor is 30°.34–37 The number of such intra-molecular hydrogen bonds (between N–H⋯O) is shown in the inset of Fig. 7A for the production run-length. The inset in Fig. 7A shows the radial distribution function (RDF), g(r), of the pyrene rings calculated using the equation,
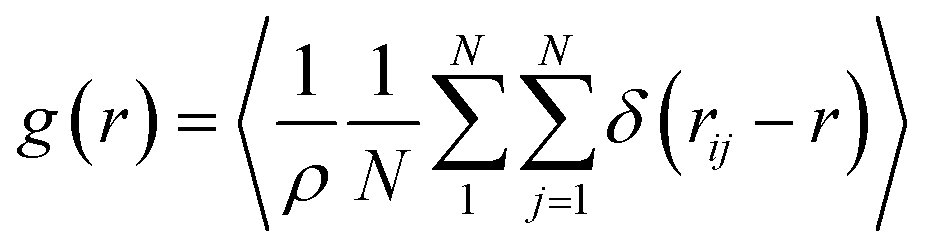 | (1) |
 | (2) |
 | ||
| Fig. 7 (A) Density profile of the PyKC layer across the normal from MD simulation. Distinct interfaces of PyKC–water are visible showing the compartmentalization of the hydrogel and water. The N/O atoms of the peptide conjugates stay within the layer and bring few water molecules inside the core of the layer. Water molecules residing continuously in the core region are shown within the vertical lines denoted by the area (a) and labelled trapped water (TW). Inset: RDF (g(r)) of pyrene–pyrene shows the first peak located at 0.4 nm where π–π stacking of pyrene rings operates. The number of intra-molecular hydrogen bonds between N and O of the peptide conjugate is shown for the production run. These hydrogen bonds contribute to stabilizing the molecular packing of the PyKC hydrogel. The number of hydrogen bonds among trapped water (TW–TW) molecules is shown for the time for which TW remains confined within the core of the PyKC layer. (B) Intermittent hydrogen bond auto-correlation function of TW and bulk water (BW) showing very slow relaxation of the TW. The slow hydrogen bond relaxation time (τHB) and high activation energy (ΔG#break) of hydrogen bond breaking of TW compared to those of BW indicate the possibility of less water transport across the hydrogel. | ||
Thus PyKC forms dimers through a disulfide linkage and the dimers self-assemble to form a water insoluble hydrogel. The self-aggregation is stabilized through the strong π–π interactions of the pyrene rings and intramolecular hydrogen bonds between the nitrogen and oxygen atoms of the peptide conjugates. Since peptide conjugates are hydrophilic in nature, they bring few water molecules. These water molecules remain confined via hydrogen bonds among trapped water (TW) molecules. The hydrogen bonds among the trapped water molecules and the hydrophobicity of the pyrene rings slow down water transport across the PyKC layer resulting in an unusual confinement in the system.
The insolubility of the hydrogel and its exceptional compartmentalizing ability can be utilized in various fields like regenerative medicine, tissue engineering, materials science, etc. As a preliminary application, we envisioned that the very tight packing of the nano-fibres can be used to create a protective envelope for proteins from denaturation. Proteins are prone to get denatured even due to minute changes in the environment and thereby lose their activity. Protecting proteins and biomolecules from the environment as well as from various denaturants is a challenging task. Various methods, including immobilization of proteins on a solid support, chemical modification, using molecular chaperons or polymeric hydrogels, etc. have been applied for this purpose.42–49 However, the reported methods are usually low in efficiency and could not provide a long life to the proteins under antagonistic conditions. We realized that, if a protein can be entrapped in the PyKC-hydrogel, (a) it can be protected from any external denaturant due to the confinement properties of the hydrogel, and (b) because of the tightly knitted network, there may not be enough space for the entrapped proteins to unwind themselves in response to external stimuli like heat.
