
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHelical supramolecular polymer nanotubes with wide lumen for glucose transport: towards the development of functional membrane-spanning channels†
Chenyang
Zhang
a,
Xiaoli
Deng
a,
Chenxi
Wang
b,
Chunyan
Bao
 b,
Bing
Yang
a,
Houyu
Zhang
a,
Shuaiwei
Qi
a and
Zeyuan
Dong
*a
b,
Bing
Yang
a,
Houyu
Zhang
a,
Shuaiwei
Qi
a and
Zeyuan
Dong
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Qianjin Street, Changchun, China. E-mail: zdong@jlu.edu.cn
bShanghai Key Laboratory of Functional Materials Chemistry, Institute of Fine Chemicals, East China University of Science and Technology, No. 130 Meilong Road, Shanghai, China
First published on 30th July 2019
Abstract
The manipulation of strong noncovalent interactions provides a concise and versatile strategy for constructing highly ordered supramolecular structures. By using a shape-persistent building block consisting of phenanthroline derivatives and two quadruply hydrogen-bonding AADD moieties, a type of precise helical supramolecular polymer (HSP) nanotube has been developed. The helical conformation of the supramolecular polymers has been proved via various techniques, showing significantly expanded topologies of supramolecular polymers. From the production of new topological structures of supramolecular polymers, predictable properties and functions have arisen. In this study, the helical folding of supramolecular polymers gave rise to the generation of specific wide lumen structures that can be directly visualized via TEM, and the resulting HSP nanotubes can puncture the lipid bilayer membrane to facilitate the transportation of glucose.
Introduction
Natural membrane protein channels puncture the cellular lipid bilayers to transport substances into cells, and thus play important roles in cellular functions including cell metabolism, ion homeostasis, and signal transduction. These protein nanochannels have proved not only to be associated with many diseases1 like ataxias, deafness, epilepsies, and paralyses, but also to be essentially promising for technological applications, such as sensing, separation, sequencing, delivery, and catalysis.2–6 However, they are generally elusive and rebellious because of their sensitive conformation.7 Therefore, numerous artificial nanochannels have been developed so as to simulate the action mode of membrane protein channels.8–11 In fact, it is of biological importance to build a biomimetic nanochannel with desired properties, which will not only help to understand the nature of biological pores but also to provide advanced porous materials. Recently, artificial ion channels have been extensively studied,8,9 but the construction of synthetic nanochannels with wide lumen, e.g., for transporting glucose, remains challenging. Actually, it is a delicate mimicry that the biological fuel molecule glucose can be efficiently conveyed across the lipid bilayers. Besides a few examples on glucose transporters,12,13 artificial nanochannels with glucose transport ability have very seldom been reported.14,15 A typical work was introduced by the Ghadiri group, in which the self-assembly of cyclic peptides formed a nanochannel with large lumen structure that can transport glucose across the lipid bilayers.15 Obviously, synthetic nanochannels with glucose transport features are very eagerly awaited for the development of functional membrane-spinning channels.Supramolecular polymer systems have provided an ideal framework for the construction of artificial nanochannels, since great progress had been made in the field of supramolecular polymers.16–20 As long as the strong noncovalent interactions can be manipulated, the shapes and topologies of supramolecular polymers can be controllable as diverse as those of traditional polymers. In fact, the topological structures of supramolecular polymers have seldom been investigated at present due to the difficulty in controlling the direction of strong noncovalent interactions.21 Although some helical supramolecular structures have been studied in recent years,22–24 helical supramolecular polymer (HSP) nanotubes have not been reported yet. Taking advantage of the special features of self-assembled structures, the lumen structure of helical supramolecular polymers would be readily achieved through controllable self-assembly of rationally designed building blocks. Keeping this strategy in mind, we herein reported a kind of HSP nanotubes by controlling the spiral self-assembly of shape-persistent building blocks consisting of phenanthroline derivatives and two quadruply hydrogen-bonding AADD (ureidopyrimidinone, UPy) moieties. The resulting supramolecular polymers possessed predictable wide lumen structure with a diameter of beyond 2.0 nm. More importantly, HSPs are capable of spanning the lipid bilayer membrane and transporting the relatively big molecule glucose (Fig. 1).
