
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA switchable iron-based coordination polymer toward reversible acetonitrile electro-optical readout†
Esther
Resines-Urien
 a,
Enrique
Burzurí
*a,
Estefania
Fernandez-Bartolome
a,
Miguel Ángel
García García-Tuñón
b,
Patricia
de la Presa
c,
Roberta
Poloni
d,
Simon J.
Teat
e and
Jose Sanchez
Costa
*a
a,
Enrique
Burzurí
*a,
Estefania
Fernandez-Bartolome
a,
Miguel Ángel
García García-Tuñón
b,
Patricia
de la Presa
c,
Roberta
Poloni
d,
Simon J.
Teat
e and
Jose Sanchez
Costa
*a
aIMDEA Nanociencia, C/Faraday 9, Campus de Cantoblanco, Madrid, 28049, Spain. E-mail: enrique.burzuri@imdea.org; jose.sanchezcosta@imdea.org
bInstituto de Cerámica y Vidrio, CSIC, C/Kelsen s/n, Madrid, 28240, Spain
cInstituto de Magnetismo Aplicado, UCM-ADIF-CSIC, A6 22.500 km, Las Rozas, 28230, Spain
dUniversité Grenoble Alpes, CNRS, SIMAP, Grenoble, 38000, France
eAdvanced Light Source (ALS), Berkeley Laboratory, 1 Cyclotron Road, Berkeley, CA 94720, USA
First published on 20th June 2019
Abstract
Efficient and low cost detection of harmful volatile organic compounds (VOCs) is a major health and environmental need in industrialized societies. For this, tailor-made porous coordination polymers are emerging as promising molecular sensing materials thanks to their responsivity to a wide variety of external stimuli and could be used to complement conventional sensors. Here, a non-porous crystalline 1D Fe(II) coordination polymer acting as a porous acetonitrile host is presented. The desorption of interstitial acetonitrile is accompanied by magneto–structural transitions easily detectable in the optical and electronic properties of the material. This structural switch and therefore its (opto)electronic readout are reversible under exposure of the crystal to acetonitrile vapor. This simple and robust iron-based coordination polymer could be ideally suited for the construction of multifunctional sensor devices for volatile acetonitrile and potentially for other organic compounds.
The increasing emissions of volatile organic compounds (VOCs) and their resulting impact on air and water quality has become one of the major environmental concerns of our age.1 Some VOCs are identified as highly toxic or carcinogenic and may have an impact on human health as well as on the natural ecosystem.2 VOCs are emitted during the use of many everyday household products which makes the control of their emission particularly difficult and critical. Highly sensitive analytical techniques are currently employed for the accurate quantification of VOCs. However, these techniques have some drawbacks, such as their low portability, constrained selectivity and high cost.3
To overcome these limitations the emergence of “porous materials” such as Metal–Organic Frameworks (MOFs)4 and Covalent Organic Frameworks (COFs)5 has provided meritorious results. These materials contain the voids necessary to absorb guest molecules, thereby changing their properties. Recently, more attention has been paid to materials where in the absence of pores an exchange might still take place through the process of diffusion throughout the crystal lattice.6 This process may produce an easy-to-measure response due to the variation of the macroscopic properties of these materials, in agreement with the definition of a chemosensor.7 Specifically, these chemosensors could display an easily detectable change in almost any physicochemical property, such as in the luminescent emission,8,9 the electrical conductivity,10 the magnetic behavior and the colour change.11,12
Here we present a novel non-porous crystalline 1D Fe(II) coordination polymer acting as a porous material hosting acetonitrile. We show that the release of interstitial acetonitrile from the crystal with increasing temperature is accompanied by sharp magneto-structural transitions that can be detected as a change of color, from yellow to orange, and as an abrupt high-current resonance in the electron transport through the crystal. Interestingly, the initial structure and therefore color and electrical transition are recovered once the crystal is exposed again to acetonitrile. This coordination polymer can therefore be used as a reversible and precise (opto)electronic detector for accurately monitoring ambient acetonitrile and potentially other VOCs that can be accommodated in the structure. The ∞{[Fe(H2O)2(CH3CN)2(pyrazine)](BF4)2·(CH3CN)2} complex, hereafter (1·2CH3CN), was synthesized by reaction of pyrazine with Fe(BF4)2·6H2O carried out in acetonitrile (see the ESI†). After one day, yellow needle-shaped crystals appeared, which crystallized in the orthorhombic space group Cmca (Table S2†).