Based on this hypothesis, as a proof of concept, two different enzymes, viz., Chromobacterium viscosum lipase (CV-lipase) and Candida rugosa lipase (CR-lipase), were tested. The enzymes were carefully chosen based on the facts that (a) the protein sequences do not contain any cysteine residue, (b) treatment with TCEP/GSH/DTT did not affect their activity, and (c) they are non-proteolytic in nature. Enzymes were trapped inside the hydrogels and dispersed in required buffers. The FETEM image of the hydrogel containing the enzyme showed the presence of the enzyme molecules trapped in the gel network (Fig. 8A). After seven days, the gels were re-dissolved in TCEP and the released proteins showed very similar CD to the native protein (Fig. 8B and S36†) which demonstrates that the proteins could retain their folded conformation inside the hydrogel. In order to overrule the possibility of any leakage of the trapped enzymes from the hydrogel, a BODIPY-labelled lipase was prepared, trapped in the hydrogel and subjected to release study by placing the hydrogel in bulk water as in the case of the other dyes described before. Similar to the dye release results, only ∼5% leakage was observed during the initial hour which remained unchanged for at least 7 days (Fig. S37†). For enzyme activity studies, several enzyme trapped hydrogel samples were prepared for each of these enzymes and as controls, similar samples of the free enzymes were also prepared in the absence of the gelator. At different time intervals, the enzyme activities of the gel-entrapped enzymes and the control samples were measured. Interestingly, the activity of both free lipases decreased significantly with time and they lost their activities completely within 6 days. However, for the gel trapped enzymes, there is an initial loss of activity (∼7–10%) in the first 24 h and thereafter no significant change was observed up to 7 days (Fig. 9A). Importantly, they retained up to ∼80% of their activities even after 30 days.
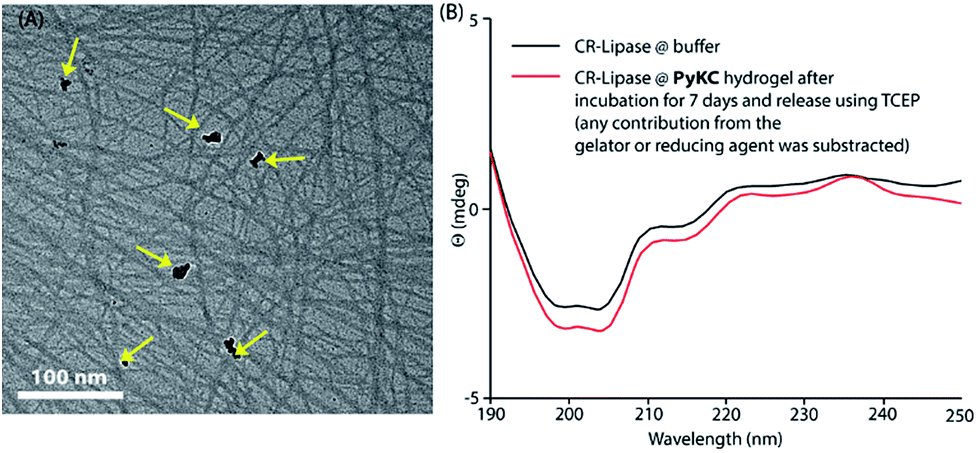 | ||
