
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic Au(I)/Au(III) arylation with the hemilabile MeDalphos ligand: unusual selectivity for electron-rich iodoarenes and efficient application to indoles†
Jessica
Rodriguez
 a,
Abdallah
Zeineddine
a,
E. Daiann
Sosa Carrizo
b,
Karinne
Miqueu
b,
Nathalie
Saffon-Merceron
c,
Abderrahmane
Amgoune
*a and
Didier
Bourissou
*a
a,
Abdallah
Zeineddine
a,
E. Daiann
Sosa Carrizo
b,
Karinne
Miqueu
b,
Nathalie
Saffon-Merceron
c,
Abderrahmane
Amgoune
*a and
Didier
Bourissou
*a
aCNRS/Université Paul Sabatier, Laboratoire Hétérochimie Fondamentale et Appliquée (LHFA, UMR 5069), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France. E-mail: dbouriss@chimie.ups-tlse.fr
bCNRS/UNIV PAU & PAYS ADOUR, Institut des Sciences Analytiques et de Physico-Chimie pour l’Environnement et les Matériaux, (IPREM UMR 5254), Hélioparc, 2 Avenue du Président Angot, 64053 Pau Cedex 09, France
cInstitut de Chimie de Toulouse (FR 2599), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France
First published on 18th June 2019
Abstract
The ability of the hemilabile (P,N) MeDalphos ligand to trigger oxidative addition of iodoarenes to gold has been thoroughly studied. Competition experiments and Hammett correlations substantiate a clear preference of gold for electron-enriched substrates both in stoichiometric oxidative addition reactions and in catalytic C–C cross-coupling with 1,3,5-trimethoxybenzene. This feature markedly contrasts with the higher reactivity of electron-deprived substrates typically encountered with palladium. Based on DFT calculations and detailed analysis of the key transition states (using NBO, CDA and ETS-NOCV methods in particular), the different behavior of the two metals is proposed to result from inverse electron flow between the substrate and metal. Indeed, oxidative addition of iodobenzene is associated with a charge transfer from the substrate to the metal at the transition state for gold, but opposite for palladium. The higher electrophilicity of the gold center favors electron-rich substrates while important back-donation from palladium favors electron-poor substrates. Facile oxidative addition of iodoarenes combined with the propensity of gold(III) complexes to readily react with electron-rich (hetero)arenes prompted us to apply the (MeDalphos)AuCl complex in the catalytic arylation of indoles, a challenging but very important transformation. The gold complex proved to be very efficient, general and robust. It displays complete regioselectivity for C3 arylation, it tolerates a variety of functional groups at both the iodoarene and indole partners (NO2, CO2Me, Br, OTf, Bpin, OMe…) and it proceeds under mild conditions (75 °C, 2 h).
Introduction
The reluctance of gold complexes to cycle between the Au(I) and Au(III) oxidation states1 has long impeded the development of Au(I)/Au(III) catalysis for the formation of carbon–carbon and carbon-element bonds. However, spectacular progress has been achieved recently so that gold redox catalysis now appears as a new paradigm in arylation reactions,2 with particular interest for transformations in which gold displays reactivity and/or selectivity complementary to the other transition metals. The main challenge in this field is certainly to generate aryl gold(III) species ready to participate in cross-coupling. To do so, several strategies have progressively emerged, as schematically illustrated in Fig. 1A. The first and main approach consists in using aryl-silanes, aryl-boronic acids or even simple arenes with external oxidants, typically F+ sources or I(III) compounds (route a). This methodology has been used successfully in a number of catalytic oxidative coupling reactions, in particular for the arylation of (hetero)arenes3 and for the oxy (and amino) arylation of alkenes.4 Starting from 2013, aryl diazonium salts (and diaryl iodonium salts) have been developed as alternative coupling partners (route b). Using this approach, the key aryl gold(III) species are most commonly generated under light activation, with or without a photosensitizer (thermal and base-activation are also possible), which circumvents the use of strong oxidants. The efficiency and generality of this method have already been substantiated in a number of catalytic C–C and C–E (E = P, S, halogen) coupling reactions, in particular thanks to dual photoredox and gold catalysis.5 Very recently, a third approach relying on purposely-designed ligands has been introduced (route c). Chelating and hemilabile ligands were shown to enable oxidant-free coupling reactions with aryl halide electrophiles (Fig. 1B).6–8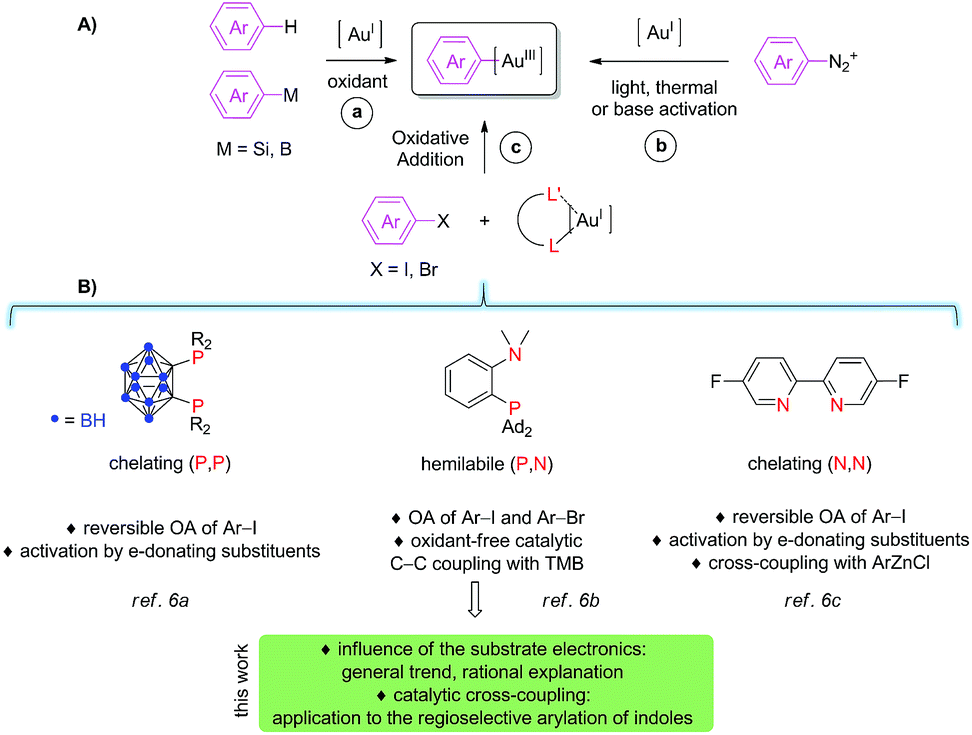 | ||
| Fig. 1 (A) Schematic representation of the different means to access aryl gold(III) species to participate in cross-coupling reactions; (B) bidentate ligands shown to promote oxidative addition of aryl halides to gold. | ||
Our group first discovered that o-carboranyl diphosphines emulate oxidative addition to gold. The bending induced by the chelating (P,P) ligand activates and preorganizes the metal fragment so that oxidative addition of iodoarenes6a as well as strained carbocycles9 proceeds readily. In the course of these investigations, we noticed an unusual substituent effect.10 Indeed, iodoarenes featuring electron-donating (EDG) para-substituents were found to react with the (P,P) gold(I) complex significantly faster than those bearing electron-withdrawing groups (EWG), in marked contrast with that classically observed with other transition metals.11 We then extended the ligand-based approach to MeDalphos, a hemilable ligand.6b,12 With the (P,N) gold complex, oxidative addition proved to be even more favored. The scope of iodoarenes was further extended and bromoarenes could be activated as well. In addition, we showed that the ensuing (P,N) aryl gold(III) complexes do engage in C–C coupling with 1,3,5-trimethoxybenzene (TMB) used as a model electron-rich arene. The cross-coupling reaction between iodoarenes and TMB was amenable to catalysis, using 5 mol% of the (P,N) gold complex. The (P,N) aryl gold(III) species were then reported by Maynard and Spokoyny to readily undergo stoichiometric aryl-S coupling with unprotected peptides and proteins, providing a very efficient and chemoselective mean for cysteine bioconjugation.13 Last but not least, a bipyridine ligand was shown very recently by McGrady, Bower and Russell to also promote oxidative addition to gold.6c With this chelating (N,N) ligand, iodoarenes are reversibly activated and again, electron-rich substrates were found to react unusually faster than electron-poor ones. The resulting (N,N) aryl gold(III) complex undergoes transmetallation and C–C reductive elimination upon reaction with an aryl zinc reagent, providing evidence for all the elementary steps of a gold-promoted Negishi-type coupling.