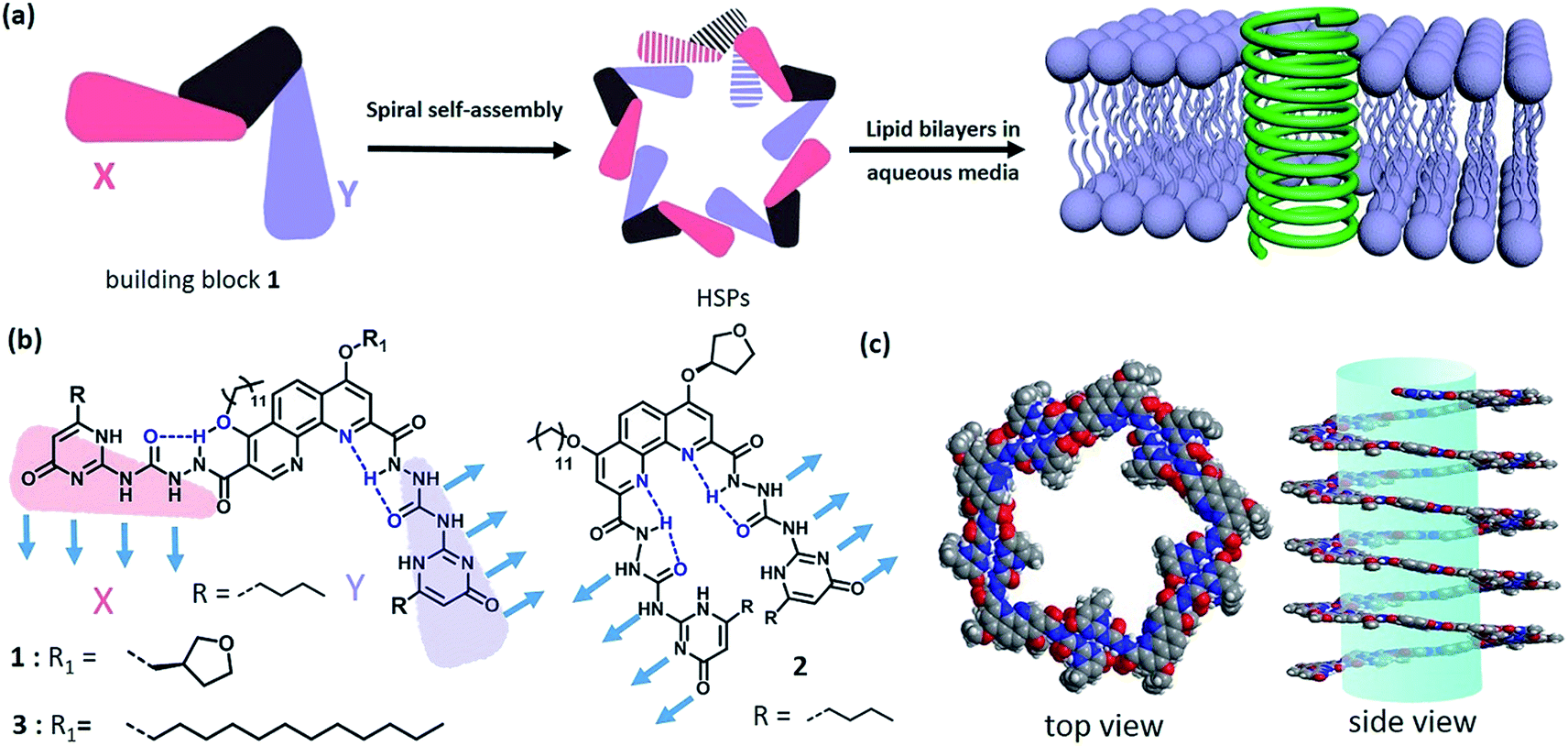 | ||
| Fig. 1 (a) Representation of the shape-persistent building block 1 preferentially self-assembling into helical supramolecular polymers and the further formation of transmembrane channels in the presence of lipid bilayers. (b) The molecular structures of building blocks 1–3. (c) Molecular modelling of helical supramolecular polymers from the self-assembly of 1via density functional theory (DFT) calculations (X3LYP/3-21G*). | ||
Results and discussion
Design and synthesis of building block 1
In order to construct the HSPs, we rationally designed a shape-persistent building block 1 based on the development of hydrazide foldamers25–28 (Scheme S1†). In the structure of 1, robust intramolecular hydrogen-bonding interactions spatially stabilize the molecular shape, thus governing the direction of the AADD·DDAA moieties bound each other. Although it seems difficult to empirically rule out random structures of supramolecular polymers with irregular sequences (binding modes XX, YY, and XY), the formation of HSPs with alternating XY sequences will dominate in this case (Fig. S1†). This hypothesis is based on the previous theory in which molecular self-assembly at equilibrium is prone to form boundary-defined supramolecular structures owing to the symmetry in the self-assembling geometry.29 The helical conformation of HSPs is a boundary-defined one-dimensional architecture. It is anticipated that, the self-assembly equilibrium of building block 1 will preferentially form HSPs rather than irregular supramolecular polymers. The formation of HSPs was essentially attributed to spatial control of the direction of noncovalent interactions between the building block 1 moieties. When the direction of noncovalent interactions was changed, like using building block 2 that is a geometric isomer, the helical structure was impossible. Instead, linear supramolecular polymer chains will be produced by self-assembly of 2 (Fig. S2†). According to the design concept, we thus synthesized these two building blocks 1 and 2. The structures of 1 and 2 were completely characterized (see ESI†).Self-assembly of building block 1
The quadruply hydrogen-bonding AADD·DDAA with a flat structure shows quite a strong binding ability.21 From Fig. S3,† we observed the chemical shifts of the UPy moieties and the shield effect of the aromatic protons in CDCl3, indicating that 1 can self-assemble into supramolecular polymers. Expectedly, 1 can reach a self-assembly equilibrium under very low concentrations in solution (Fig. S4†). The molar absorption coefficient did not change with the concentration of 1, indicating that the supramolecular polymers remained stable in dilute solution (10 μM). NMR titration results