In this structure there is one crystallographically independent iron(II) ion, which is coordinated octahedrally with two H2O molecules, two acetonitrile (CH3CN) molecules and two pyrazine molecules. Pyrazine acts as a bridging ligand between two neighboring iron ions, resulting in the formation of 1D polymeric chains along the a axis (Fig. 1). The Fe–N (pyrazine) distance is 2.225 Å and the Fe–N (CH3CN) distance is 2.163 Å. These distances are characteristic of high spin (HS) iron(II).7,13
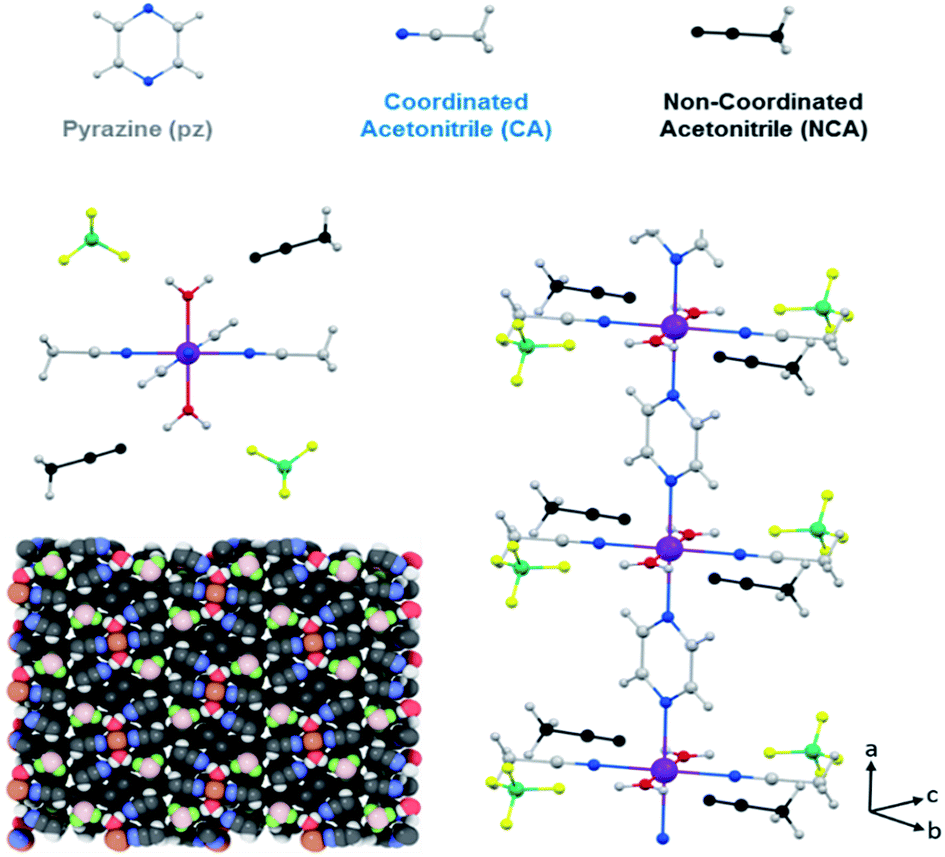 | ||
| Fig. 1 Crystal structure of 1·2CH3CN, where Fe is represented in purple, C in grey, N in blue, O in red, B in green, F in yellow, H in white and the uncoordinated acetonitrile in black. | ||
There is no direct inter-chain interaction although they interact via the BF4− counterion, which forms a hydrogen bond with a water molecule of one chain, with an F–H (H2O) bond length of 1.795 Å (F–O: 2.655 Å), and a van der Waals interaction with a coordinated acetonitrile of the adjacent chain, with a F–H (CH3CN) distance of 2.270 Å (F–C: 3.066 Å). These interactions occur along the b axis, but there is no interaction along the c axis. The interstitial acetonitrile forms a hydrogen bond with the water molecule, with an O–H distance of 1.862 Å (O–N: 2.744 Å), and a BF4− counterion, with a F–H distance of 2.667 Å (F–C: 3.193 Å).
Fig. 2a shows the optical reflectivity (OR) of a 1·2CH3CN crystal measured while heating it from 288 K up to 373 K. Initially, the OR remains constant and no change in the color of the crystal is observed as seen in Fig. S1.† Interestingly, the OR sharply increases at around 305 K and the 1·2CH3CN crystal changes color to a brighter shade of yellow, as seen in Fig. S2.† This change is also clearly seen in Video S1.† The OR remains approximately constant from 306 K up to 316 K and thereafter gradually decreases as the color of the crystal changes from yellow to orange (see Fig. 2b and S1†). At 355 K, the 1·2CH3CN crystal is completely orange and the OR stabilizes to a constant value. Upon cooling the sample back to room temperature no change in color was observed, suggesting that the change in the optical reflectivity is not temperature reversible.