From all these recent reports, it is clear that ligand design offers new perspectives in gold chemistry, in particular for Au(I)/Au(III) catalysis, but the field is still in its infancy. Indeed, we know very little about oxidative addition to gold and catalytic applications with aryl halides as substrates have to be developed beyond the proof-of-concept arylation reaction of TMB. In the present study, we aimed to gain more insight into the seemingly specific trend of gold towards substituted aryl halides. We tried in particular to address the following questions: Does the (P,N) gold complex also display a preference for electron-rich substrates? Does the substituent influence observed for the oxidative addition step transpose to the catalytic cross-coupling? What is the reason for the difference between gold and the other transition metals, palladium in particular? In addition, we sought to illustrate the synthetic interest of the (P,N) gold complex in catalysis and thoroughly investigated the direct catalytic arylation of indoles. The results obtained along these two lines are reported hereafter.
Results and discussion
Relative reactivities of substituted iodoarenes in the oxidative addition to the (P,N) gold complex
As we pointed out for the (P,P) gold complex,6a the influence of the iodoarene electronics on the rate of oxidative addition to gold is unique and intriguing. As mentioned above, a similar tendency was recently observed by McGrady, Bower, Russell et al. with a 2,2′-bipyridyl, (N,N) chelated gold complex.6c This raises the question of the generality of this trend and if so, of the reason for such a specific behavior of gold. We thus examined the relative reactivities of a series of para-substituted iodoarenes with the (P,N) gold complex. To start with, the scope of the reaction was further extended (Scheme 1). Electron-poor substrates featuring a p-CF3 as well as p-CN and p-COMe substituents – two potentially coordinating groups – were found to undergo oxidative addition to gold within the time of mixing. With the electron-rich p-NMe2 substituted iodobenzene, a complex mixture of gold complexes was obtained, which might be due to competitive N coordination to gold and/or redox processes.14 In contrast, p-Br iodobenzene reacted smoothly with complete selectivity for C–I oxidative addition. | ||
| Scheme 1 Oxidative addition of a series of para-substituted iodoarenes to the (P,N) gold complex. | ||
The rate of Ar–I oxidative addition to the (P,N) gold complex is much faster than with the (P,P) ligand, preventing a direct comparison of the substrates based on their reaction times (all reactions are complete by the time the medium reaches room temperature). We thus resorted to competition experiments and first performed a few test reactions to determine whether the electron-rich/electron-poor character of the substrate matters or not. Two competition experiments were carried out between p-(MeO)C6H4I as electron-rich substrate and p-(MeCO)C6H4I or p-(F3C)C6H4I as electron-poor substrate (Scheme 2).14 In each case, equimolar amounts of the two substituted iodoarenes were mixed and added in 10-fold molar excess to a solution of the (P,N) gold complex to ensure that the concentration of the two halides remains approximately constant over the reaction (Scheme 2). The ratio of the resulting gold(III) complexes was determined by 31P NMR spectroscopy. In both cases, the major product formed corresponds to the gold(III) complex P1 ensuing from oxidative addition of the electron-rich p-OMe iodobenzene (in 64/36 ratio with p-COMe and 87/13 ratio with p-CF3 iodobenzene). To confirm this trend, another competition reaction was performed with p-(F3C)C6H4I and p-MeC6H4I. Here also, the major product corresponds to the oxidative addition of the iodobenzene bearing the electron-donating group (in 84/16 ratio).
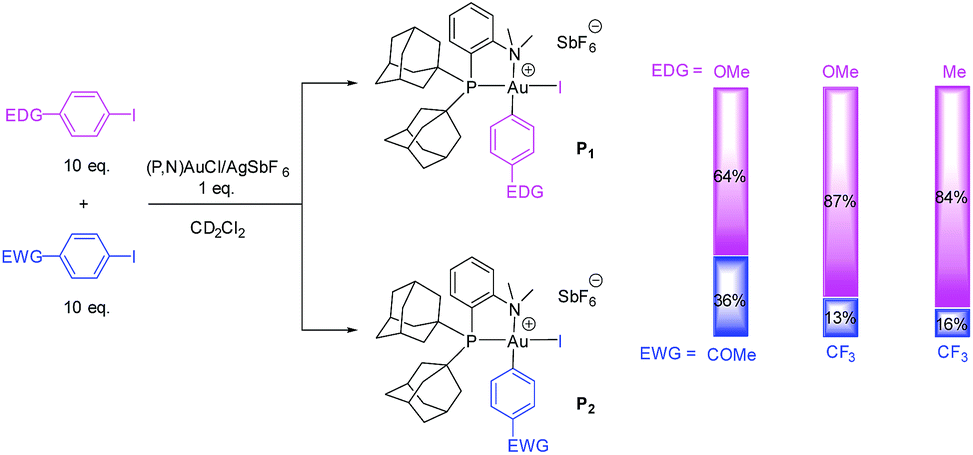 | ||
| Scheme 2 Test competition reactions between EDG and EWG para-substituted iodoarenes in the oxidative addition to the (P,N) gold complex. | ||
These encouraging results prompted us to run a Hammett type analysis. To this end, iodobenzene was reacted with the (P,N) gold complex in competition with several para-substituted iodoarenes (p-OMe, p-Me, p-Br and p-CF3) following the same protocol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the two iodoarenes, 10-fold molar excess with respect to Au). Under these pseudo-first-order conditions, the relative rate constants for the oxidative addition can be derived from the ratio of the gold(III) products (P1vs.P2). The Hammett correlation against σP values15 (Fig. 2) reveals a good linear correlation (R2 = 0.93). The negative slope (ρ = −1.09) confirms the preference of the gold complex to react with electron-rich iodoarenes. This contrasts with the general trend of group 10 metals to react faster with electron-poor substrates,11 as reflected in the positive ρ values reported for palladium. The magnitude of the ρ value for gold is in the lower range of those reported for palladium (1.5–2.3).16 The negative sign of the ρ value found for the gold complex indicates a build-up of positive charge on the aromatic ring upon oxidative addition of the iodoarene and thus suggests an electrophilic behavior of gold.
