
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetic hydricity of silane hydrides in the gas phase†
Jiahui
Xu
 ,
Allison E.
Krajewski
,
Yijie
Niu
,
G. S. M.
Kiruba
and
Jeehiun K.
Lee
*
,
Allison E.
Krajewski
,
Yijie
Niu
,
G. S. M.
Kiruba
and
Jeehiun K.
Lee
*
Department of Chemistry and Chemical Biology, Rutgers The State University of New Jersey, New Brunswick, NJ 08901, USA. E-mail: jee.lee@rutgers.edu
First published on 17th July 2019
Abstract
Herein, gas phase studies of the kinetic hydricity of a series of silane hydrides are described. An understanding of hydricity is important as hydride reactions figure largely in many processes, including organic synthesis, photoelectrocatalysis, and hydrogen activation. We find that hydricity trends in the gas phase differ from those in solution, revealing the effect of solvent. Calculations and further experiments, including H/D studies, were used to delve into the reactivity and structure of the reactants. These studies also represent a first step toward systematically understanding nucleophilicity and electrophilicity in the absence of solvent.
Introduction
Hydride transfers play an important role in many areas of chemistry.1 In organic chemistry, hydride transfer is a key step in many invaluable reactions, including the Cannizzaro, the Meerwein–Ponndorf–Verley, and the Tischenko reactions. Furthermore, metal hydrides such as lithium aluminum hydride and sodium borohydride are indispensable in synthetic chemistry for reducing ketones and aldehydes to alcohols.2 Nicotinamide adenine dinucleotide phosphate (NADPH) and flavin adenine dinucleotide (FADH2) are important enzymatic cofactors that are hydride donors for many biological processes.3 Recent interest in metal-free hydride donors as catalysts for hydrogen activation further solidifies the importance of hydride transfer reactions.4–8Thermodynamic hydricity is the standard Gibbs free energy change for the dissociation of a hydride donor RH to R+ and H−. These values are always positive and therefore, lower values indicate a better hydride donor.6 Kinetic hydricity is usually expressed in terms of a nucleophilicity factor N, which is an empirical value obtained from rate constants for the reaction of hydride donors with reference acceptors.6,9,10 Stronger hydride donors have a higher N value.
The development of the nucleophilicity parameter N has been accomplished largely by Mayr and coworkers, who have demonstrated that the rates of reactions of electrophiles and nucleophiles can be described by a linear-free energy relationship (eqn (1)).9–11
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k = sN(N + E) [at 20 °C] k = sN(N + E) [at 20 °C] | (1) |
“sN” is a nucleophile-specific slope parameter, “N” is a nucleophilicity parameter and “E” is an electrophilicity parameter. k is a normalized rate constant for the nucleophile–electrophile reactions. Mayr's studies of hydride donor ability are one type of reaction examined, toward his developing a general nucleophilicity–electrophilicity scale.12 As Mayr's work has been done in the solution phase, solvent is a potentially complicating factor.
To fill these major gaps in knowledge of general intrinsic nucleophilicity and electrophilicity, as well as specifically, hydricity, we have studied, in the gas phase, the reactions of a series of silane hydrides with substituted benzhydryl cations. While there have been some prior studies of nucleophilicity and electrophilicity in the gas phase, including a Mayr-like study on the association of benzhydryl cations with amines, the systematic application to hydricity has not heretofore been conducted.13–26 Knowledge of the intrinsic kinetic hydricities of metal-free hydrides will be useful for understanding the role of solvent in reactivity. These results are of import both for further developing the utility of hydrides for organic and catalytic applications, as well as for generally understanding nucleophilicity and electrophilicity in the absence of solvent.