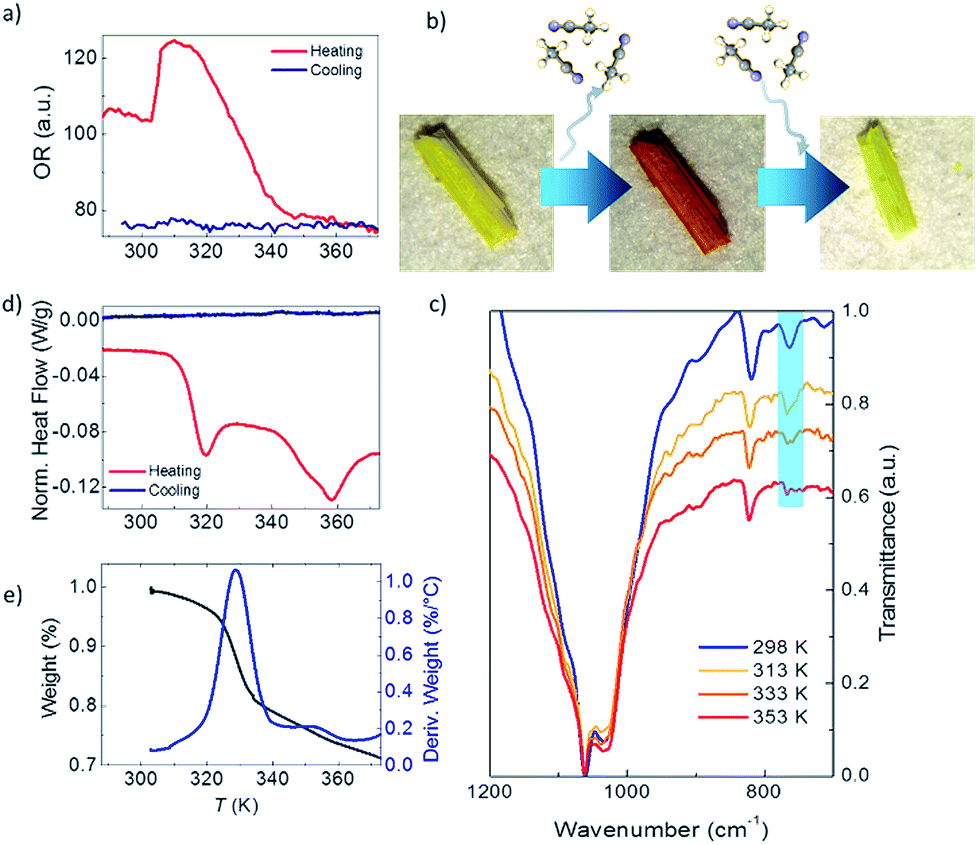 | ||
| Fig. 2 (a) 1·2CH3CN optical reflectivity measurements between 288 K and 373 K. (b) Optical image of a 1·2CH3CN crystal at room temperature (yellow), after heating and subsequent loss of acetonitrile molecules (1, orange) and after exposure to acetonitrile (yellow). (c) 1·2CH3CN infrared spectra at different temperatures. (d) DSC measurements of a 1·2CH3CN crystal between 288 K and 373 K. (e) TGA curve measured between 303 K and 373 K (black line). The blue line represents the derivative weight. | ||
The infrared IR spectrum of 1·2CH3CN is measured in the same range of temperature to determine whether the changes in the OR are related to structural changes in the molecules. The low energy spectrum is shown in Fig. 2c. See Fig. S3 to S6† for the full energy scale spectrum.