further supported the strong self-assembly feature of 1, as evidenced by the observation that the chemical shifts of the protons of 1 were concentration-independent (Fig. S3†). Moreover, dynamic light scattering (DLS) experiments demonstrated that aggregates with gradually increasing sizes were observed as the concentration increased, implying the self-assembled process of 1 (Fig. S5†). The supramolecular structure formed by the self-assembly of 1 was studied by electron spray ionization mass spectrometry (ESI-MS). Several supramolecular oligomers, like pentamers and hexamers, were detected in the MS spectrum (Fig. S6†), which indicated the chain-growth mode of supramolecular polymers. Gel permeation chromatography (GPC) analysis (Fig. S7†) found the production of supramolecular polymers (Mn = 2.37 × 105, PDI = 1.17) and oligomers.Modelling of the self-assembled structure formed by building block 1 (see ESI†) reveals a helically folding supramolecular polymer with a diameter of 7.8 ± 0.2 nm. Such a large diameter will allow one to confirm the self-assembling structure of 1 by TEM. As observed in Fig. 2a, the self-assembly of 1 resulted in the formation of ultralong supramolecular polymers with uniform diameters (8.6 ± 0.9 nm), which is close to the diameter of the hypothetical HSPs model (Fig. 1c). More importantly, the lumen structures of the designed HSPs were clearly visualized with a cavity diameter of ca. 2.6 nm (Fig. 2b), thus providing a solid proof on the formation of hollow structure of HSPs. AFM measurements were also employed to identify the structure of HSPs. As seen in Fig. 2c, the AFM patterns were very similar to that of the TEM (Fig. 2a), and the height of the HSP fibers was measured to be 8.2 ± 0.5 nm, which is consistent with the diameter of the HSPs. Additionally, the left-handed and right-handed helical patterns were explicitly detected (Fig. 2d), demonstrating the helical conformation of the HSPs. Moreover, similar HSP fibers can be found by SEM (Fig. S8†). These observations clearly proved the preferential formation of HSPs in our case, which is strongly supported by previous theory.27 Self-assembly of 1 at equilibrium is prone to form boundary-defined helical structures because the high geometric symmetry will dominate in the self-assembling structures. Therefore, self-assembly of 1 might preferentially form HSP nanotubes rather than irregular structures so as to avoid further aggregation and even precipitation.
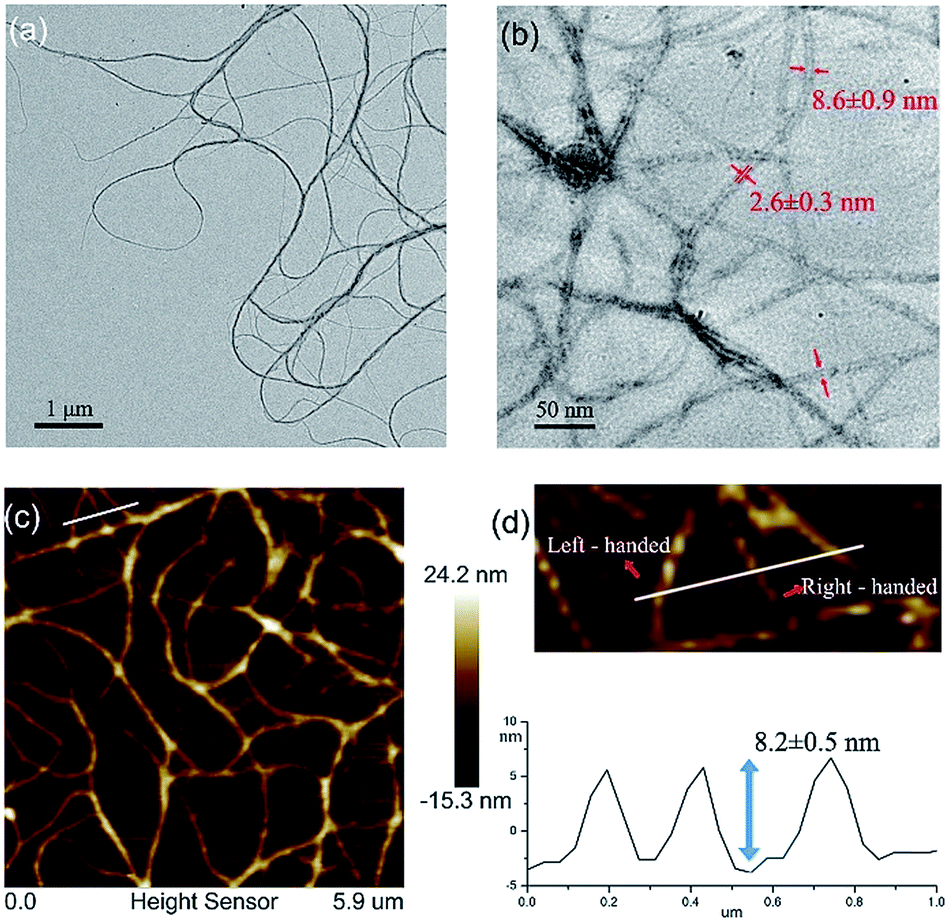 | ||
| Fig. 2 (a) A TEM image of HSPs; (b) lumen structures of HSPs visualized via TEM; (c) an AFM image of HSPs; and (d) height profile and helical patterns of HSPs. | ||