:1 mixture of the two iodoarenes, 10-fold molar excess with respect to Au). Under these pseudo-first-order conditions, the relative rate constants for the oxidative addition can be derived from the ratio of the gold(III) products (P1vs.P2). The Hammett correlation against σP values15 (Fig. 2) reveals a good linear correlation (R2 = 0.93). The negative slope (ρ = −1.09) confirms the preference of the gold complex to react with electron-rich iodoarenes. This contrasts with the general trend of group 10 metals to react faster with electron-poor substrates,11 as reflected in the positive ρ values reported for palladium. The magnitude of the ρ value for gold is in the lower range of those reported for palladium (1.5–2.3).16 The negative sign of the ρ value found for the gold complex indicates a build-up of positive charge on the aromatic ring upon oxidative addition of the iodoarene and thus suggests an electrophilic behavior of gold.
 | ||
| Fig. 2 Hammett correlation for the oxidative addition of different para-substituted iodoarenes to the (P,N) gold complex. | ||
The (P,N) gold complex thus displays the same reverse electronic trend towards iodoarenes than the (P,P) and (N,N) chelated complexes, suggesting it is a general feature of gold complexes to react faster with electron-rich substrates. This prompted us to further examine the oxidative addition path computationally to try to decipher possible underlying factors.
DFT analysis of the oxidative addition path to gold, and comparison with palladium
Our previous DFT studies have substantiated the key role of the chelating (P,N) ligand.6b It lowers the activation barrier of oxidative addition and stabilizes the ensuing aryl gold(III) species thanks to coordination of the nitrogen atom to gold. The weakly coordinating SbF6− counter-anion was also shown to play no major role. Its displacement by iodobenzene proceeds with a very low activation barrier and the rate-determining step is the oxidative addition to gold. As a result, the gold preference for electron-rich iodoarenes is unlikely to originate from the initial displacement of SbF6− at gold, but rather stands in the oxidative addition process itself.The structure of the associated transition state was thus further analyzed (Fig. 3A).14 Charge analysis reveals an electron transfer from iodobenzene to gold (by 0.16 e according to Bader charges computed at the transition state, Atom-In-Molecules calculations), in line with the electrophilic behavior of the gold complex observed experimentally. Consistently, NBO (Natural Bond Orbital) and CDA (Charge Decomposition Analysis) indicate that donor–acceptor interactions from PhI to Au largely prevail (Tables S5 and S6†).14 Most significant is the donation from an iodine lone pair to the vacant σ*(Au–P) orbital which is associated with a stabilization energy of ca 40 kcal mol−1. The sum of stabilizing energies for PhI → Au interactions amounts to ca 72 kcal mol−1 whereas Au → PhI back-donations sum to only ∼13 kcal mol−1. The donation/back-donation ratio (d/b) as estimated by CDA is 2.67. Thus, despite the electron-donation from the nitrogen atom, the gold atom remains highly electrodeficient and the oxidative addition of the Csp2–I bond is associated with a charge transfer from PhI to Au.17 Note that recent detailed analyses of gold carbonyl complexes have also pointed out the high electrophilicity of cationic gold(I) complexes. Here, strong σ(CO) to gold charge transfer results in an uncommon high-energy shift of the CO stretching frequency due to C ← O polarization.18
 | ||
| Fig. 3 Reaction profiles computed for the oxidative addition of iodobenzene to the real (P,N) gold complex (A) and to a model (P,P) palladium complex (B). Calculations performed at the B97D(SMD-DCM)/SDD+f(Au,Pd), SDD(I),6-31G**(other atoms) level of theory. Structure of the transition state of oxidative addition (bond lengths in Å, bond angles in °). Contour plot of the NOCV deformation density Δρorb and the associated stabilizing energy (ΔEorb(ρ), in kcal mol−1) for the predominant orbital interaction between PhI and the Au/Pd metal fragments. The charge flow is red → blue (Δρ < 0 in red and Δρ > 0 in blue). Analysis performed at ZORA-BP86-D3/TZ2P level of theory. The contour value for density is 0.001 a.u. | ||
Oxidative addition of iodobenzene to a model chelating diphosphine Pd complex was also theoretically investigated to draw comparison between the two metals (Fig. 3B). In line with previous calculations,19 the Csp2–I bond activation at palladium is very favored thermodynamically and proceeds with a very low activation barrier (∼1 kcal mol−1). The structure of the corresponding transition state was analyzed in detail. Of note, the charge transfer is opposite to that found with gold (electron transfer of 0.46 e from Pd to PhI according to the Bader charges). The NBO picture is also quite different from that encountered with the gold complex. The PhI → Pd and Pd → PhI donor–acceptor interactions are much more balanced. The sum of stabilizing energies are 71 and 30 kcal mol−1, respectively (NBO) and the d/b ratio is 0.73 (CDA). This reflects a significantly higher contribution of Pd to PhI back-donation in the oxidative addition process, in line with the observed charge transfer from the metal to the substrate. We surmised that the reverse electron flow in the oxidative addition transition state is responsible for the different behavior of the two metals. Important back-donation from palladium favors electron-deprived substrates whereas the electrophilicity of the gold center favors electron-rich substrates. This difference probably stems from the intrinsic properties of the two metals, as well as from the charge of the reacting metal species (cationic vs. neutral). Chelation and bending of the L2Au+ fragment has been reported to enhance back-donation from gold20 but the cationic and electro-deficient character of the metal remains and makes the difference with neutral L2Pd moieties.
To further analyze the different behavior of gold and palladium, the NOCV (Natural Orbital for the Chemical Valence) extension of the EDA (Energy Decomposition Analysis) approach was used and the charge flow in the transition state of oxidative addition was analyzed (Table S7, Fig. S30†).14,21 For both gold and palladium, the electrostatic term contributes to 49–52% of the interaction between the metal fragment and substrate, while the orbital term accounts for 40–42%. The primary orbital interaction is associated with substrate to metal donation for gold, but metal to substrate back-donation for palladium. The associated deformation densities Δρ are represented in Fig. 3, substantiating inverse charge flows of the electron density (red → blue). The overall charge flow estimated by Hirshfeld partitioning confirms the electron-accepting behavior of gold (0.08 e transfer from PhI) but electron-donating behavior of palladium (0.38 e transfer to PhI).