The room temperature spectrum shows the bands associated with the different constituents of the polymer, acetonitrile, coordinated water molecules and pyrazine, and the broad band of the BF4− anions at 1002 cm−1. Interestingly, a gradual disappearance of the band at 766 cm−1 is clearly observed as the temperature increases. The vanishing band can be assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bend overtone14,15 that is present in the uncoordinated acetonitrile of the 1·2CH3CN. Its disappearance therefore suggests that acetonitrile desolvation16 from the crystal occurs as the temperature increases. On the other hand, the structural integrity of the crystal is conserved, which is an indication that the acetonitrile groups coordinated to the Fe(II) remain in the structure. To further prove this, we have compared the 1·2CH3CN IR spectra with those of the analogue ∞{[Fe(CH3CN)4(pyrazine)](ClO4)2}1 with a similar structure.13 The difference is that no uncoordinated acetonitrile molecules are present in its lattice. The band at 766 cm−1 does not appear in the spectra at any temperature (Fig. S7†). Therefore, the presence and vanishing of the bands in 1·2CH3CN can be unambiguously assigned to the desorption of uncoordinated acetonitrile with increasing temperature. The process is not temperature reversible and the IR bands do not reappear when cooling down to room temperature, in agreement with the OR measurements (Fig. S6†). These results are consistent with the loss of these non-coordinated acetonitrile molecules from the structure, hereinafter named 1.
N bend overtone14,15 that is present in the uncoordinated acetonitrile of the 1·2CH3CN. Its disappearance therefore suggests that acetonitrile desolvation16 from the crystal occurs as the temperature increases. On the other hand, the structural integrity of the crystal is conserved, which is an indication that the acetonitrile groups coordinated to the Fe(II) remain in the structure. To further prove this, we have compared the 1·2CH3CN IR spectra with those of the analogue ∞{[Fe(CH3CN)4(pyrazine)](ClO4)2}1 with a similar structure.13 The difference is that no uncoordinated acetonitrile molecules are present in its lattice. The band at 766 cm−1 does not appear in the spectra at any temperature (Fig. S7†). Therefore, the presence and vanishing of the bands in 1·2CH3CN can be unambiguously assigned to the desorption of uncoordinated acetonitrile with increasing temperature. The process is not temperature reversible and the IR bands do not reappear when cooling down to room temperature, in agreement with the OR measurements (Fig. S6†). These results are consistent with the loss of these non-coordinated acetonitrile molecules from the structure, hereinafter named 1.
To further explore the reversibility of the optical and structural transitions induced by the loss of acetonitrile from the crystal, a drop of acetonitrile is added to the “dry” orange crystal at room temperature after one temperature cycle. The drop is thereafter allowed to dry under ambient conditions to eliminate any non-reabsorbed acetonitrile from the surface. Strikingly, the color and optical reflectivity return to the original yellowish characteristic color as seen in Fig. 2b and S1.† In addition, the absorption band at 766 cm−1 reappears in the IR spectrum (Fig. S8†). The initial properties are also recovered when 1 is exposed to acetonitrile via vapor diffusion.
We performed X-ray powder diffraction (XRPD) measurements of the three phases to determine the unit cell parameters. The results are shown in Fig. 3. The pattern obtained from polycrystalline 1·2CH3CN is in good agreement with the simulated single-crystal diffractogram. The unit cell parameters extracted using HighScore Plus17 show an excellent match with the single-crystal values (see Table S3 and Section 6 for details†). Sample 1·2CH3CN was then heated for 5 hours at 50 °C to ensure that the interstitial acetonitrile was completely desorbed from the material. Then the spectral acquisition was performed (see Fig. 3). The first observation is that the collected spectrum is different from the spectrum previously collected for 1·2CH3CN. The appearance of new peaks suggests a new crystalline phase associated with the evacuation of acetonitrile molecules. In addition, the lattice parameters from this pattern are smaller than those in the initial 1·2CH3CN. This is in agreement with the decrease of volume predicted by DFT calculations (Fig. S9–S11†) and with the decrease of the macroscopic size of the crystals (see Fig. S12†).