The formation of HSP nanotubes essentially depends on spatial control of the direction of the noncovalent interactions. Like building block 1, a geometric isomer 2 can also self-assemble into supramolecular polymers. Due to the intramolecular hydrogen bonds of building block 2, the direction of the intermolecular non-covalent interactions changed. A linear structure was dominantly formed instead of a helical one. As observed in ESI-MS (Fig. S9†), several oligomers like octamers formed by the self-assembly of 2 were detected, suggesting that the generation of supramolecular polymers is straightforward. Furthermore, the solubility of 2 in chloroform or DMSO was found to be much lower than that of 1, indicating that the linear supramolecular polymers can further self-assemble into aggregates and even precipitates. As shown in AFM (Fig. S10†), fully different patterns from 2 were observed in contrast to 1, suggesting that the self-assembled structure of 2 was disordered. These observations suggested that the formation of HSP nanotubes was ascribed to the reasonable control of the direction of noncovalent interactions, such as using building block 1 rather than 2.
The handedness of the HSP nanotubes can be induced by appending a (S)-(+)-3-hydroxytetrahydrofuran chiral group (Fig. 1 and Scheme S1†). As shown in the CD spectra (Fig. 3a), self-assembly of 1 gave a negative CD signal at 378 nm, and it was consistent with the absorption wavelength of the UV-vis spectrum. Compared to 1, however, a reference compound 3 (Fig. 1b) with an identical scaffold but without any chiral group did not show any CD signals in the CD spectrum (Fig. S11†). This result indicated that the helically left-handed bias was induced in the supramolecular polymers from the chiral groups (Fig. S12†). In addition, the CD signals remarkably decreased when the temperature increased or polar solvents were added (Fig. 3b, S13 and S14†), suggesting that the noncovalent interactions such as hydrogen bonding sustained the structure of the HSPs. Moreover, the red shift of 10 nm at the maximum absorbance peaks between CD (378 nm) and UV-vis (368 nm) spectroscopies indicated that the helical folding of supramolecular polymers was mainly driven by π stacking inside the HSP nanotubes. This result was also underpinned by the observation that the chemical shifts of the aromatic protons in 1 are highly shielded in the NMR spectrum (Fig. S14†). All these results were consistent with the model of the self-assembled structure by the density functional theory (DFT) calculations, suggesting that the supramolecular polymers were formed in helical conformations driven by π stacking. In contrast, the self-assembly of 2 did not show any signal of helical handedness in the CD spectra (Fig. S15†), indicating the absence of helical structures.