Results
Experimental results: hydride abstraction from silanes
We have examined hydride abstraction from silanes by benzhydryl cations; the general reaction is shown in Fig. 1.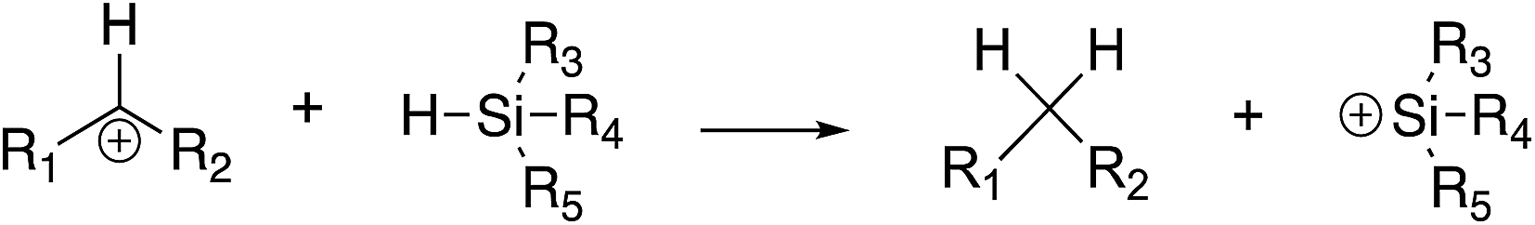 | ||
| Fig. 1 General hydride transfer reaction studied herein. | ||
For this model to succeed in the gas phase, the reaction must be exergonic. This poses some challenges in design since the parent benzhydryl cation (R1 = R2 = Ph) is quite stable. At B3LYP/6-31+G(d), the benzhydryl cation has a calculated hydride affinity (ΔG) of 245 kcal mol−1, while a silyl cation with methyl groups (R3 = R4 = R5 = methyl) has a computed hydride affinity of 252 kcal mol−1. The reaction between these two species would therefore be endergonic. Although our experiments measure kinetic, not thermodynamic, hydricity, our gas phase conditions are such that we do not generally observe endergonic reactions (more details in Experimental section).13,27–31
We first allowed Me2PhSiH and iPr3SiH to react with the parent benzhydryl cation, as calculations (Table 1) indicated that these reactions should be exergonic in the gas phase. These reactions are also known to proceed in solution, which would allow for direct comparison between gas and solution phase reactivity.10
Unfortunately, we found that although these reactions are calculated to be slightly exergonic in the gas phase, we do not observe reaction under our experimental conditions. We hypothesize that the reactions may in reality be slightly endergonic. The reactions may also be favored by solvation of the silyl cation, which is not possible in vacuo.
To drive these reactions further, we added electron withdrawing groups to the benzhydryl cation. The reactions between a series of fluoride-substituted benzhydryl cations and silanes shown in Fig. 2 were examined.
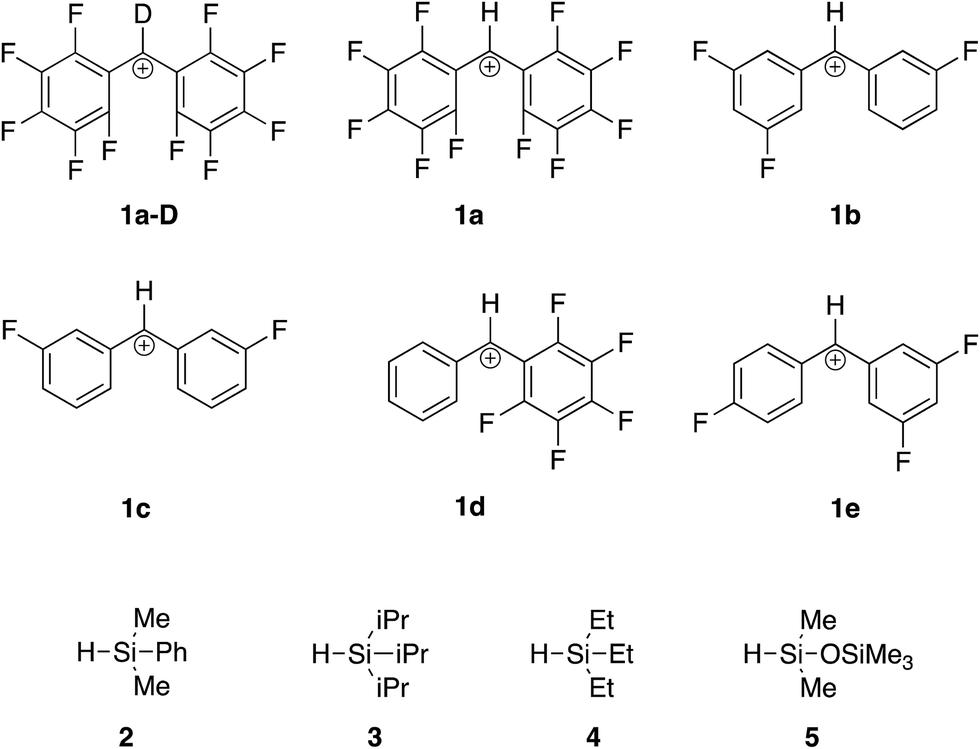 | ||
| Fig. 2 Benzhydryl cations and silanes studied herein. | ||
Briefly, to conduct this experiment, we vaporize the alcohol 1-OH into our Fourier transform mass spectrometer (Fig. 3). Reaction with hydronium ions should generate the desired benzhydryl cation. We then allow the cation to react with the silane hydrides. The rate constants for the reactions between 1 and the various silane hydrides are reported in Table 2.