| Fig. 8 (A) FETEM image of a 1 wt% hydrogel prepared from PyKC in 20 mM Tris buffer (pH 8) containing CV-lipase. The yellow arrows indicate the enzymes embedded in the network. (B) CD spectra of CV-lipase under free and gel-trapped conditions to identify any denaturation due to encapsulation and incubation at room temperature for 7 days. | ||
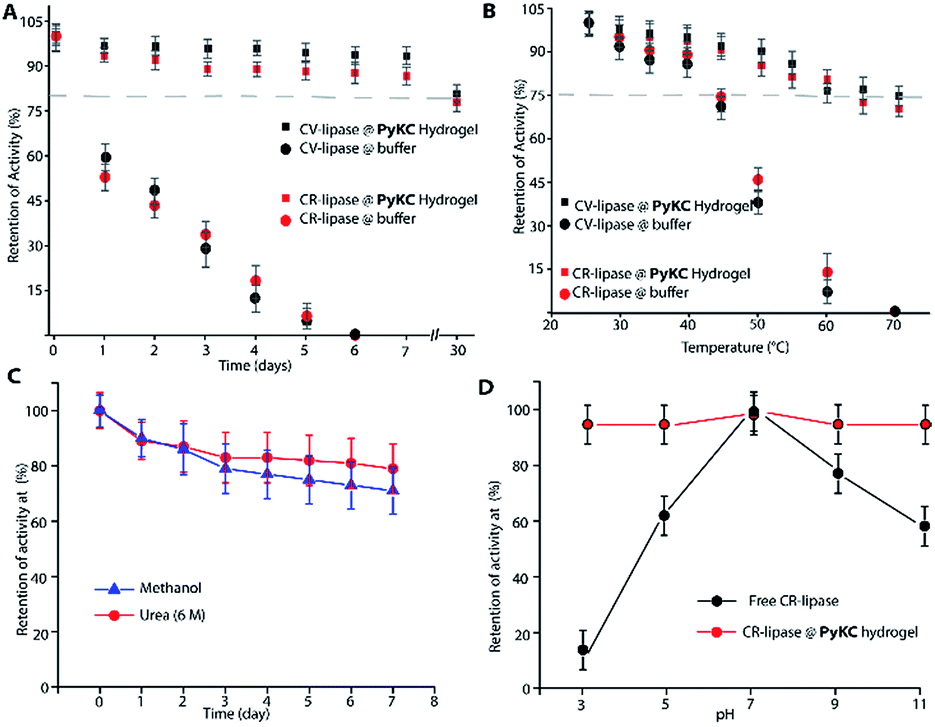 | ||
| Fig. 9 (A) Retention of the catalytic activities (%) of the gel-trapped and free enzymes at different time intervals when incubated at room temperature. (B) Retention of the catalytic activities (%) of CV and CR lipases as a function of temperature under PyKC hydrogel-trapped and free conditions. (C) Retention of activity (%) at different time intervals by the gel-trapped CR-lipase when dispersed is methanol or 6 M urea solutions. (D) Comparison of retention of activity by the CR-lipase under free and gel-trapped conditions when exposed to buffer solutions of different pH for 1 h. | ||
Similar experiments were performed for both enzymes where each sample was heated at a particular temperature for 1 h prior to cooling to room temperature and activity measurements. As expected, the free enzymes lost their activities drastically with increase in temperature and at 60 °C, only ∼10% activities were observed while the gel-trapped enzyme retained 75% activity even at 70 °C (Fig. 9B). Moreover, maintaining the temperature at 40 °C for seven days resulted in only ∼18% loss in activity for the gel-trapped CR-lipase (Fig. S38†). Exposure to other denaturing agents like 6 M aqueous urea solution or methanol (alcohols are known inhibitors for lipases50) could not impart any significant changes on the activities of the enzymes as only 10–15% loss was noted after seven days of exposure while the free enzymes were found inactive in the presence of these external agents (Fig. 9C). Moreover, suspending the hydrogel in buffers of different pH for 1 h did not make any difference in the activity of the trapped-enzyme (Fig. 9D). However, the free enzymes showed the typical pH response of lipases with the highest activity at pH 7.50 These results prove our hypothesis and show that the hydrogel can be used as an effective protecting envelope for biomolecules. We are in the process of expanding the methodology with other biomolecules and results will be reported in due course.