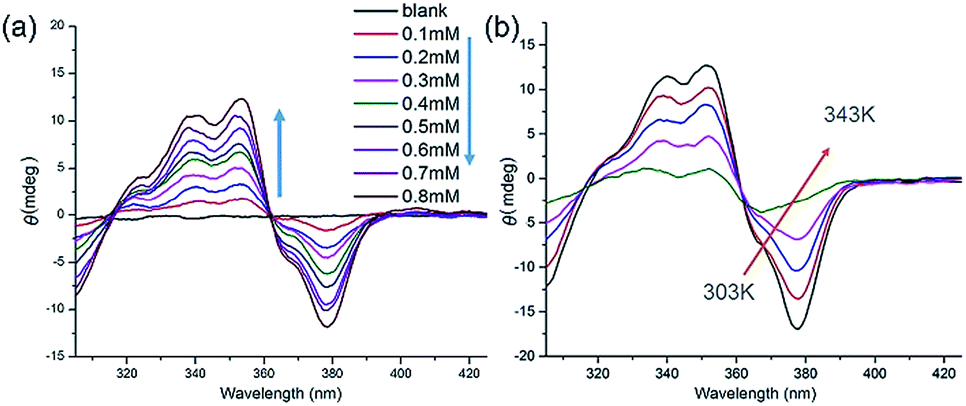 | ||
| Fig. 3 (a) Concentration-dependent CD spectra of 1 in chloroform from 0.01 mM to 0.8 mM. (b) Temperature-varied CD spectra of 1 in chloroform from 303 K to 343 K. | ||
Transmembrane transport properties of HSPs
The co-assembly of membrane proteins and lipid bilayers opens the landscape of supramolecular structures and functions, which inspired us to use a similar way to control the molecular self-assembly as well as to investigate the structure-directed properties. In the presence of the lipid bilayer membrane, supramolecular polymers formed by self-assembly of 1 are prone to penetrate the lipid membrane under the assistance of dodecyl side chains. After the addition of the lipid bilayer, the CD intensity was significantly enhanced (Fig. S16†), similarly, both of UV-vis and fluorescence spectra showed increased intensity in the presence of the lipid bilayer (Fig. S17 and S18†). These enhancements in the intensities of the CD, UV-visible and fluorescence spectra are mainly attributed to the enhanced solubility of a phenanthroline derivative by the addition of EYPC lipids, suggesting that the HSPs prefer to stand in the lipid bilayer membrane rather than precipitate out in water.The transmembrane transport properties of the HSP channels were studied according to previous methods.30,31 The co-assembly morphology of the HSPs and lipid bilayers was observed by fluorescence microscopy (Fig. 4a). No difference was detected between the giant unilamellar vesicle (GUV) with or without 1 in the bright field images. However, under the fluorescence mode, GUV formed by pure lipid vesicles was undetectable, while the GUV mixed with 1 exhibited a bright blue signal as well as the same shape as the vesicle in the bright field. This result suggested that HSPs from 1 were embedded in the lipid bilayer, whereas the vesicles remain intact.
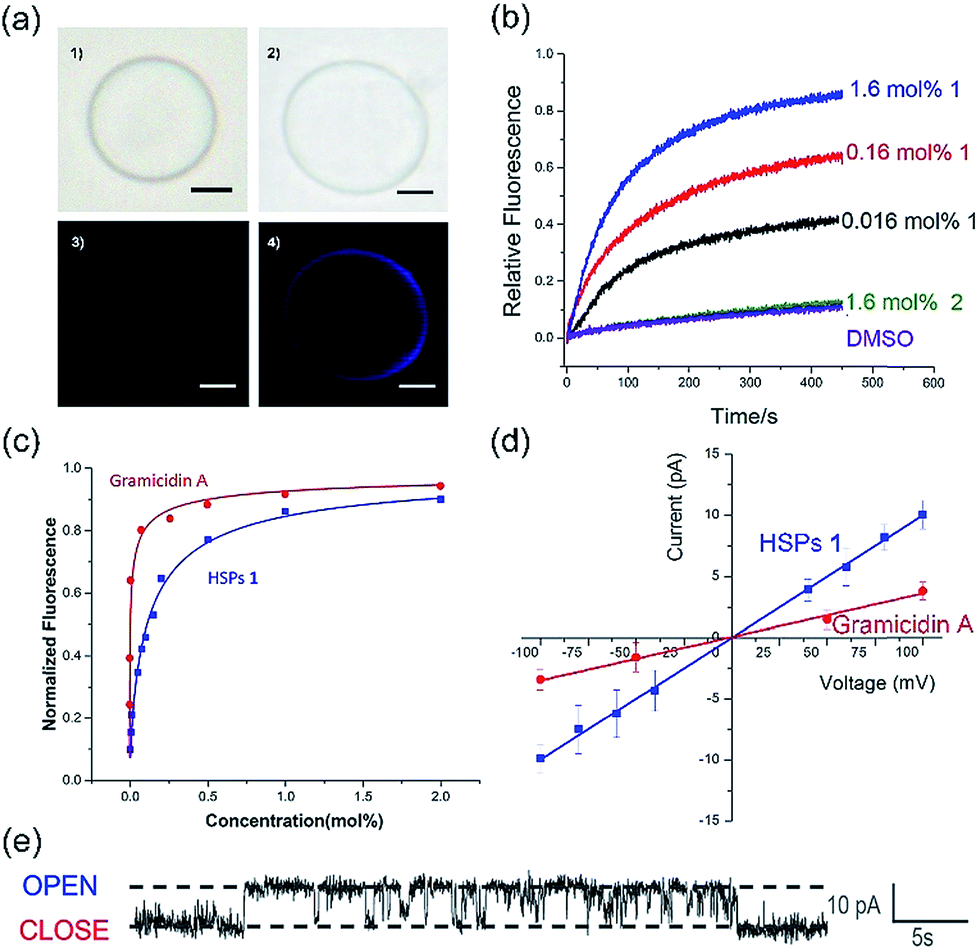 | ||
| Fig. 4 (a) Images of giant unilamellar vesicles (GUVs) in (1) bright field and (3) fluorescence mode; images of GUVs with 1 in (2) bright field and (4) fluorescence mode (scale bar = 5 μm). (b) Normalized ion transport activities of 1 and 2 under the HPTS assay (building block to lipid ratio). (c) Fitted curve of the transport activities for 1 and gA by the Hill equation (building block to lipid ratio). (d) I–V plots of 1 and gA from electrophysiology tests. (e) Channel signals of 1 from the planar lipid bilayer experiments at 100 mV in 1 M KCl. | ||
To assess the transport activity of HSPs, large unilamellar vesicles (LUVs) labeled with fluorescent probes were used. The building blocks 1 and 2 were respectively added to EYPC LUVs suspension (pH at 7.0 in and out of the vesicle) that encapsulated a pH-sensitive dye, 8-hydroxypyrene-1,3,6-trisulfonate (HPTS). Then, some sodium hydroxide solution was added to the suspension, resulting in a concentration difference of protons between the inside and outside of the vesicles. By monitoring the change in the fluorescence intensity of the suspension, the ion transfer efficiency can be reflected. After recalibrating the resulting fluorescence intensity, we can obtain the fractional activity (Fig. 4b). The result found that the geometric isomer 2 without forming a lumen structure did not increase the ionic flux, whereas under the identical conditions 1 showed very high ion transport activity. Our results suggested that the emergence of a lumen structure is essential for transmembrane ion transport.