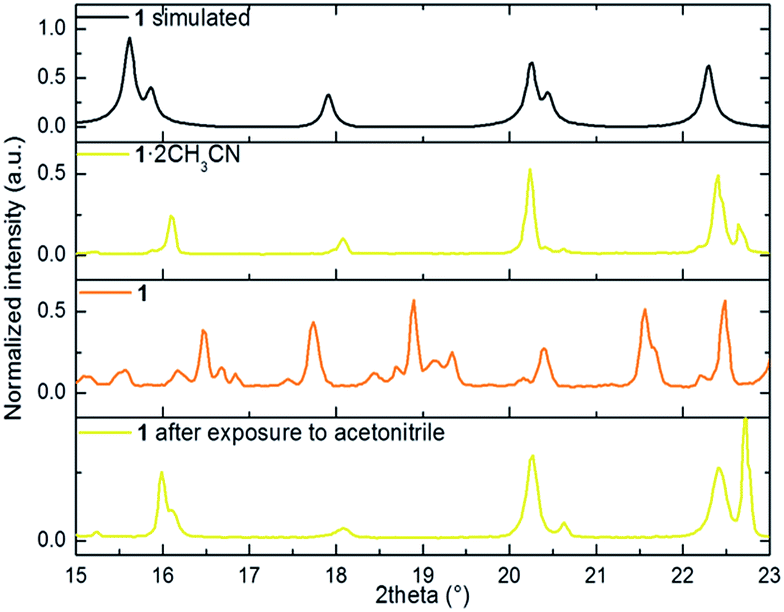 | ||
| Fig. 3 portion of the simulated powder X-ray diffraction patterns from the CIF file, 1·2CH3CN, 1 and 1 after exposure to acetonitrile. | ||
Finally, with the aim of recovering the initial phase, the polycrystalline sample 1 was exposed to a saturated atmosphere of acetonitrile vapors for 10 hours. After confirmation of a pure phase by both FT-IR and naked-eye visible color change (from orange to yellow), the material was analyzed. The acquired pattern reflects an almost pure 1·2CH3CN phase (see Table S3†). This study was complemented by elemental analysis characterization of the three phases. The result shows good agreement between the calculated and the estimated mass after evacuation and reinsertion of the host acetonitrile molecules (see Section 2 of the ESI†).
Further proof of the structural origin of the color and OR transitions is provided by differential scanning calorimetry (DSC) measurements between 288 K and 373 K. Two clear endothermic peaks, i.e. the crystal absorbs energy, can be observed during the heating process (Fig. 2d). The first one is centered around 315 K and is consistent with the first change in the optical reflectivity. The second peak is centered at 358 K and occurs at the same temperature when the OR becomes lowest and constant and the crystal has completely changed from yellow to dark orange (see Fig. 2a and S1†). The endothermic nature of the peaks is indicative of a crystalline phase transition involving breaking of molecular bonds or the melting/evaporation of molecular species present in the crystal. No peaks, either endothermic or exothermic, were observed when cooling the sample from 373 K back to 288 K, as also observed in the OR. Interestingly, both structural transitions are also translated into magnetic anomalies in the magnetic susceptibility at the same temperatures (see the Discussion in the ESI†).
To complete the structural characterization, thermogravimetric analysis (TGA) of 1·2CH3CN was performed between room temperature and 873 K (see Fig. 2e and S13†). A first weight loss is triggered at around 315 K (first transition in OR and DSC) and ends at 345 K (the OR and color become constant). The total weight loss in this temperature range is 15.73% and approximately fits with the loss of two acetonitrile molecules (16.08%) per formula unit. This confirms that only the Fe-uncoordinated acetonitrile molecules evaporate from the structure at this temperature.
The effect of the structural transitions on the electronic properties of 1·2CH3CN has been studied in the same range of temperatures. Fig. 4a shows the current I measured across a single 1·2CH3CN crystal in a nitrogen atmosphere at a fixed V = 1 V while increasing the temperature from 288 K to 373 K (red curve) and subsequently decreasing it to the initial temperature (blue curve). Additional measurements of different crystals reproducing the same transport features can be found in Fig. S14.† Initially the crystal behaves almost like an insulator with current levels below 10−11 A, in agreement with the large band gap (∼3 eV) computed by Density Functional Theory (DFT) for this polymer (see the ESI†). As the temperature is increased, a sharp increase of the current of about 2 orders of magnitude begins at 306 K followed by a softer decay back to low current levels. At higher temperatures, a second wider high-current peak appears between 343 and 353 K. Note that the order of magnitude and position of the peaks with respect to temperature are roughly reproducible in all the measured crystals (see Fig. S14†). Fig. 4b shows the current–voltage IV characteristics measured at different temperatures around the first peak in conductance. The current follows the same dependence on temperature observed in Fig. 4a.