 | ||
| Fig. 3 Generation of benzhydryl cations and subsequent reaction with silane hydrides. | ||
| 1a-D | 1a | 1b | 1c | 1d | 1e | |
|---|---|---|---|---|---|---|
| 2 | 5.62 ± 0.17 | 4.13 ± 0.26 | 3.47 ± 0.22 | 3.16 ± 0.27 | 2.76 ± 0.17 | 1.69 ± 0.45 |
| 3 | 3.31 ± 0.11 | 2.82 ± 0.12 | 2.09 ± 0.04 | 1.90 ± 0.11 | 1.78 ± 0.05 | 0.81 ± 0.06 |
| 4 | 3.30 ± 0.27 | 2.60 ± 0.19 | 1.84 ± 0.09 | 1.16 ± 0.20 | 1.27 ± 0.11 | 0.57 ± 0.09 |
| 5 | 0.49 ± 0.07 | 0.42 ± 0.11 | 0.14 ± 0.08 | 0.13 ± 0.01 | 0.08 ± 0.02 | Too slow |
Further interpretation of these data will follow, in the “Discussion” section. We next describe additional studies delving into reaction mechanisms.
Reaction details: reaction paths
Two concerns that arose in the course of our studies were whether (i) the desired hydride transfer, and not a proton transfer type reaction was taking place; and (ii) the structure of the reactant cation was in fact a benzhydryl cation, and not a tropylium.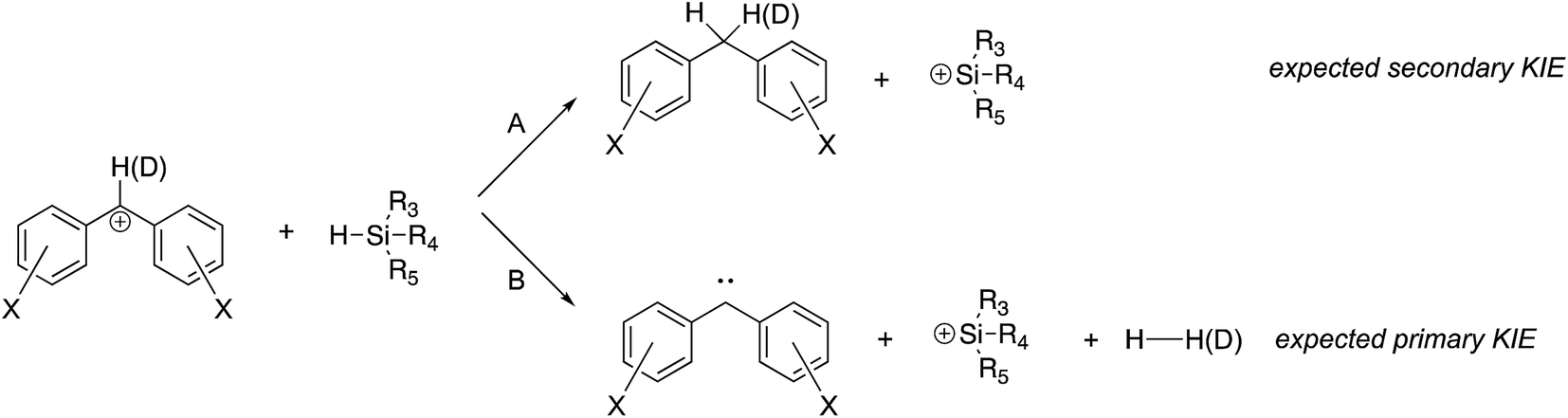 | ||
| Fig. 4 Hydride abstraction versus proton abstraction. | ||
To lend further insight into which pathway we see, we compared the rate constant for reaction of the deuterated perfluorobenzhydryl cation 1a-D with that of the undeuterated compound 1a. As shown in Fig. 4, if hydride transfer occurs (Path A), the carbon with the H(D) should undergo a hybridization change from sp2 to sp3. This should yield a secondary KIE of less than 1. If the H(D) is abstracted (Path B), then the KIE should be normal and primary, a value greater than 1.
Comparison of the rate constants for the reaction of cation 1aversus that of the deuterated 1a-D (Table 2) indicate an inverse secondary KIE of ∼0.8; we therefore believe hydride abstraction is indeed taking place.