In order to investigate the transport activities, the EC50 and Hill coefficient n were calculated by fitting the Hill plots of the channels (Fig. 4c and S19†). A natural short peptide Gramicidin A (gA) forms a dimer channel across the lipid bilayers. However, the self-assembled transmembrane channel approximately consists of 50 repeating units of 1. The EC50 value was defined as the concentration of channels (instead of the traditional monomer concentration) for 50% transport activity. The EC50 values given by the Hill analysis were 0.007 mol% and 0.001 mol% (channel to lipid ratio) for channel 1 and gA, respectively. Like natural ion channel gA, the HSP nanotubes from 1 exhibited a highly efficient transport ability. The Hill coefficient n was related to the dispersed states of the active channels in the experiments. When the Hill coefficient n > 1, the Hill coefficient corresponded to the number of monomers per active channels. The Hill coefficient n for gA was 1.7, which is consistent with the transmembrane morphology of gA in active channels. The Hill coefficient n of channel 1 was calculated to be 0.78. The Hill coefficient n < 1 indicated that the formation of the HSP channels was exergonic and spontaneous, implying that the building blocks prefer to form stable transmembrane HSP nanotubes to avoid precipitating in the aqueous media.29
The single-channel electrophysiology of 1 was investigated on the planar lipid bilayer by patch clamp, since the single channel current recordings are one of the most convincing pieces of evidence for biomimetic channels. In this experiment, the planar lipid bilayer composed of 1,2-diphytanoyl-sn-glycero-3-phosphocholine (diPhyPC) was formed on a 200 μm micropore and separated two connected compartments (cis and trans chambers). The compartments were both filled with 1 ml KCl solution (1 M). HSP nanotubes in DMSO were added to the cis compartment, the clamped voltages of +100 mV were applied to both sides of the lipid bilayer. When the supramolecular channels were formed in the lipid bilayer, the current signal appeared and was recorded by the computer. As shown in Fig. 4e, HSP nanotubes from 1 displayed a clear rectangular current signal, which proved that 1 can form transmembrane supramolecular channels in the lipid bilayers. The channel conductance g can be calculated around 98 pS utilizing the I–V plot, larger than 38 pS for gA under the identical conditions (Fig. 4d and S21†). Our study found that the current signal and conductance increased with the increase of channel diameter, and the helix-based nanochannels maintained a certain regularity of their own on the conductance (Fig. S22†). However, the Hill equation cannot match the actual situation, and an attempt to estimate the lumen diameter failed for this system of HSP nanotubes (ESI†).