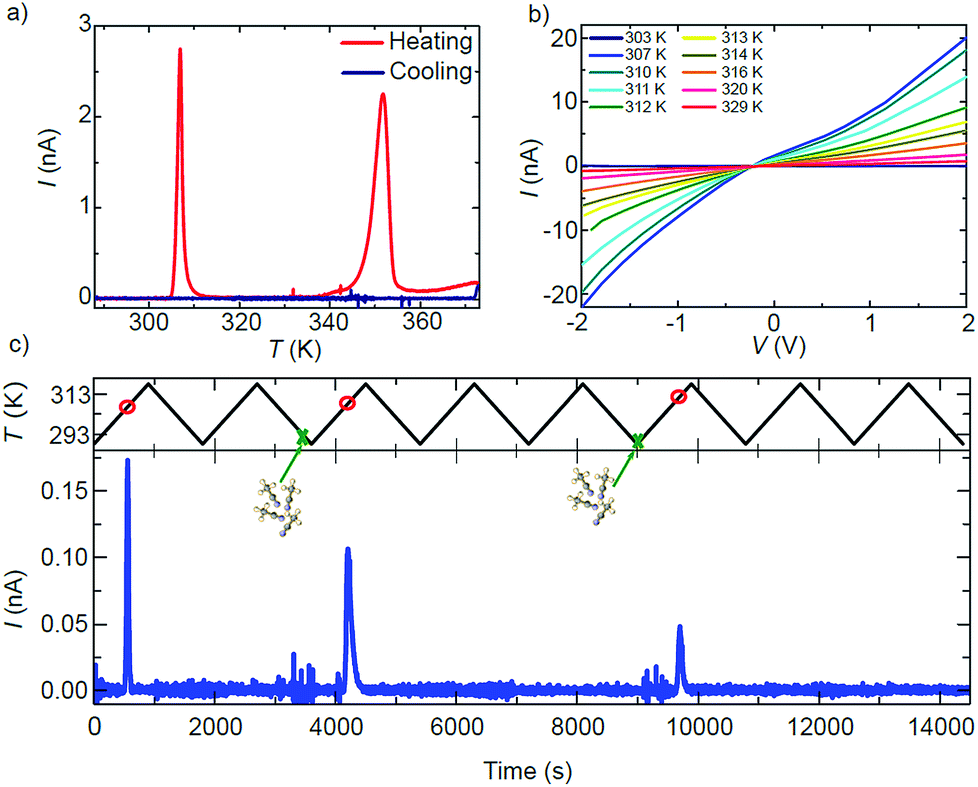 | ||
| Fig. 4 (a) Electrical current I vs. temperature T measured for a single 1·2CH3CN crystal. Two sharp high-conductance peaks appear centered at 306 K and 353 K with increasing T. The peaks do not appear when the temperature is decreased to the initial value. (b) Current–voltage IV characteristics measured at different temperatures around the first conductance peak. (c) Proof-of-concept sensor of acetonitrile. A resonance in the current appears at a specific temperature (red circles) only in those temperature cycles where the 1·2CH3CN crystal is exposed to acetonitrile (green crosses). | ||
The shape evolves from an insulator-like behaviour to a clear non-linear dependence with bias, indicating the presence of Schottky barriers at the interface between electrodes and the material or between grain boundaries in the material. Interestingly, the current levels after the transition temperature remain higher than those before the transition (dark blue curve at 303 K in Fig. 4b).
A careful comparison of the structural and optical characterization of crystalline 1·2CH3CN shows that the high-current peaks match with respect to the temperature of the two respective anomalies in the OR, the calorimetric and the magnetic response. The sharp change in the conducting properties of the crystal is therefore univocally connected to the structural transitions undergone by the 1·2CH3CN crystal at around 306 K and 353 K. None of the current peaks are present when the temperature is decreased again to 288 K (blue curve in Fig. 4a) and do not appear if the temperature is increased again, as also observed in the optical reflectivity and DSC. Strikingly, the two high-conductance peaks reappear in the charge transport measurement at identical temperatures after the 1·2CH3CN crystal is exposed to acetonitrile (vapor or liquid), as seen in Fig. S15.† The changes in the conductance are therefore reversible upon acetonitrile absorption–desorption in the 1·2CH3CN lattice. A simple conductance measurement can in principle be used as a method to detect ambient acetonitrile. A proof-of-concept of such a sensor is shown in Fig. 4c. The current is measured across a single 1·2CH3CN crystal while cycling the temperature between 288 K and 318 K, i.e. around the first peak in conductance. A peak in the current appears at roughly the same temperature (red circles) in those cycles where the crystal is exposed to acetonitrile vapor (green crosses), whereas it is absent in the rest of the cycles. That is, the emission of acetonitrile is detected only if the acetonitrile molecule has been previously inserted into the crystal. We note that the absorption of acetonitrile by the crystal is also immediately sensed by the conductance through the crystal. This is seen as distinct noise in the current before the high conductance peaks in Fig. 4c. A second example is shown in Fig. S16.†
The origin of the sharp change in conductance is intriguing since MOFs and coordination polymers are typically insulators or bad conductors18,19 except for a few remarkable cases.20–24
A possible explanation for the first high-conductance peak can be connected to the structural change and the augmented mobility of the interstitial acetonitrile in the 1·2CH3CN crystals, as proved by IR spectroscopy and DSC measurements. Reordering of small molecules in porous materials is known to induce structural changes in the chain-like architecture that in turn may induce transitions in the magnetic behavior25 and possibly the charge transport properties. On the other hand, the rapid drop in current with increasing temperature after the transition seems to rule out static changes in the electronic structure of the polymer, like a change in the band gap. This scenario is in agreement with our DFT calculations that show a sizeable change in the structure of the polymer after removal of the acetonitrile molecules, but a negligible change in the band gap, consistent with weakly interacting molecules (see Fig. S9 and S10†).