Relative reactivities of iodoarenes in the gold-catalyzed cross-coupling with TMB
We were then keen to examine if the reverse electronic trend observed with gold in the oxidative addition step may be extrapolated to the catalytic cross-coupling of iodoarenes. For this purpose, competition experiments analogous to those described above have been performed to compare the relative reactivities of para-substituted iodoarenes in the cross-coupling with TMB catalyzed by the (P,N) gold complex. The reactions were carried out under the previously reported conditions (5 mol% Au complex, 1.05 eq. AgSbF6, 1 eq. K3PO4, 75 °C, 2 h), except that we used a 10-fold excess of the para-substituted iodoarene and iodobenzene with respect to TMB. The ratios of the resulting biaryl products [P1]/[P2] were determined by GC at complete consumption of TMB and used to build the Hammett correlation shown in Fig. 4. | ||
| Fig. 4 Competition cross-coupling reaction of para-substituted iodoarenes with TMB and associated Hammett correlation. | ||
As observed for the oxidative addition step, the reactivity of iodoarenes towards cross-coupling with TMB increases with increasing the electron donating ability of the para-substituent (Table S2†).14 The slope of the Hammett correlation is negative and similar in magnitude to that found for the oxidative addition step (ρ value = −1.09 vs. −0.92).22,23 Thus, the unusual electronic trend observed in the oxidative addition under stoichiometric conditions is transposed to the cross-coupling reaction under catalytic conditions. The favoured activation and coupling of iodoarenes featuring electron-donating substituents as observed with the (P,N) gold complex is quite unique. It markedly contrasts with the behavior of group 10 metals and provides an efficient alternative to the gold-catalyzed coupling reactions under oxidative and/or photoredox conditions, for which electron-rich aryl electrophiles are challenging substrates.3–5
Synthetic application of gold-catalyzed cross-coupling to the arylation of indoles
We were then interested in applying our gold-catalyzed arylation methodology to an important and challenging transformation with the aim to assess how gold compare with the other transition metals and eventually complement their performance. In this perspective, we targeted the preparation of aryl-substituted indoles, which are important structural motifs found in many natural products and pharmaceuticals.24 The arylation of indoles with transition metal catalysts has garnered tremendous efforts over the last two decades and Pd clearly occupies the forefront position (Scheme 3).25,26 The main challenge of the transformation is the control of regioselectivity in the absence of directing groups. Most transition metals tend to intrinsically favour C2 arylation,25,27 while direct arylations of indoles at the C3 position remain scant.28,29 | ||
| Scheme 3 Transition metal catalyzed methodologies for the regioselective arylation of indoles. | ||
Over the last few years, gold-catalyzed methodologies have started to be developed for the arylation of indoles. These transformations are based on the oxidative coupling of N-substituted indoles with aryl silanes,30 aryl boronates3f or perfluoro arenes3d (see ESI and Scheme S1† for a detailed state-of-the-art). An appealing feature of these gold-catalyzed reactions lies in the mild conditions in which they operate, typically between room temperature and 110 °C, while Pd, Rh and Ru-catalyzed reactions generally require prolonged heating at 120 °C or above. Moreover, gold selectively promotes C3 arylation in the absence of directing groups and is thus very complementary to the other transition metals (Scheme 3). However, the gold-catalyzed arylation of indoles remains relatively limited in scope. Due to compatibility issues associated with the use of a strong oxidant in stoichiometric amount, the indole is generally protected with an electron-withdrawing group at N (such as Ts, SiiPr3 or Piv). Electron-deprived arylating reagents are also usually preferred, the presence of strong electron-donating substituents commonly leading to greater extents of side-reactions. Note that merging gold and photoredox catalysis2 offers another way to achieve Au(I)/Au(III) cross-couplings, but attempts to apply this methodology to the arylation of indoles have been unsuccessful so far due to competing azo coupling.5e
The promising preliminary result we obtained upon arylation of N-phenyl pyrrole (61% yield of coupling with 9/1 selectivity in favor of the β isomer)6b prompted us to investigate the (P,N) gold catalyst for the coupling of indoles and iodoarenes. As reported hereafter, it proved to be very efficient, general and robust, giving access to a variety of C3-arylated indoles with complete regioselectivity within 2 hours at 75 °C.