Glucose transport of HSPs
Taking advantage of the large size diameter of the HSP nanotubes from 1, the capacity of glucose transport was investigated. We set tested experiments to study the transport of glucose by an enzyme-coupled assay (Fig. 5a). LUVs entrapped with glucose were used. 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) in the outside can be converted to ABTS˙+ by utilizing glucose as the substrate for Glucose Oxidase (GOX) and Horseradish Peroxidase (HRP). The production of ABTS˙+ was monitored at 405 nm under UV-vis spectra. Owing to the extremely high catalytic activities of GOX and HRP, the amount of ABTS˙+ directly corresponds to the glucose leakage. As shown in Fig. 5b, the HSP nanotubes from 1 showed excellent glucose leakage efficiency. The glucose entrapped in LUVs was completely released in 30 minutes at 4.0 mol% (building block to lipid ratio) in the presence of channels, and their transport capacity was concentration-dependent. However, no detectable glucose leakage efficiency was found in the presence of isomer 2. Notably, we found that the transmission rate of ion transport (Fig. 4b) is much faster than that of glucose transport (Fig. 5b) during the measurement of channel functions of HSP nanotubes from 1. This result implied that the lumen structures were involved in the transport process.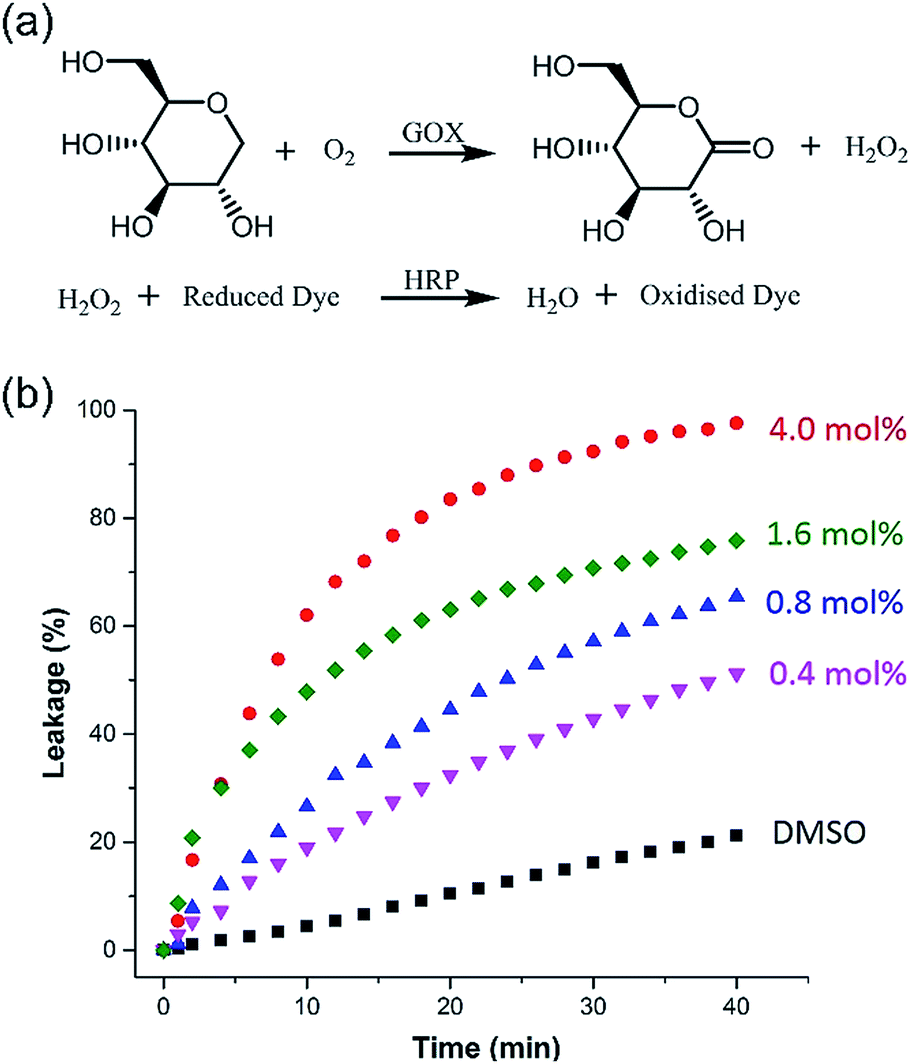 | ||
| Fig. 5 (a) The catalytic mechanism of the GOX and HRP enzyme-coupled assay. ABTS was the substrate for HRP in this experiment. (b) Kinetic curves of glucose leakage in the presence of different amounts of channels (pure solvent DMSO was used as benchmark). | ||
Conclusions
In conclusion, strong noncovalent interactions have been manipulated to realize controllable self-assembly by using a shape-persistent building block consisting of phenanthroline derivatives and two quadruply hydrogen-bonding AADD moieties, thus giving rise to the formation of highly ordered HSP nanotubes. The helical conformation of the supramolecular polymers has been proved by both helical patterns in the AFM image and the helicity bias via chiral induction. The topologies of the supramolecular polymers can be enriched, like those of traditional polymers. Certainly, the development of topological structures of supramolecular polymers will lead to the emergence of new and predictable properties and functions that supramolecular structures can demonstrate. In this case, the helical folding of supramolecular polymers gave rise to the production of specific wide lumen structures that can be directly visualized by TEM, and the resulting HSP nanotubes can puncture the lipid bilayer membrane to facilitate the transportation of glucose. The successful fabrication of HSP nanotubes not only indicated the feasibility of precisely controlling molecular self-assembly via rational design, but it also significantly expanded the topology of supramolecular polymers. Such artificial nanochannels will be considered for technological applications in due course.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 21574054 and 21722403) and the Program for JLU Science and Technology Innovative Research Team (JLUSTIRT) (No. 2019TD-36).References
- M. A. Aydman, J. R. Silva and J. Cui, Chem. Rev., 2012, 112, 6319–6333 CrossRef PubMed
.