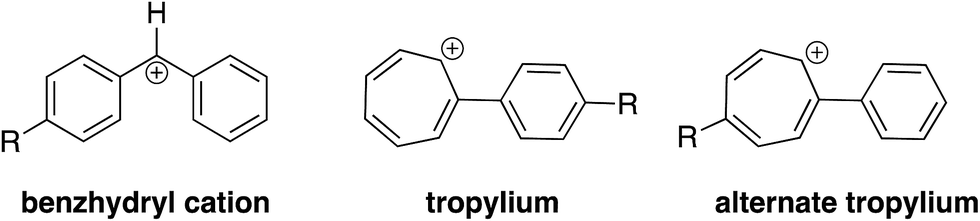 | ||
| Fig. 5 Benzhydryl cation versus tropylium structures. | ||
We first calculated the free energy change associated with the isomerization of the benzhydryl cation to the tropylium (Table 3). In all cases, the isomerization is endergonic (albeit by just 0.5 kcal mol−1 for 1b).
We also sought to determine structure experimentally, through reactions with toluene. It is known that the benzyl cation reacts with toluene in the gas phase, while the tropylium remains unreactive (Fig. 6).32–36
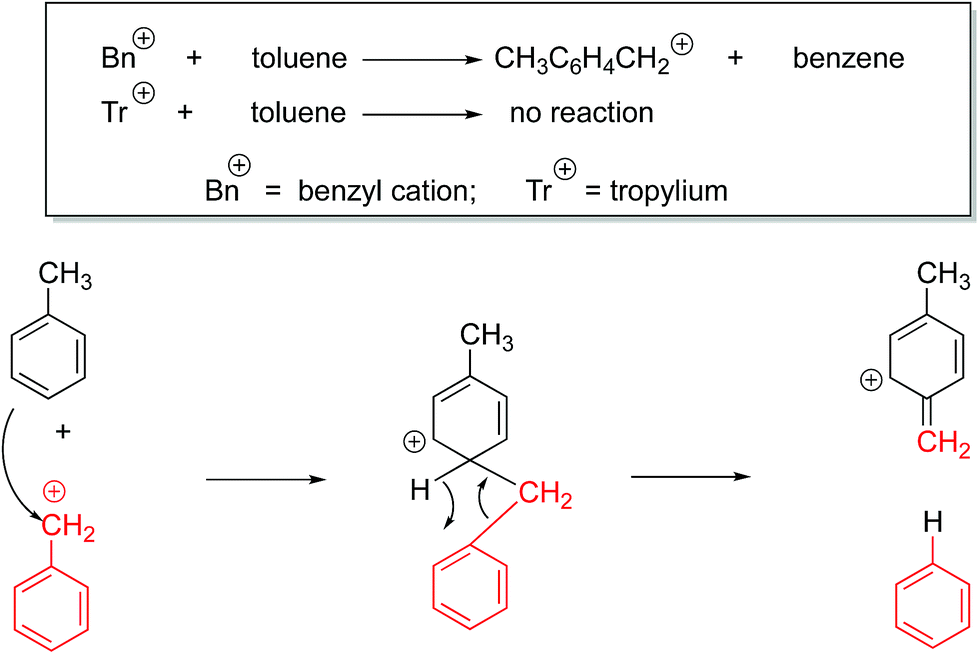 | ||
| Fig. 6 Characteristic benzyl cation reaction with toluene in the gas phase. | ||
While benzyl cation and tropylium display the divergent reactivity shown in Fig. 6, to our knowledge, such reactions have not been studied with benzhydryl cations. As a control, we examined the reaction of benzyl cation with toluene under our conditions, and we do produce the expected CH3C6H4CH2+ cation. However, when we allow the cations derived from the parent benzhydrol to react with toluene, we observe no reaction. We also tried the cations derived from the perfluorobenzhydrol; these also show no reaction with toluene.
These results could either mean that we have tropylium structures, or that the reactivity of benzhydryl cations with toluene is not the same as that of benzyl cations with toluene. We conducted calculations on the parent benzhydryl cation plus toluene reaction, to ascertain the barriers to reaction. Calculations indicate that the barrier to nucleophilic attack of the benzhydryl cation by toluene is relatively higher, by roughly 20 kcal mol−1, than that of the benzyl cation with toluene. Unlike benzyl cation, the benzhydryl cation has a quite sterically hindered center, which might slow down any reaction. Thus, the lack of reaction between toluene and the cations derived from benzhydrol would seem inconclusive; we cannot say that we do not have the benzhydryl cation structure as the reaction with toluene may simply be unfavored.