To start with, the coupling of N-methyl indole with iodobenzene was tested under the catalytic conditions used for the arylation of TMB and N-phenyl pyrrole (1 eq. of each substrate, 5 mol% of (P,N)AuCl complex, 1.05 eq. of AgSbF6, 1 eq. of K3PO4, o-DCB/MeOH 50:1, 75 °C, 2 h).6b GC-MS analysis of the crude mixture revealed the formation of a single regioisomer of phenyl-substituted N-methyl indole but only in trace amount. Fast screening of the reaction conditions enabled significant improvement. Using two equivalents of N-methyl indole and replacing AgSbF6 by AgOTf, the coupling product was obtained in 99% yield (Scheme 4).31 The TfO− counter-anion is more coordinating than SbF6− and may stabilize the gold(III) intermediate by the time the indole reacts. 1H NMR spectroscopy indicated the exclusive formation of the C3-arylated product 1.32 This regioselectivity is consistent with the C–H activation of the indole at gold proceeding via electrophilic aromatic substitution.33
 | ||
| Scheme 4 Scope of N-substituted indoles. Cross-coupling of iodobenzene and indoles catalyzed by the (P,N) gold complex. Yields determined using calibrated GC-MS with n-dodecane as internal standard. | ||
Next, we examined the applicability of these catalytic conditions to the reaction of iodobenzene with different N-substituted indoles (Scheme 4). The reaction works well with N-phenyl and N-benzyl indoles to afford the corresponding aryl products 2 and 3 in high yields (95 and 82%, respectively) and with complete C3 regioselectivity. Noticeably, the N-tosyl and N-triisopropylsilyl protected substrates used standardly in gold-catalyzed oxidative arylations of indoles3d,30 did not work in our conditions (the N-Ts indole is inert while the N-SiiPr3 undergoes desilylation). No coupling product was detected either with N-acetyl indole. Indoles not depleted in electron density are needed for the reaction to take place, underpinning the electrophilic mechanism of the indole auration.28c Selective arylation of unprotected indoles is challenging. It often requires harsh conditions and gives mixtures of regioisomers. It was thus of interest to test our methodology with N–H indole. The coupling proceeded with moderate yield (51%), but gratifyingly the C3-arylated product 4 was obtained without by-products stemming from N or C2 arylation. This regioselectivity is complementary to those usually observed upon Pd- and Cu-catalyzed arylation of N–H indoles.34,35 The operating conditions for the Au(I)/Au(III) arylation of indoles are remarkably mild for such reactions (2 h at 75 °C, while Pd catalysts typically require heating at 120–150 °C for ∼24 h).27b
The scope of iodoarenes was then examined with N-methyl indole as partner (Scheme 5). Pleasingly, the arylation proceeds with good yields and complete C3 selectivity with both electron-rich (8–9) and electron-deficient (10–12) para-substituted iodobenzenes. More sterically hindered substrates such as 1- and 2-iodonaphthalenes as well as ortho-methoxy iodobenzene also react smoothly and regioselectively (16–18). Notably, bifunctional iodoarenes featuring C–Br or C–OTf bonds display complete chemoselectivity. In contrast to Pd-catalysts that usually react with C–I, C–Br and C–OTf bonds with limited selectivity, only the C–I bond is activated at gold under these conditions. The corresponding C3-arylated indoles 13 and 14 are obtained in good to excellent yields and offer the possibility for subsequent cross-coupling transformations. Boronate moieties are also compatible with the gold-catalyzed arylation, as shown by the preparation of compound 15.36 The scope of iodoarenes was further studied with N–H indole. With both electron-rich (19) and electron-deficient (20) substrates, the arylation proceeds with moderate yield but complete C3 selectivity. Preliminary tests with 2-iodopyridine and 3-iodothiophene show that the reaction is also feasible with heteroaryl iodides (21 and 22), although the corresponding C3-arylated indoles were obtained in moderate yields. Note that in some cases, the use of 2,6-di-tert-butylpyridine (DTBP) instead of K3PO4 as the base afforded better results (for example indole 9 was obtained in 86% yield with DTBP but only 37% with K3PO4). X-ray diffraction analyses of compounds 10 and 13 unambiguously confirmed the regiochemical outcome of the indole arylation.14
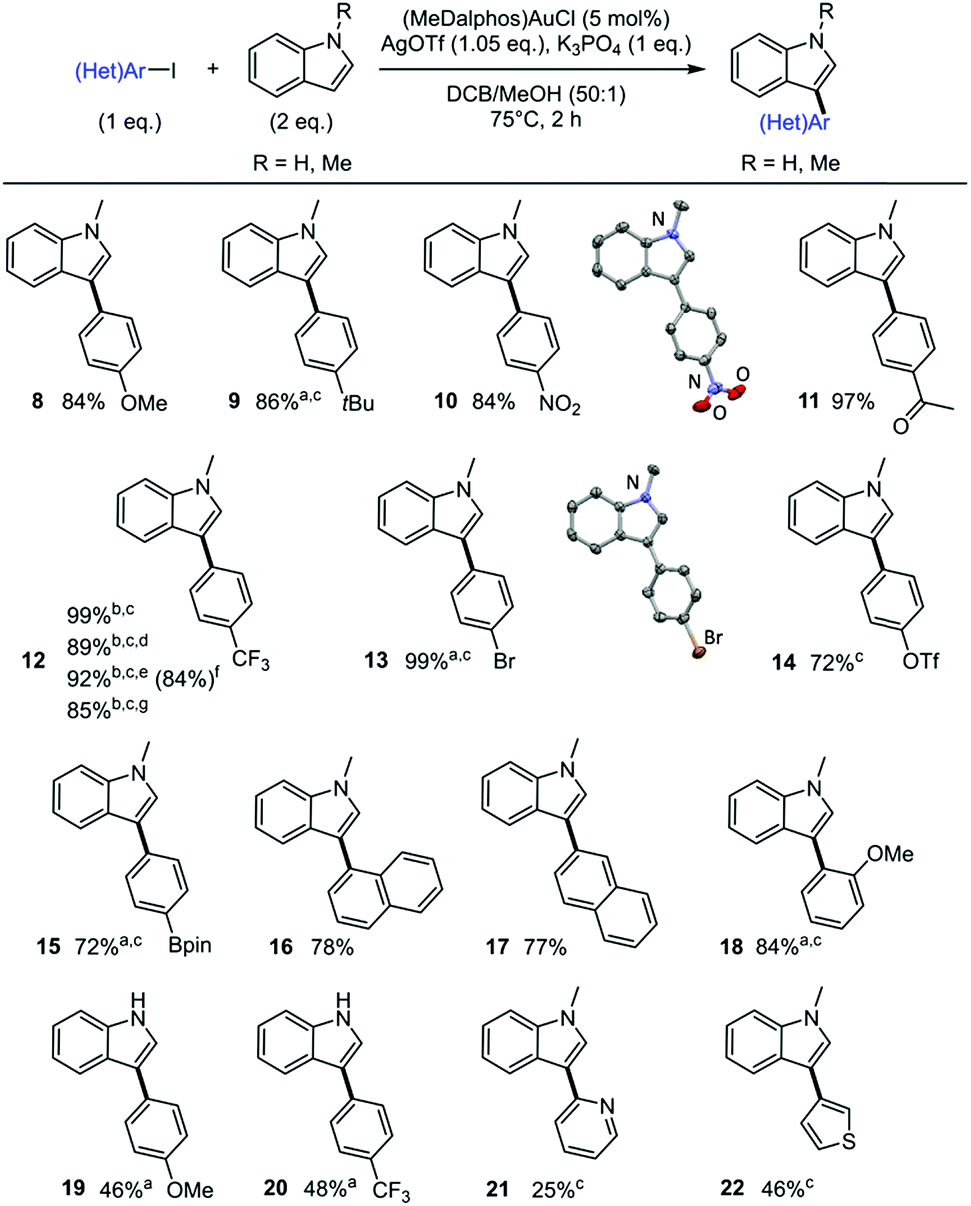 | ||
| Scheme 5 Scope of iodoarenes. Cross-coupling of iodoarenes and N-methyl indole catalyzed by the (P,N) gold complex. Unless otherwise stated, yields determined using calibrated GC-MS with n-dodecane as internal standard. Crystallographic structures of indoles 10 and 13 as inserts. Reaction conditions: 0.1 M of aryl iodide. aDTBP (2 eq.) instead of K3PO4. bDTBP (1 eq.) and N-methyl indole (1 eq.). cYields determined by NMR with n-dodecane as internal standard. dAll reagents weighted in air. eN-methyl indole (1 g), 2 mol% (MeDalphos)AuCI, 4.5 h, 1.3 M of aryl iodide. flsolated yield in parenthesis. gN-methyl indole (1 g), 1 mol% (MeDalphos)AuCI, overnight, 1.4 M of aryl iodide. | ||
The robustness and scalability of the reaction was assessed using 4-iodobenzotrifluoride as substrate. The corresponding C3-arylated indole 12 was obtained in excellent yield using only 1 eq. of N-methyl indole. The reaction displays low air and moisture sensitivity. It allows the use of commercial chemicals without further purification and does not require inert atmosphere. The synthetic applicability of the process was further substantiated by carrying out gram scale experiments using lower catalytic loadings. The C3-arylated indole 12 was for example obtained in 85% yield when performing the reaction overnight with 1 mol% of (MeDalphos)AuCl.