Conclusions
In summary, a unique supramolecular hydrogel is reported which remains insoluble in water and other aqueous or water soluble systems. Importantly, unlike polymeric cross-linked gels, no exchange of solute or solvent is allowed from and to the hydrogel. The hydrogel remained insoluble for a prolonged time in bulk aqueous medium as well as in a variety of other solutions. Notably, the hydrogel showed unusual confinement properties as none of the tested solutes or solvents could be exchanged to and from the hydrogel. Experimental evidence along with theoretical calculations reveals that the gelator molecule dimerizes and the dimers pack themselves to form a layer of PyKC and water which leads to the formation of a tightly knitted network of thin fibres and consequently the hydrogel. The unique confinement ability of the hydrogel emerged due to the combined effect of π–π stacking of pyrene rings and hydrogen bonds in peptide conjugates and confined water. The insolubility of the hydrogel in water can potentially originate from the sub-diffusive behavior of the water molecules which continuously remain confined within the core of the PyKC layer via hydrogen bonds and the hydrophobic barrier by the pyrene rings. The tight packing of the fibres within the hydrogel network and the unusual compartmentalizing properties of the hydrogel are efficiently used to confine and protect enzymes from denaturation in the presence of various denaturing agents and stimuli. The presented system is one of its kind and the insolubility property goes against the present understanding of supramolecular hydrogelation of small molecules. The unique confinement property opens many new doors toward future biomedical applications which are not possible with the present set of such soft materials.Author contribution
D. D. conceived the idea and supervised research work and data analysis. N. S. synthesized the molecules and carried out most of the experiments. B. P., S. A., and B. K. D. carried out experiments related to purification, FESEM, TEM and NMR measurements and analyzed the data. A. S. and A. D. carried out the MD simulation studies. The manuscript was prepared by D. D., A. D., and N. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DD acknowledges financial support from SERB (EMR/2016/000857), India, UKIERI (DST/INT/UK/P-119/2016), the Alexander von Humboldt Foundation, Germany, and the DST-FIST program. AD acknowledges financial support from IITJ/SEED/20140016.Notes and references
- A. Stoddart, Nat. Rev. Mater., 2017, 2, 17018 CrossRef.
- H. W. Ooi, S. Hafeez, C. A. van Blitterswijk, L. Moroni and M. B. Baker, Mater. Horiz., 2017, 4, 1020–1040 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC.
- C. P. Goodrich, M. P. Brenner and K. Ribbeck, Nat. Commun., 2018, 9, 4348 CrossRef PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- L.-Y. Chu, R. Xie, X.-J. Ju and W. Wang, Smart Hydrogel Functional Materials, Springer, Beijing, 2013 Search PubMed.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS PubMed.
- J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC.
- W. Y. Seow and C. A. E. Hauser, Mater. Today, 2014, 17, 381–388 CrossRef CAS.
- N. Singh, M. Kumar, J. F. Miravet, R. V. Ulijn and B. Escuder, Chem.–Eur. J., 2017, 23, 981–993 CrossRef CAS PubMed.
- A. M. Jonker, D. W. P. M. Löwik and J. C. M. van Hest, Chem. Mater., 2012, 24, 759–773 CrossRef CAS.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- W. Liyanage, K. Vats, A. Rajbhandary, D. S. W. Benoit and B. L. Nilsson, Chem. Commun., 2015, 51, 11260–11263 RSC.
- J. Gao, H. Wang, L. Wang, J. Wang, D. Kong and Z. Yang, J. Am. Chem. Soc., 2009, 131, 11286–11287 CrossRef CAS PubMed.
- Y. Wan, Z. Wang, J. Sun and Z. Li, Langmuir, 2016, 32, 7512–7518 CrossRef CAS PubMed.
- F. Zhao, Y. Gao, J. Shi, H. M. Browdy and B. Xu, Langmuir, 2011, 27, 1510–1512 CrossRef CAS PubMed.
- N. Singha and D. Das, India Pat., 201831035121, 2018.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- J. D. Ehrick, S. K. Deo, T. W. Browning, L. G. Bachas, M. J. Madou and S. Daunert, Nat. Mater., 2005, 4, 298 CrossRef CAS PubMed.
- B. Pramanik, N. Singha and D. Das, ACS Appl. Polym. Mater., 2019, 1, 833–843 CrossRef CAS.
- N. Singha, P. Gupta, B. Pramanik, S. Ahmed, A. Dasgupta, A. Ukil and D. Das, Biomacromolecules, 2017, 18, 3630–3641 CrossRef CAS PubMed.