Overall, we do have reason to believe that our cations are benzhydryl in structure. First, the generation method should favor the benzhydryl cation, not the tropylium. We produce the cation from the corresponding benzhydryl alcohol, via protonation. Sen Sharma and Kebarle established that abstraction of chloride from benzyl chloride in the gas phase yields the benzyl cation, not the tropylium.33 This reaction is similar to our generation method, which should favor the benzhydryl cation structure. Tropylium is usually produced by direct electron impact, which we are not using.34–41 Furthermore, both computational and experimental evidence indicates that under thermoneutral conditions, rearrangement from benzyl cation to tropylium is unlikely.33,34,41–43
Second, in work done for an orthogonal project, we find that our measured gas phase acidity of perfluorobenzhydryl cation 1a-D is the same, whether we generate the cation in the gas phase or from solution.44 That is, if we generate 1a-D as described herein, or from solution (via electrospray), we find that both species have the same acidity. Our calculations indicate that tropylium should be about 15 kcal mol−1 more acidic than benzhydryl cation. Therefore, the similar acidity for the two differentially generated cations implies both have the same structure.44 Electrospray ionization (ESI) is widely accepted as a gentle method which captures the ions that already exist in solution.45 Since in solution, these ions are believed to be benzhydryl cations, it would stand to reason that the ions brought into the mass spectrometer by ESI would also be benzhydryl in structure.10
We should also note that ultimately, while the evidence taken together points to a benzhydryl cationic structure, the identity of the structure does not strictly affect the results herein. The rate constants are used to ascertain the silane hydride nucleophilicities, and if the cations are of varying structure, it will not affect those nucleophilicity trends.
Discussion
With rate constants (Table 2) in hand, we looked to Mayr eqn (1), to ascertain the kinetic hydricity, or nucleophilicity, of the silanes. At this early point in our research, rather than derive any specific parameter values, we instead made a “Mayr-type” graph, plotting the log of the rate constant for the reaction between the series of nucleophiles and electrophiles (y axis) versus the log of the rate constant of the electrophiles with 3 as our reference nucleophile (x axis) (Fig. 7). | ||
| Fig. 7 Plot of benzhydryl cation and silane data. The y-axis represents the log of the rate constant for the reaction of each electrophile with each nucleophile. The x-axis is the log of the rate constant of each electrophile with 3. | ||
The results show that the trend for silane nucleophilicity in the gas phase is 5 < 4 < 3 < 2. The trend in solution (methylene chloride) is 3 < 5 < 2 < 4.10 This change in trend between the gas phase and solution is of interest, since such changes usually reveal the role of solvent. One possible reason for the differences between the gas phase and the solution phase could be solvation of the silyl cation product. In solution, the most nucleophilic silane hydride is Et3SiH (4), while the least nucleophilic species is iPr3SiH (3). In the gas phase, iPr3SiH (3) is more nucleophilic than Et3SiH (4). The higher nucleophilicity of iPr3SiH in the gas phase could be due to the stability that the product iPr3Si+ cation gains from the polarizable isopropyl groups. In solution, those bulky isopropyl groups may inhibit solvation of iPr3Si+ relative to Et3Si+, which results in the reversal of the hydricity of the parent neutral compounds. This is reminiscent of the well-known reversal of acidity of methanol versus tert-butanol in solution versus the gas phase.46 In order to lend insight into this possible explanation, we calculated the free energy of solvation in methylene chloride for the silyl cations. We find that the triethylsilyl cation (generated from 4) has a ΔGsolvation of −52 kcal mol−1, while the triisopropyl cation (generated from 3) has a ΔGsolvation of −49 kcal mol−1. Therefore, the triethylsilyl cation is slightly better solvated, reflecting the observed trend. The solvation model does not include specific solvation, which would be expected to further enhance the differences between the two cations.47
Another possible reason for the differences between the gas phase and solution silane nucleophilicity, suggested by a referee, is that the dichloromethane solvent may form weak Cl–Si interactions, such that a CH2Cl2–SiR3H species might be the true hydride donor in solution.
Hydricity is of importance for understanding reactions in which silane hydrides serve as hydride sources. One intriguing example is the use of silane hydrides to reduce N-heterocyclic carbene (NHC)-activated carbon dioxide.7,8 Utilization of carbon dioxide has become of paramount importance, given the rising carbon dioxide concentration in our atmosphere. Radian, Zhang and Ying found that CO2 activated by NHCs (to form an imidazolium carboxylate) will react with silane hydrides to form methanol.7,8 In terms of substrates that overlap with ours, they found that hydride transfer by Me2PhSiH (2) is more effective than Et3SiH (4). We find this of interest, as our gas phase nucleophilicity trend is consistent with this result, while the solvent nucleophilicity trend is not. That is, our gas phase results on hydricity may be useful for predictive power in applications utilizing silane hydrides.