Then the influence of the substitution pattern at the indole moiety was investigated. The coupling of iodobenzene with a variety of N-methyl indoles was tested (Scheme 6). No coupling product was obtained from 2-methyl indole, indicating that the reaction is very sensitive to steric hindrance at this position. Conversely, indoles methylated at either C4, C5, C6 or C7 position readily couple with iodobenzene to give the corresponding C3-arylated products 24–27 in good yields. Particularly noteworthy is the 4-methyl substrate in which the proximity of the methyl substituent at C4 does not hamper the arylation in the C3 position. It is also worth noting that common functional groups are well tolerated on the indole partner, as illustrated by the preparation of the CO2Me, Br, OMe and Bpin-substituted products 28–31.
 | ||
| Scheme 6 Scope of substituted N-methylindoles. Cross-coupling of iodobenzene and indoles catalyzed by the (P,N) gold complex. Yields determined by 1H NMR with n-dodecane as internal standared. aDTBP (1eq.). | ||
A robustness screen was also conducted to further assess the tolerance of the catalytic reaction towards various functional groups. The arylation of N-methyl indole with iodobenzene was undertaken in the presence of one molar equivalent of additives. The yield of 1 was determined by GC-MS analysis, as well as the amount of additive and starting material (SM) remaining (see Table S3 in the ESI†).37 The results show that alkenes (1-octene), alcohols (phenol, methanol), amides (acetanilide) and nitriles (benzonitrile) are well tolerated: the coupling product 1 is obtained in >87% yield, the recovery yields of excess substrate (N-methyl indole) and additive are >94% and >74%, respectively. In contrast, the presence of acids (benzoic acid) or amines (p-toluidine) completely quenched the reactivity, while the addition of aldehydes (cyclohexanecarboxaldehyde) or ketones (cyclohexanone) gives rise to side-reactions with consumption of both the N-methyl indole and the additive. Compatibility of heterocycles was also studied. While imidazole inhibited the reaction, the presence of thiophene and benzofurane did not alter the catalytic arylation of the indole.
Conclusions
Judicious choice of ligand has been recently demonstrated to emulate oxidative addition of iodo- and bromoarenes to gold, a basic elementary step long considered inadequate to access aryl gold(III) complexes. Among the three sets of suitable ligands, only the hemilabile (P,N) MeDalphos ligand has been shown to date to pave the way for Au(I)/Au(III) catalysis from aryl halides. The present study advances our knowledge of the factors controlling this new gold reactivity and its synthetic interest is illustrated in an important catalytic transformation, namely the C3 arylation of indoles.In the presence of AgSbF6, the (MeDalphos)AuCl complex reacts instantaneously and quantitatively with a variety of iodoarenes at room temperature. To assess the influence of electronics on the oxidative addition, competition experiments have been performed with para-substituted substrates, revealing a clear preference for electron-rich iodoarenes. Thus, whatever the ligand, (P,P), (N,N) or (P,N), gold displays in oxidative addition a reactivity trend opposite to that typically encountered with palladium. Of note, similar competition experiments could be carried out with the (P,N) ligand under catalytic conditions. The cross-coupling of 1,3,5-trimethoxybenzene with para-substituted iodoarenes was found to follow the same trend, showing that the unique preference of gold for electron-rich substrates can be transposed from the elementary step to a catalytic transformation.
Analysis of the oxidative addition process by DFT calculations has provided a plausible rationale for the different behavior of gold and palladium. Indeed, the respective transition states display opposite electron flows according to NBO, CDA and ETS-NOCV analyses. Despite the donor bidentate ligand, the cationic (L,L′)Au+ complexes are highly electrophilic and significant PhI → Au charge transfer is observed at the oxidative addition transition state. Conversely, metal to PhI back-donation is prevalent for neutral L2Pd(0) complexes, which explains why electron-deprived substrates are more reactive.
Taking advantage of the facile oxidative addition of iodoarenes and of the propensity of the ensuing aryl gold(III) complexes to react with electron-rich (hetero)arenes via electrophilic C–H activation, we have applied the (MeDalphos)AuCl complex to the catalytic arylation of indoles. The transformation proved to be very efficient, general and robust. As the recently developed gold-catalyzed oxidative arylation of indoles, it displays complete regioselectivity for C3 arylation and thus nicely complements the Pd-catalyzed transformations that tend to favor C2 arylation. The (MeDalphos)AuCl complex operates under mild conditions (75 °C, 2 h) without any external oxidant. As a result, it does not require protection of the N atom of the indole and tolerates a variety of functional groups at both the iodoarene and indole partners (NO2, CO2Me, Br, OTf, Bpin, OMe…).
This study underscores the unique properties of (P,N) gold complexes towards oxidative addition and highlights their potential in Au(I)/Au(III) catalysis with aryl halide electrophiles. Future studies will aim to further develop this chemistry by taking advantage of the easy access and rich reactivity of aryl gold(III) complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Centre National de la Recherche Scientifique and the Université de Toulouse is gratefully acknowledged. J. R. thanks the Fundación Ramón Areces for a post-doctoral fellowship and the European Commission for a MCIF (Gold3Cat-799606). The “Direction du Numérique” of the Université de Pau et des Pays de l′Adour, MCIA (Mésocentre de Calcul Intensif Aquitain) and CINES under allocation A005080045 made by Grand Equipement National de Calcul Intensif (GENCI) are acknowledged for computational facilities. E. D. Sosa Carrizo thanks CDAPP for funding part of his post-doctoral contract.Notes and references
- (a) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed; (b) T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006–3009 CrossRef CAS PubMed; (c) M. Livendahl, P. Espinet and A. M. Echavarren, Platinum Met. Rev., 2011, 55, 212–214 CrossRef; (d) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Ackermann, J. De Buck Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133–147 CrossRef CAS; (e) M. Livendahl, C. Goehry, F. Maseras and A. M. Echavarren, Chem. Commun., 2014, 50, 1533–1536 RSC.
- For reviews on gold-catalyzed oxidative couplings, see: (a) P. Garcia, M. Malacria, C. Aubert, V. Gandon and L. Fensterbank, ChemCatChem, 2010, 2, 493–497 CrossRef CAS; (b) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2011, 17, 8248–8262 CrossRef CAS PubMed; (c) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236–8247 CrossRef CAS PubMed; (d) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC. For reviews dealing with oxidant-free gold-catalysis and dual photoredox/gold catalysis, see: (e) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed; (f) M. O. Akram, S. Banerjee, S. S. Saswade, V. Bedi and N. T. Patil, Chem. Commun., 2018, 54, 11069–11083 RSC; (g) M. Zidan, S. Rohe, T. McCalluma and L. Barriault, Catal. Sci. Technol., 2018, 8, 6019–6028 RSC; (h) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- For selected references, see: (a) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644–1648 CrossRef CAS PubMed; (b) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, J. Am. Chem. Soc., 2014, 136, 254–264 CrossRef CAS PubMed; (c) T. J. A. Corrie, L. T. Ball, C. A. Russell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 245–254 CrossRef CAS PubMed; (d) M. P. Robinson and G. C. Lloyd-Jones, ACS Catal., 2018, 8, 7484–7488 CrossRef CAS; (e) X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636–15639 CrossRef CAS PubMed; (f) Q. Wu, C. Du, Y. Huang, X. Liu, Z. Long, F. Song and J. You, Chem. Sci., 2015, 6, 288–293 RSC; (g) Y. Hua, P. Asgari, T. Avullala and J. Jeon, J. Am. Chem. Soc., 2016, 138, 7982–7991 CrossRef CAS PubMed; (h) M. Hofer, A. Genoux, R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1021–1025 CrossRef CAS PubMed; (i) W. Li, D. Yuan, G. Wang, Y. Zhao, J. Xie, S. Li and C. Zhu, J. Am. Chem. Soc., 2019, 141, 3187–3197 CrossRef CAS PubMed.