- J. Hoche, H.-C. Schmitt, A. Humeniuk, I. Fischer, R. Mitrić and M. I. S. Röhr, Phys. Chem. Chem. Phys., 2017, 19, 25002–25015 RSC.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- F. Eisenhaber, P. Lijnzaad, P. Argos, C. Sander and M. Scharf, J. Comput. Chem., 1995, 16, 273–284 CrossRef CAS.
- A. Debnath, K. G. Ayappa, V. Kumaran and P. K. Maiti, J. Phys. Chem. B, 2009, 113, 10660–10668 CrossRef CAS PubMed.
- M. Kanduč, A. Schlaich, E. Schneck and R. R. Netz, Langmuir, 2016, 32, 8767–8782 CrossRef PubMed.
- S. Baoukina, D. Rozmanov and D. P. Tieleman, Biophys. J., 2017, 113, 2750–2761 CrossRef CAS PubMed.
- M. L. Berkowitz, D. L. Bostick and S. Pandit, Chem. Rev., 2006, 106, 1527–1539 CrossRef CAS PubMed.
- R. Rey, K. B. Møller and J. T. Hynes, J. Phys. Chem. A, 2002, 106, 11993–11996 CrossRef CAS.
- C. P. Lawrence and J. L. Skinner, J. Chem. Phys., 2003, 118, 264–272 CrossRef CAS.
- J. D. Eaves, J. J. Loparo, C. J. Fecko, S. T. Roberts, A. Tokmakoff and P. L. Geissler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13019–13022 CrossRef CAS PubMed.
- A. Luzar and D. Chandler, Nature, 1996, 379, 55–57 CrossRef CAS.
- A. Luzar and D. Chandler, Phys. Rev. Lett., 1996, 76, 928–931 CrossRef CAS PubMed.
- A. Chandra, Phys. Rev. Lett., 2000, 85, 768–771 CrossRef CAS PubMed.
- S. Balasubramanian, S. Pal and B. Bagchi, Phys. Rev. Lett., 2002, 89, 115505 CrossRef PubMed.
- A. Srivastava and A. Debnath, J. Chem. Phys., 2018, 148, 094901 CrossRef.
- C. J. Epstein and C. B. Anfinsen, J. Biol. Chem., 1962, 237, 2175–2179 CAS.
- K. Jiang, L. S. Schadler, R. W. Siegel, X. Zhang, H. Zhang and M. Terrones, J. Mater. Chem., 2004, 14, 37–39 RSC.
- J. Forde, E. Tully, A. Vakurov, T. D. Gibson, P. Millner and C. Ó’Fágáin, Enzyme Microb. Technol., 2010, 46, 430–437 CrossRef CAS PubMed.
- S. Ganguli, K. Yoshimoto, S. Tomita, H. Sakuma, T. Matsuoka, K. Shiraki and Y. Nagasaki, J. Am. Chem. Soc., 2009, 131, 6549–6553 CrossRef CAS PubMed.
- H. Takahashi, S.-i. Sawada and K. Akiyoshi, ACS Nano, 2011, 5, 337–345 CrossRef CAS PubMed.
- Q. Tao, A. Li, Z. Zhang, R. Ma and L. Shi, ACS Biomater. Sci. Eng., 2017, 3, 3141–3145 CrossRef CAS.
- J. M. Beierle, K. Yoshimatsu, B. Chou, M. A. A. Mathews, B. K. Lesel and K. J. Shea, Angew. Chem., Int. Ed., 2014, 53, 9275–9279 CrossRef CAS PubMed.
- L. Bayne, R. V. Ulijn and P. J. Halling, Chem. Soc. Rev., 2013, 42, 9000–9010 RSC.
- D. Das and P. K. Das, Langmuir, 2003, 19, 9114–9119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, characterization of synthesized compounds, one scheme, thirty three figures and two tables. Movie S1: the water insolubility of a PyKC hydrogel containing rhodamine B. See DOI: 10.1039/c9sc01754b |
| This journal is © The Royal Society of Chemistry 2019 |