Ultimately, the kinetic hydricity of the studied silanes are different in the gas phase and in solution. Such results show the importance of gas phase studies, to establish how solvent is affecting our understanding of reactivity.
In terms of electrophilicity, the gas phase trend for the benzhydryl cations is 1e < 1d < 1c < 1b < 1a, with 1a being most electrophilic. Mayr and coworkers have examined 1b and 1c in solution; the electrophilicity trend for these cations is the same as that in the gas phase. This is consistent with the Mayr interpretation that electrophile solvation energies change proportionally with electrophilic reactivities; we see this proportionality in the absence of solvent. Furthermore, a survey of other fluorinated benzhydryl cations studied by Mayr indicates that fluorine substitution on the 3-position increases electrophilicity the most; F substitution on the 5 position also enhances electrophilicity, but not as much as on the 3-position; and F substitution on the 4-position tends to have a negative effect on electrophilicity.12 These trends are consistent with that which we see in the gas phase.11,12,25,48
In terms of absolute rate constants, the reactions are faster in the gas phase than in solution. For the reaction of cation 1b with Et3SiH 4, the solution phase rate constant is 6.04 × 107 M−1 s−1. In the gas phase, this reaction rate constant is 1.11 × 1011 M−1 s−1. For the reaction of cation 1c with 4, the solution phase rate constant is 2.51 × 107 M−1 s−1, while the gas phase k is 0.70 × 1011 M−1 s−1. Thus, for these reactions, the rates are roughly 2000 times faster in the gas phase.
This work indicates the importance of examining properties in the gas phase; comparison of intrinsic reactivity with solution phase behavior can ferret out the details of media effects.
Conclusions
This work represents the first attempt toward developing a gas phase hydricity scale. Kinetic hydricity is of import both for general understanding of nucleophilicity and electrophilicity, as well as for improved design of hydrides for energy applications. Our studies indicate that the abstraction of hydride from a series of silane hydrides follows a trend that differs from that in solution, revealing the effect of solvent. Our work also delves into the reactivity and structure of our cations, using H/D studies, calculations, and further experimentation. Having established that hydricity can be studied as described herein, we are moving toward expanding the cations and silane hydrides studied. Overall, the ability to independently investigate electrophilicity and nucleophilicity in the gas phase would allow us to ascertain the solvent effects by direct comparison to solution phase electrophile and nucleophile data. Such studies would be a breakthrough in the understanding of organic reactivity, because many of the phenomena observed in electrophile-nucleophile studies in solution cannot be adequately interpreted.49Experimental
All of the benzhydryl cation precursors (the benzhydryl alcohols) were commercially available, except for the deuterated substrate described herein, which was synthesized following literature procedure.50Measurement of rate constants was carried out in a Fourier transform ion cyclotron resonance mass spectrometer (FTMS). The FTMS has a dual cell setup (described previously).27,28,51–53 The magnetic field is 3.3 T; the baseline pressure is 1 × 10−9 Torr. The solid benzhydrol precursors were introduced into the cell via a heatable solids probe, while the silanes were introduced via a system of heatable batch inlets. Water was pulsed into the cell, and ionized by an electron beam (typically 20 eV, 6 μA, 0.5 s) to generate hydronium ions. The benzhydryl cations were generated by reaction of the benzhydrol with the hydronium ions. The cations were then selected, and transferred from one cubic cell to another via a 2 mm hole in the middle trapping plate. Reaction with silanes was then tracked. Experiments were conducted at ambient temperature. The typical protocol for obtaining gas phase rate constants has been described previously.27,28,51,54,55 The experiments are run under pseudo-first-order conditions with the neutral silane hydride reactant in excess, relative to the reactant cations. Reading the pressure of the neutral compounds from the ion gauges is not always accurate; therefore, we “back out” the neutral substrate pressure from fast control reactions (described previously).27,55–59 Under our conditions, the ionic species are generated, then argon gas is pulsed in, so that the ions are cooled. Thus, only thermoneutral and exergonic reactions are observable.13,60
All calculations were performed using density functional theory (B3LYP/6-31+G(d)) as implemented in Gaussian 09 and 16.61–66 All ground state geometries were fully optimized and frequencies were calculated; no scaling factor was applied. The optimized structures had no negative frequencies. The transition structures corresponding to the reaction of toluene with benzyl cation, and toluene with benzhydryl cation, correspond to partially optimized structures in which the distance between carbocation (benzyl/benzhydryl) and carbon atom para to the methyl carbon of toluene was constrained to 2.0 A. The partially optimized transition structure resulted in the one imaginary frequency corresponding to the bond formation between the carbocation (benzyl/benzhydryl) and toluene. The temperature for the calculations was set to be 298 K. Calculations in methylene chloride were also conducted at B3LYP/6-31+G(d), using a polarized continuum model; specifically, we utilized the SMD variation of IEFPCM of Truhlar and co-workers.67
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the NSF and the NSF Extreme Science and Engineering Discovery Environment (XSEDE) for support. We are also grateful to Professors Herbert Mayr and Steven Kass for incredibly insightful discussions.References
- C. I. F. Watt, Adv. Phys. Org. Chem., 1988, 24, 57–112 CrossRef CAS.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Part B: Reaction and Synthesis, Springer US, USA, 2007 Search PubMed.