- S. Litvinchuk, H. Tanaka, T. Miyatake, D. Pasini, T. Tanaka, G. Bollot, J. Mareda and S. Matile, Nat. Mater., 2007, 6, 576–580 CrossRef CAS PubMed
- D. L. Gin and R. D. Noble, Science, 2011, 332, 674–676 CrossRef CAS PubMed
- M. Mayer and J. Yang, Acc. Chem. Res., 2013, 46, 2998–3008 CrossRef CAS PubMed
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS PubMed
- A. G. Cioffi, J. Hou, A. S. Grillo, K. A. Diaz and M. D. Burke, J. Am. Chem. Soc., 2015, 137, 10096–10099 CrossRef CAS PubMed
- R. Nussinov, C. J. Tsai and J. Liu, J. Am. Chem. Soc., 2014, 136, 17692–17701 CrossRef CAS PubMed
- S. Howorka, Nat. Nanotechnol., 2017, 12, 619–630 CrossRef CAS PubMed
- B. Gong and Z. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef CAS PubMed
- W. Si, L. Chen, X. B. Hu, G. Tang, Z. Chen, J. L. Hou and Z. T. Li, Angew. Chem., Int. Ed., 2011, 50, 12564–12568 CrossRef CAS PubMed
- C. Lang, W. Li, Z. Dong, X. Zhang, F. Yang, B. Yang, X. Deng, C. Zhang, J. Xu and J. Liu, Angew. Chem., Int. Ed., 2016, 55, 9723–9727 CrossRef CAS PubMed
- P. R. Westmark, S. J. Gardiner and B. D. Smith, J. Am. Chem. Soc., 1996, 118, 11093–11100 CrossRef CAS
- H. Cho and Y. Zhao, Chem. Commun., 2011, 47, 8970–8972 RSC
- H. Cho and Y. Zhao, Langmuir, 2011, 27, 4936–4944 CrossRef CAS PubMed
- J. R. Granja and M. R. Ghadiri, J. Am. Chem. Soc., 1994, 116, 10785–10786 CrossRef CAS
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed
- Y. Liu, Y. Yu, J. Gao, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6576–6579 CrossRef CAS PubMed
- Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS PubMed
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed
- J. H. Hirschberg, L. Brunsveld, A. Ramzi, J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS PubMed
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef CAS PubMed
- X. Huang, S. Jiang, X. Wang, B. Zhang and M. Liu, J. Am. Chem. Soc., 2004, 126, 1322–1323 CrossRef CAS PubMed
- X. Zhao, X. Z. Wang, X. K. Jiang, Y. Q. Chen, Z. T. Li and G. J. Chen, J. Am. Chem. Soc., 2003, 125, 15128–15139 CrossRef CAS PubMed
- J. S. Ferguson, K. Yamato, R. Liu, L. He, X. C. Zeng and B. Gong, Angew. Chem., Int. Ed., 2009, 48, 3150–3154 CrossRef CAS PubMed
- S. Saha, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2018, 57, 13542–13544 CrossRef CAS PubMed
- H. M. Keizer, R. P. Sijbesma and E. W. Meijer, Eur. J. Org. Chem., 2004, 12, 2553–2555 CrossRef
- K. V. Workum and J. F. Douglas, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 73, 031502 CrossRef PubMed
- C. Lang, X. Deng, W. Wang, S. Qi, X. Zhang, C. Zhang, Z. Dong and J. Liu, Angew. Chem., Int. Ed., 2017, 56, 12668–12671 CrossRef CAS PubMed
-
S. Matile and N. Sakai, in Analytical Methods in Supramolecular Chemistry, Wiley-VCH, Weinheim, 2012, p. 711 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02336d |
| This journal is © The Royal Society of Chemistry 2019 |