- For selected references, see: (a) L. T. Ball, M. Green, G. C. Lloyd-Jones and C. A. Russell, Org. Lett., 2010, 12, 4724–4727 CrossRef CAS PubMed; (b) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Chem.–Eur. J., 2012, 18, 2931–2937 CrossRef CAS PubMed; (c) M. J. Harper, E. J. Emmett, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2017, 139, 12386–12389 CrossRef CAS PubMed; (d) A. D. Melhado, W. E. Brenzovich, A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885–8887 CrossRef CAS PubMed; (e) W. E. Brenzovich, D. Benitez, A. D. Lackner, H. P. Shunatona, E. Tkatchouk, W. A. Goddard and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 5519–5522 CrossRef PubMed; (f) G. Zhang, L. Cui, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 1474–1475 CrossRef CAS PubMed.
- For selected references, see: (a) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505–5508 CrossRef CAS PubMed; (b) M. N. Hopkinson, B. Sahoo and F. Glorius, Adv. Synth. Catal., 2014, 356, 2794–2800 CrossRef CAS; (c) S. Kim, J. Rojas-Martin and F. D. Toste, Chem. Sci., 2016, 7, 85–88 RSC; (d) V. Gauchot and A.-L. Lee, Chem. Commun., 2016, 52, 10163–10166 RSC; (e) V. Gauchot, D. R. Sutherland and A.-L. Lee, Chem. Sci., 2017, 8, 2885–2889 RSC; (f) B. Dong, H. Peng, S. E. Motika and X. Shi, Chem.–Eur. J., 2017, 23, 11093–11099 CrossRef CAS PubMed; (g) J. Xie, K. Sekine, S. Witzel, P. Krämer, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 16648–16653 CrossRef CAS PubMed; (h) S. Witzel, K. Sekine, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2018, 54, 13802–13804 RSC.
- (a) M. Joost, A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654–14657 CrossRef CAS PubMed; (b) A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565–572 CrossRef PubMed; (c) M. J. Harper, C. J. Arthur, J. Crosby, E. J. Emmett, R. L. Falconer, A. J. Fensham-Smith, P. J. Gates, T. Leman, J. E. McGrady, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2018, 140, 4440–4445 CrossRef CAS PubMed.
- For oxidant-free gold-catalyzed halogen exchange, C–N and C–O coupling with chelating aryl halides, see: (a) J. Serra, C. J. Whiteoak, F. Acuna-Pares, M. Font, J. M. Luis, J. Lloret-Fillol and X. Ribas, J. Am. Chem. Soc., 2015, 137, 13389–13397 CrossRef CAS PubMed; (b) J. Serra, T. Parella and X. Ribas, Chem. Sci., 2017, 8, 946–952 RSC.
- Some aryl gold(III) species deriving from aryl diazonium salts have also been isolated thanks to the use of ancillary chelating ligands, see: (a) A. Tlahuext-Aca, M. N. Hopkinson, C. G. Daniliuc and F. Glorius, Chem.–Eur. J., 2016, 22, 11587–11592 CrossRef CAS PubMed; (b) L. Huang, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2016, 52, 6435–6438 RSC; (c) E. O. Asomoza-Solís, J. Rojas-Ocampo, R. A. Toscano and S. Porcel, Chem. Commun., 2016, 52, 7295–7298 RSC; (d) A. Tabey, M. Berlande, P. Hermange and E. Fouquet, Chem. Commun., 2018, 54, 12867–12870 RSC.
- (a) M. Joost, L. Estévez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236–5240 CrossRef CAS PubMed; (b) J. H. Teles, Angew. Chem., Int. Ed., 2015, 54, 5556–5558 CrossRef CAS PubMed.
- Little is known yet about the effects of aryl electronics in Au(III) and Au(I)/Au(III) reactivity. See: (a) ref. 3b ; (b) K. Kang, S. Liu, T. Xu, D. Wang, X. Leng, R. Bai, Y. Lan and Q. C. Shen, Organometallics, 2017, 36, 4727–4740 CrossRef CAS; (c) L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann, Chem.–Eur. J., 2018, 24, 8893–8903 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, Universtity science books ed., Sausalito, CA, 2009 Search PubMed; (b) K. C. Lam, T. B. Marder and Z. Lin, Organometallics, 2007, 26, 758–760 CrossRef CAS.
- R. J. Lundgren, K. D. Hesp and M. Stradiotto, Synlett, 2012, 17, 2443–2458 Search PubMed.
- M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, J. Am. Chem. Soc., 2018, 140, 7065–7069 CrossRef CAS PubMed.
- See ESI.†.
- H. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef.
- (a) C. Amatore and F. Pflüger, Organometallics, 1990, 9, 2276–2282 CrossRef CAS; (b) L. A. Perego, P.-A. Payard, B. Haddou, I. Ciofini and L. Grimaud, Chem.–Eur. J., 2018, 24, 2192–2199 CrossRef CAS PubMed.
- Same calculations have been performed on the (N,N) and (P,P) chelated gold complexes6a,c which displayed preference for electron-rich aryl iodides. Similar results were obtained with electron transfer from iodobenzene to the (L,L)Au+ fragment at the transition state of oxidative addition, and prevalent substrate to metal donation.14.
- (a) D. Sorbelli, L. Belpassi, F. Tarantelli and P. Belanzoni, Inorg. Chem., 2018, 57, 6161–6175 CrossRef CAS PubMed; (b) C. A. Gaggioli, L. Belpassi, F. Tarantelli and P. Belanzoni, Chem. Commun., 2017, 53, 1603–1606 RSC; (c) G. Bistoni, S. Rampino, N. Scafuri, G. Ciancaleoni, D. Zuccaccia, L. Belpassi and F. Tarantelli, Chem. Sci., 2016, 7, 1174–1184 RSC.
- (a) H. M. Senn and T. Ziegler, Organometallics, 2004, 23, 2980–2988 CrossRef CAS; (b) M. Ahlquist, P. Fristrup, D. Tanner and P.-O. Norrby, Organometallics, 2006, 25, 2066–2073 CrossRef CAS; (c) See also ref. 11b.
- (a) M. Joost, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2014, 53, 14512–14516 CrossRef CAS PubMed; (b) A. Zeineddine, F. Rekhroukh, E. D. Sosa Carrizo, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 1306–1310 CrossRef CAS PubMed.
- For recent computational studies of gold complexes using the ETS-NOCV approach, see: (a) P. Jerabek, H. W. Roesky, G. Bertrand and G. Frenking, J. Am. Chem. Soc., 2014, 136, 17123–17135 CrossRef CAS PubMed; (b) E. P. A. Couzijn, Y.-Y. Lai, A. Limacher and P. Chen, Organometallics, 2017, 36, 3205–3214 CrossRef CAS; (c) R. Bhattacharjee and A. Datta, Chem.–Eur. J., 2018, 24, 13636–13646 CrossRef CAS PubMed; (d) A. Couce-Rios, A. Lledós, I. Fernández and G. Ujaque, ACS Catal., 2019, 9, 848–858 CrossRef CAS.