- D. Voet and J. G. Voet, Biochemistry, John Wiley & Sons, Inc., New York, 4th edn, 2010 Search PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Chem. Soc. Rev., 2018, 47, 2809–2836 RSC.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS.
- S. N. Riduan, Y. Zhang and J. Y. Ying, ChemCatChem, 2013, 5, 1490–1496 CrossRef CAS.
- H. Mayr and M. Patz, Angew. Chem., Int. Ed., 1994, 33, 938–957 CrossRef.
- M. Horn, L. H. Schappele, G. Lang-Wittkowski, H. Mayr and A. R. Ofial, Chem.–Eur. J., 2013, 19, 249–263 CrossRef CAS PubMed.
- H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS.
- H. Mayr, Mayr's Database of Reactivity Parameters, 2017, http://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/.
- S. Gronert, Chem. Rev., 2001, 101, 329–360 CrossRef CAS.
- W. N. Olmstead and J. I. Brauman, J. Am. Chem. Soc., 1977, 99, 4219–4228 CrossRef CAS.
- J. L. Wilbur and J. I. Brauman, J. Am. Chem. Soc., 1994, 116, 9216–9221 CrossRef CAS.
- C. H. DePuy, S. Gronert, A. Mullin and V. M. Bierbaum, J. Am. Chem. Soc., 1990, 112, 8650–8655 CrossRef CAS.
- M. J. Pellerite and J. I. Brauman, J. Am. Chem. Soc., 1980, 102, 5993–5999 CrossRef CAS.
- J. A. Dodd and J. I. Brauman, J. Am. Chem. Soc., 1984, 106, 5356–5357 CrossRef CAS.
- M. J. Pellerite and J. I. Brauman, J. Am. Chem. Soc., 1983, 105, 2672–2680 CrossRef CAS.
- J. A. Dodd and J. I. Brauman, J. Phys. Chem., 1986, 90, 3559–3562 CrossRef CAS.
- B. D. Wladkowski, J. L. Wilbur and J. I. Brauman, J. Am. Chem. Soc., 1994, 116, 2471–2480 CrossRef CAS.
- B. D. Wladkowski and J. I. Brauman, J. Phys. Chem., 1993, 97, 13158–13164 CrossRef CAS.
- K. Tanaka, G. I. Mackay, J. D. Payzant and D. K. Bohme, Can. J. Chem., 1976, 54, 1643–1659 CrossRef CAS.
- D. K. Bohme and L. B. Young, J. Am. Chem. Soc., 1970, 92, 7354–7358 CrossRef CAS.
- C. Denekamp and Y. Sandlers, Angew. Chem., Int. Ed., 2006, 45, 2093–2096 CrossRef CAS.
- A. Streitwieser Jr, Solvolytic Displacement Reactions, MacGraw-Hill, New York, 1962 Search PubMed.
- M. A. Kurinovich and J. K. Lee, J. Am. Chem. Soc., 2000, 122, 6258–6262 CrossRef CAS.
- M. A. Kurinovich, L. M. Phillips, S. Sharma and J. K. Lee, Chem. Commun., 2002, 2354–2355 RSC.
- K. Wang, M. Chen, Q. Wang, X. Shi and J. K. Lee, J. Org. Chem., 2013, 78, 7249–7258 CrossRef CAS.
- A. Z. Michelson, M. Chen, K. Wang and J. K. Lee, J. Am. Chem. Soc., 2012, 134, 9622–9633 CrossRef CAS PubMed.