An alternative but related explanation for dynamic changes in the conductance has been proposed for porous silicon and metal oxide detectors exposed to acetonitrile26–28 and other polar solvents with high dielectric constants. The increase in current is not related to static changes in the bandgap. Instead, it can be explained in terms of dynamic changes in the dielectric constant of the material after infiltration of the acetonitrile that in turn modifies the charge distribution in the material. The subsequent rapid decrease of the conductance can be explained in this case as a dynamic capacitive effect over time or by the fast evaporation of acetonitrile due to its low vapor pressure. This effect has also been observed in MOFs29–31 and CNT–polymer hybrid materials.19 The fast evaporation is clearly observed in Video S2.† An increase of the surface brightness of the crystal is followed by the release of acetonitrile gas seen as blurriness in the image. To check this scenario we have measured the complex AC admittance across a crystal while heating it. We observe a clear peak in the real conductance and the imaginary susceptance at around 303 K (see the ESI†). These peaks can be well described by a sharp increase of the capacitance and probably the resistance of the material, induced either by a change in the dimensions of the lattice, predicted by DFT and observed experimentally, or by a change in the dielectric constant, both as a result of the release of interstitial acetonitrile.
The second high-conductance peak also seems to be related to the second structural transition observed in the optical reflectivity and the DSC measurements. Interestingly, the peak appears exactly at the acetonitrile boiling point temperature (355 K), indicating that the change in conductance is again linked to the reordering of acetonitrile.
Conclusions
To summarize we have presented a new non-porous crystalline one-dimensional coordination polymer acting as a porous material hosting acetonitrile. The material presents a reversible magneto-structural transition with desorption/absorption of acetonitrile molecules in the crystalline structure. The change in the structure is in turn translated into a distinct response in the electron transport through the material and its optical properties at well-defined temperatures close to ambient conditions. This family of non-porous materials can therefore be a versatile platform to fabricate tailor-made detectors for specific volatile organic compounds with a versatile variety of read-out options from optical and magnetic to electron transport measurements. The 1D and nanoscale nature of the Fe coordination polymer backbone has the potential for organic molecule detection with high spatial resolution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JSC acknowledges funds from the Spanish MINECO through the National Research Project (CTQ2016-80635-P), the Ramon y Cajal Research Program (RYC-2014-16866), the Comunidad de Madrid (PEJD-2017-PRE/IND-4037) and the NANOMAGCOST (P2018/NMT-4321). EB acknowledges funds from the MSCA-IF European Commission programme (746579) and Programa de Atracción del Talento Investigador de la Comunidad de Madrid (2017-T1/IND-5562). IMDEA Nanociencia acknowledges support from the ‘Severo Ochoa’ Programme for Centres of Excellence in R&D (MINECO, Grant SEV-2016-0686). This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. We acknowledge the XALOC-ALBA synchrotron source under project 2018012561. PP acknowledges financial support from the Spanish MINECO project MAT2015-67557-C2-1-P. DFT calculations were performed using resources granted by the GENCI under the CINES Grant Nos. A0020907211 and A0040907211. Additionally, the Froggy platform of the CIMENT infrastructure was employed. We thank Dr J. Perles and Dr M. Ramírez for collection and analysis of the X-ray data at the SIDI (Universidad Autonoma de Madrid).Notes and references
- E. Environment Agency, https://www.eea.europa.eu/data-and-maps/indicators/main-anthropogenic-air-pollutant-emissions/assessment-4.
- U. S. Environmental Protection Agency, https://www.epa.gov/indoor-air-quality-iaq/volatile-organic-compounds-impact-indoor-air-quality.
- A. Lewis and P. Edwards, Nature, 2016, 535, 29–31 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- E. Fernandez-Bartolome, J. Santos, A. Gamonal, S. Khodabakhshi, L. J. McCormick, S. J. Teat, E. C. Sañudo, J. S. Costa and N. Martín, Angew. Chem., Int. Ed., 2019, 2310–2315 CrossRef CAS PubMed.