- ρ values from 0.29 to 1.44 have been reported for Pd-catalyzed cross-couplings of para-substituted iodoarenes: (a) C. Consorti, G. Ebeling, F. Flores, F. Rominger and J. Dupont, Adv. Synth. Catal., 2004, 346, 617–624 CrossRef CAS; (b) D. Zim, S. M. Nobre and A. L. Monteiro, J. Mol. Catal. A: Chem., 2008, 287, 16–23 CrossRef CAS; (c) D. E. Stephens, J. Lakey-Beitia, A. C. Atesin, T. A. Ateşin, G. Chavez, H. D. Arman and O. V. Larionov, ACS Catal., 2015, 5, 167–175 CrossRef CAS PubMed.
- A small negative ρ value (−0.25) was reported for the Ullmann Cu-catalyzed coupling of iodoarenes with diarylamines: A. J. Paine, J. Am. Chem. Soc., 1987, 109, 1496–1502 CrossRef CAS.
- N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159–184 CrossRef CAS PubMed.
- (a) E. M. Beck and M. J. Gaunt, Top. Curr. Chem., 2010, 292, 85–121 CrossRef CAS PubMed; (b) N. Lebrasseur and I. Larrosa, in Advances in Heterocyclic Chemistry, ed. A. Katritzky, Academic Press, USA, 2012, vol. 105, pp. 309−351 Search PubMed; (c) A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403–2435 CrossRef CAS; (d) L. Ping, D. S. Chung, J. Bouffard and S. G. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC.
- For transition-metal-free processes using highly electrophilic arylating substrates (i.e. diazonium salts) or highly reactive metalating reagents (such as potassium tert-butylate or lithium tetramethylpiperidine), see: (a) L. Ackermann, M. Dell'Acqua, S. Fenner, R. Vicente and R. Sandmann, Org. Lett., 2011, 13, 2358–2360 CrossRef CAS PubMed; (b) Y.-P. Zhang, X.-L. Feng, Y.-S. Yang and B.-X. Cao, Tetrahedron Lett., 2016, 57, 2298–2302 CrossRef CAS; (c) T. Truong and O. Daugulis, J. Am. Chem. Soc., 2011, 133, 4243–4245 CrossRef CAS PubMed; (d) J. Chen and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 3951–3955 CrossRef CAS PubMed.
- (a) X. Wang, B. S. Lane and D. Sames, J. Am. Chem. Soc., 2005, 127, 4996–4997 CrossRef CAS PubMed; (b) X. Wang, D. V. Gribkov and D. Sames, J. Org. Chem., 2007, 72, 1476–1479 CrossRef CAS PubMed; (c) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972–4973 CrossRef CAS PubMed; (d) H. P. L. Gemoets, I. Kalvet, A. V. Nyuchev, N. Erdmann, V. Hessel, F. Schoenebeck and T. Noel, Chem. Sci., 2017, 8, 1046–1055 RSC; (e) C. Sollert, K. Devaraj, A. Orthaber, P. J. Gates and L. T. Pilarski, Chem.–Eur. J., 2015, 21, 5380–5386 CrossRef CAS PubMed.
- (a) B. S. Lane, M. A. Brown and D. Sames, J. Am. Chem. Soc., 2005, 127, 8050–8057 CrossRef CAS PubMed; (b) D. R Stuart and K. Fagnou, Science, 2007, 316, 1172–1175 CrossRef PubMed; (c) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172–8174 CrossRef CAS PubMed; (d) B. Join, T. Yamamoto and K. Itami, Angew. Chem., Int. Ed., 2009, 48, 3644–3647 CrossRef CAS PubMed; (e) K. Yamaguchi, H. Kondo, J. Yamaguchi and K. Itami, Chem. Sci., 2013, 4, 3753–3757 RSC; (f) S. Perato, B. Large, Q. Lu, A. Gaucher and D. Prim, ChemCatChem, 2017, 9, 389–392 CrossRef CAS.
- For examples of indole arylation at remote positions, see: (a) Y. Yang and Z. Shi, Chem. Commun., 2018, 54, 1676–1685 RSC; (b) J. Kalepu, P. Gandeepan, L. Ackermann and L. T. Pilarski, Chem. Sci., 2018, 9, 4203–4216 RSC; (c) X. Qiu, H. Deng, Y. Zhao and Z. Shi, Sci. Adv., 2018, 4, eaau6468 CrossRef PubMed; (d) X. Qiu, P. Wang, D. Wang, M. Wang, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 1504–1508 CrossRef CAS PubMed and references cited therein.
- (a) K. Hata, H. Ito, Y. Segawa and K. Itami, Beilstein J. Org. Chem., 2015, 11, 2737–2746 CrossRef CAS PubMed; (b) A. J. Cresswell and G. C. Lloyd-Jones, Chem.–Eur. J., 2016, 22, 12641–12645 CrossRef CAS PubMed.
- The catalytic reaction did not proceed when the K3PO4/AgOTf combination was replaced by Ag3PO4..
- Y. Zou, G. Yue, J. Xu and J. Zhou, Eur. J. Org. Chem., 2014, 5901–5905 CrossRef CAS.
- Electrophilic substitutions are known to proceed selectively at the C3 position of indoles: A. H. Jackson and P. P. Lynch, J. Chem. Soc., Perkin Trans. 2, 1987, 1215–1219 RSC.
- Pd catalysts generally show high C2 selectivity (see ref. 25). For a rare example of C3 arylation of N–H indoles, using sterically demanding magnesium salts, see ref. 28a..
- Cu catalysts generally promote N-arylation, see: (a) L. Hu, P. Guo, G. Li, J. Lan, R. Xie and J. You, J. Org. Chem., 2007, 72, 8535–8538 CrossRef; (b) S. Xu, X. Huang, X. Hong and B. Xu, Org. Lett., 2012, 14, 4614–4617 CrossRef CAS PubMed; (c) C3-regioselective arylation of N–H indoles has been observed using diaryl iodonium salts and Cu(OTf)2 as catalyst, see ref. 28c..
- For the selective coupling of B,Si-difunctional arenes with aryl diazonium salts with gold catalysts under irradiation, see ref. 5g..
- For recent discussions of such robustness screening, see: (a) K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS; (b) K. D. Collins and F. Glorius, Acc. Chem. Res., 2015, 48, 619–627 CrossRef CAS PubMed; (c) T. Gensch and F. Glorius, Science, 2016, 352, 294–295 CrossRef CAS PubMed; (d) L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572–8576 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, analytical data and NMR spectra, optimized structures, reaction profiles and bonding analysis (PDF). Crystallographic data for 10 and 13 (CIF). Xyz files (XYZ). CCDC 1894885 and 1894886. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01954e |
| This journal is © The Royal Society of Chemistry 2019 |