- M. Chen, J. P. Moerdyk, G. A. Blake, C. W. Bielawski and J. K. Lee, J. Org. Chem., 2013, 78, 10452–10458 CrossRef CAS.
- J. J. Melko, S. G. Ard, N. S. Shuman and A. A. Viggiano, Int. J. Mass Spectrom., 2013, 353, 60–66 CrossRef CAS.
- D. S. Sharma and P. Kebarle, Can. J. Chem., 1981, 59, 1592–1601 CrossRef CAS.
- J. Shen, R. C. Dunbar and G. A. Olah, J. Am. Chem. Soc., 1974, 96, 6227–6229 CrossRef CAS.
- A. Giardini-Guidoni and F. Zocchi, Trans. Faraday Soc., 1968, 64, 2342–2347 RSC.
- S. Wexler and R. Clow, J. Am. Chem. Soc., 1968, 90, 3940–3945 CrossRef CAS.
- P. N. Rylander, S. Meyerson and H. M. Grubb, J. Am. Chem. Soc., 1957, 79, 842–846 CrossRef CAS.
- J. Winkler and F. McLafferty, J. Am. Chem. Soc., 1973, 95, 7533–7535 CrossRef CAS.
- F. McLafferty and J. Winkler, J. Am. Chem. Soc., 1974, 96, 5182–5189 CrossRef CAS.
- E. W. Fu, P. P. Dymerski and R. C. Dunbar, J. Am. Chem. Soc., 1976, 98, 337–342 CrossRef CAS.
- C. Lifshitz, Acc. Chem. Res., 1994, 27, 138–144 CrossRef CAS.
- C. Cone, M. J. S. Dewar and D. Landman, J. Am. Chem. Soc., 1977, 99, 372–376 CrossRef CAS.
- S. Bourcier and Y. Hoppilliard, Eur. J. Mass Spectrom., 2003, 9, 351–360 CrossRef CAS.
- J. Xu, J. Mieres Perez, E. Sanchez-Garcia and J. K. Lee, J. Org. Chem., 2019, 84, 7685–7693 CrossRef CAS.
- D. Schröder, Acc. Chem. Res., 2012, 45, 1521–1532 CrossRef.
- J. I. Brauman and L. K. Blair, J. Am. Chem. Soc., 1970, 92, 5986–5992 CrossRef CAS.
- Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818–1827 CrossRef CAS.
- One other study of gas phase benzhydryl cation electrophilicity has been conducted, in the context of association reactions with amines (ref. 25); we cannot directly compare our data with theirs, as we have no electrophiles in common.
- H. Mayr and A. R. Ofial, Acc. Chem. Res., 2016, 49, 952–965 CrossRef CAS.
- P. Srivastava, R. Ali, S. S. Razi, M. Shahid, S. Patnaik and A. Misra, Tetrahedron Lett., 2013, 54, 3688–3693 CrossRef CAS.
- M. Liu, M. Xu and J. K. Lee, J. Org. Chem., 2008, 73, 5907–5914 CrossRef CAS.
- S. Sharma and J. K. Lee, J. Org. Chem., 2002, 67, 8360–8365 CrossRef CAS.
- S. Sharma and J. K. Lee, J. Org. Chem., 2004, 69, 7018–7025 CrossRef CAS.
- M. A. Kurinovich and J. K. Lee, J. Am. Soc. Mass Spectrom., 2002, 13, 985–995 CrossRef CAS.
- M. Liu, T. Li, F. S. Amegayibor, D. S. Cardoso, Y. Fu and J. K. Lee, J. Org. Chem., 2008, 73, 9283–9291 CrossRef CAS.
- A. Zhachkina, M. Liu, X. Sun, F. S. Amegayibor and J. K. Lee, J. Org. Chem., 2009, 74, 7429–7440 CrossRef CAS.
- X. Sun and J. K. Lee, J. Org. Chem., 2007, 72, 6548–6555 CrossRef CAS.
- W. J. Chesnavich, T. Su and M. T. Bowers, J. Chem. Phys., 1980, 72, 2641–2655 CrossRef CAS.
- T. Su and W. J. Chesnavich, J. Chem. Phys., 1982, 76, 5183–5185 CrossRef CAS.
- C. H. DePuy and V. M. Bierbaum, Acc. Chem. Res., 1981, 14, 146–153 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Rev. A.01 GAUSSIAN09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Rev. B.01 Gaussian16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates for all calculated species, full citations for references with greater than 16 authors, and representative mass spectra are available. See DOI: 10.1039/c9sc02118c |
| This journal is © The Royal Society of Chemistry 2019 |