- J. S. Costa, S. Rodríguez-Jiménez, G. A. Craig, B. Barth, C. M. Beavers, S. J. Teat and G. Aromí, J. Am. Chem. Soc., 2014, 136, 3869–3874 CrossRef CAS PubMed.
- P. Wang, J. Xu, Q. D. Zhuo, Y. S. Ma, H. J. Cheng, X. Y. Tang and R. X. Yuan, Inorg. Chem. Commun., 2016, 67, 14–16 CrossRef CAS.
- Y. Deng, Z. Y. Yao, P. Wang, Y. Zhao, Y. S. Kang, M. Azam, S. I. Al-Resayes and W. Y. Sun, RSC Adv., 2017, 7, 44639–44646 RSC.
- P. Kumar, A. Deep, K. H. Kim and R. J. C. Brown, Prog. Polym. Sci., 2015, 45, 102–118 CrossRef CAS.
- S. Rodrïguez-Jimenez, H. L. C. Feltham and S. Brooker, Angew. Chem., Int. Ed., 2016, 55, 15067–15071 CrossRef PubMed.
- E. Coronado, M. Giménez-Marqués, G. M. Espallargas and L. Brammer, Nat. Commun., 2012, 3, 828 CrossRef PubMed.
- A. Białońska, R. Bronisz, K. Darowska, K. Drabent, J. Kusz, M. Siczek, M. Weselski, M. Zubko and A. Ozarowski, Inorg. Chem., 2010, 49, 11267–11269 CrossRef PubMed.
- J. S. Loring and W. Ronald Fawcett, J. Phys. Chem. A, 1999, 103, 3608–3617 CrossRef CAS.
- E. L. Pace and L. J. Noe, J. Chem. Phys., 1968, 49, 5317–5325 CrossRef CAS.
- J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781–1795 CrossRef CAS.
- T. Degen, M. Sadki, E. Bron, U. König and G. Nénert, Powder Diffr., 2014, 29, 13–18 CrossRef.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- R. Shevate, M. A. Haque, F. H. Akhtar, L. F. Villalobos, T. Wu and K. V. Peinemann, Angew. Chem., Int. Ed., 2018, 57, 11218–11222 CrossRef CAS PubMed.
- G. Givaja, P. Amo-Ochoa, C. J. Gómez-García and F. Zamora, Chem. Soc. Rev., 2012, 41, 115–147 RSC.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- H. J. Bae, S. Bae, C. Park, S. Han, J. Kim, L. N. Kim, K. Kim, S. H. Song, W. Park and S. Kwon, Adv. Mater., 2015, 27, 2083–2089 CrossRef CAS PubMed.
- L. Sun, S. S. Park, D. Sheberla and M. Dincă, J. Am. Chem. Soc., 2016, 138, 14772–14782 CrossRef CAS PubMed.
- L. Sun, C. H. Hendon, S. S. Park, Y. Tulchinsky, R. Wan, F. Wang, A. Walsh and M. Dincă, Chem. Sci., 2017, 8, 4450–4457 RSC.
- M. del C. Gimenez Lopez, M. Clemente Leon and C. Gimenez-Saiz, Dalton Trans., 2018, 47, 10453–10462 RSC.
- F. A. Harraz, A. A. Ismail, H. Bouzid, S. A. Al-Sayari, A. Al-Hajry and M. S. Al-Assiri, Phys. Status Solidi A, 2015, 212, 1851–1857 CrossRef CAS.
- F. A. Harraz, A. A. Ismail, S. A. Al-Sayari, A. Al-Hajry and M. S. Al-Assiri, Superlattices Microstruct., 2016, 100, 1064–1072 CrossRef CAS.
- T. Wagner, S. Haffer, C. Weinberger, D. Klaus and M. Tiemann, Chem. Soc. Rev., 2013, 42, 4036–4053 RSC.
- A. Chidambaram and K. C. Stylianou, Inorg. Chem. Front., 2018, 5, 979–998 RSC.
- J. Liu, F. Sun, F. Zhang, Z. Wang, R. Zhang, C. Wang and S. Qiu, J. Mater. Chem., 2011, 21, 3775–3778 RSC.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details, tables of crystal and refinement data, experimental procedures and characterization for 1·2CH3CN, and Figures S1–S23. CCDC 1895056. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02522g |
| This journal is © The Royal Society of Chemistry 2019 |