
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical C–H oxygenation and alcohol dehydrogenation involving Fe-oxo species using water as the oxygen source†
Amit
Das‡
 ,
Jordan E.
Nutting‡
and
Shannon S.
Stahl
*
,
Jordan E.
Nutting‡
and
Shannon S.
Stahl
*
Department of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, Wisconsin 53706, USA. E-mail: stahl@chem.wisc.edu
First published on 27th June 2019
Abstract
High-valent iron-oxo complexes are key intermediates in C–H functionalization reactions. Herein, we report the generation of a (TAML)Fe-oxo species (TAML = tetraamido macrocyclic ligand) via electrochemical proton-coupled oxidation of the corresponding (TAML)FeIII–OH2 complex. Cyclic voltammetry (CV) and spectroelectrochemical studies are used to elucidate the relevant (TAML)Fe redox processes and determine the predominant (TAML)Fe species present in solution during bulk electrolysis. Evidence for iron(IV) and iron(V) species is presented, and these species are used in the electrochemical oxygenation of benzylic C–H bonds and dehydrogenation of alcohols to ketones.
Introduction
High-valent iron-oxo species are identified as key catalytic intermediates in a number of enzyme-mediated oxidation reactions,1 including those involving heme- and non-heme active sites. Iron(IV)- and iron(V)-oxo species are key intermediates that effect C–H oxygenation, among other transformations. The reactive nature of these high-valent species and their ability to carry out challenging and important synthetic transformations have motivated efforts to design and synthesize high-valent iron-oxo complexes.2 Considerable success has been achieved, both in the synthesis of well-defined high-valent complexes and in the development and application of iron-based catalysts for selective oxidation of organic molecules.3The generation of high-valent iron-oxo complexes typically proceeds via reaction of a reduced iron precursor with an oxygen-atom transfer reagent, such as iodosylbenzene, a peracid, or hydrogen peroxide (Scheme 1).4 An appealing alternative approach would utilize water as the oxygen-atom source and generate the iron-oxo species via electrochemical proton-coupled oxidation of an Fe–OH2 complex (Scheme 1, right), resembling the pathway involved in photosynthetic water oxidation.5 Electrochemical generation of iron-oxo species from reduced Fe–OH/OH2 species has been demonstrated,6 especially in the context of molecular iron-based catalysts for electrochemical water oxidation.7,8 The investigation of analogous reactivity for electrochemical oxidation of organic molecules however has been limited to electroanalytical studies9 or to the mineralization of organic pollutants.10,11 On the other hand, stoichiometric one-electron oxidants, such as cerium(IV) ammonium nitrate (CAN), have been used to promote catalytic oxidation of organic molecules by high-valent metal-oxo species using water as an oxygen-atom source.12,13 For example, Fukuzumi and Nam generated a tetrapyridylamine-ligated iron-oxo complex via oxidation of the corresponding iron(II) complex in the presence of water with CAN and showed that this complex could support catalytic oxygenation of thioethers and C–H bonds.14 Analogous catalytic reactivity was demonstrated with other non-heme iron complexes15 and has been extended to photochemical processes by employing a photocatalyst in combination with stoichiometric [CoIII(NH3)Cl]2+ or persulphate as the terminal oxidant.6c,16
 | ||
| Scheme 1 Different approaches for the generation of high-valent iron-oxo species (left) and illustration of the application of electrochemical iron-oxo generation to the selective oxidation of organic molecules (right). | ||
Electrochemical oxidation of Fe–OH2 complexes would bypass the need for stoichiometric chemical oxidants of the type noted above, as the reaction could be coupled to proton reduction at the cathode to generate H2 as the sole byproduct. Here, we report the first preparative electrochemical method for oxidation of organic molecules involving generation of a molecular metal-oxo species using water as the source of oxygen. The catalyst consists of an iron complex bearing TAML as an ancillary ligand (TAML = tetraamido macrocyclic ligand), which was pioneered by Collins and co-workers for iron-catalyzed oxidation of organic molecules with hydrogen peroxide.17 Cyclic voltammetry (CV) and spectroelectrochemical studies are used to characterize the catalyst resting state under bulk electrolysis conditions, and the TAML-ligated iron catalyst is then used in the electrochemical oxidation of a series of alcohols and benzylic C–H substrates. These results establish an important benchmark for electrochemical C–H oxidation of organic molecules.
Results and discussion
To begin our studies, we identified two molecular iron complexes previously reported to promote electrochemical water oxidation, anticipating that the high-valent iron-oxo intermediates formed in this reaction could also promote C–H hydroxylation of organic substrates. The two complexes consisted of a six-coordinate FeIII–OH2 complex, [(dpaq)FeIII(OH2)]2+ (dpaq = 2-[bis(pyridine-2-ylmethyl)]amino-N-quinolin-8-yl-acetamido), reported by Meyer and co-workers,7a and the commercially available [(TAML)FeIII(OH2)]Na complex 1 developed by Collins and co-workers.7b Preliminary studies of the (dpaq)Fe complex, however, showed that it is unstable under the buffered conditions initially chosen to explore electrochemical reactions with organic substrates (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN:H2O, pH = 6–9) (see the ESI for details†). Therefore, the ensuing efforts focused on reactions with the (TAML)Fe complex. TAML and a number of closely related analogs are strongly donating tetraanionic macrocyclic ligands that stabilize high oxidation states of iron, including FeV.17 For example, [(TAML)FeV(O)]− species generated by reaction of the FeIII precursor with meta-chloroperbenzoic acid (mCPBA)4a,g,18 or NaOCl19 or by photochemical oxidation16c promote the oxidation of sulfides, alcohols, and sp3 C–H bonds.20
:1 CH3CN:H2O, pH = 6–9) (see the ESI for details†). Therefore, the ensuing efforts focused on reactions with the (TAML)Fe complex. TAML and a number of closely related analogs are strongly donating tetraanionic macrocyclic ligands that stabilize high oxidation states of iron, including FeV.17 For example, [(TAML)FeV(O)]− species generated by reaction of the FeIII precursor with meta-chloroperbenzoic acid (mCPBA)4a,g,18 or NaOCl19 or by photochemical oxidation16c promote the oxidation of sulfides, alcohols, and sp3 C–H bonds.20
Cyclic voltammetry (CV) analysis of 1 in CH3CN reveals two (quasi)reversible redox features at 620 mV and 1200 mV vs. Ag/AgCl (Fig. 1A), which are attributed to the generation of FeIV and FeV species, respectively.7b,d,19a,21 Efforts to analyze the CV of 1 in aqueous solution showed that this complex decomposes rapidly via demetallation of the TAML ligand at pH < 6 (see the ESI for details†).22 In a mixed acetonitrile–water solution with K2HPO4 as the supporting electrolyte (pH ∼ 8.5), CV analysis reveals the presence of three irreversible redox features (Fig. 1B). The current response for the redox feature at ca. 1250 mV is significantly higher than that for the two lower potential features. Control studies show that this enhanced current response does not reflect (TAML)Fe-catalyzed oxidation of water or CH3CN under these conditions (see the ESI for details†), suggesting that it corresponds to the self-promoted ligand oxidation that has been characterized in chemical oxidation studies with (TAML)Fe complexes.17b,23 Addition of ethylbenzene to this solution reveals a further increase in current at the redox feature at 1250 mV (Fig. 1B), indicating electrocatalytic turnover of the (TAML)Fe species via reaction with ethylbenzene at this potential.
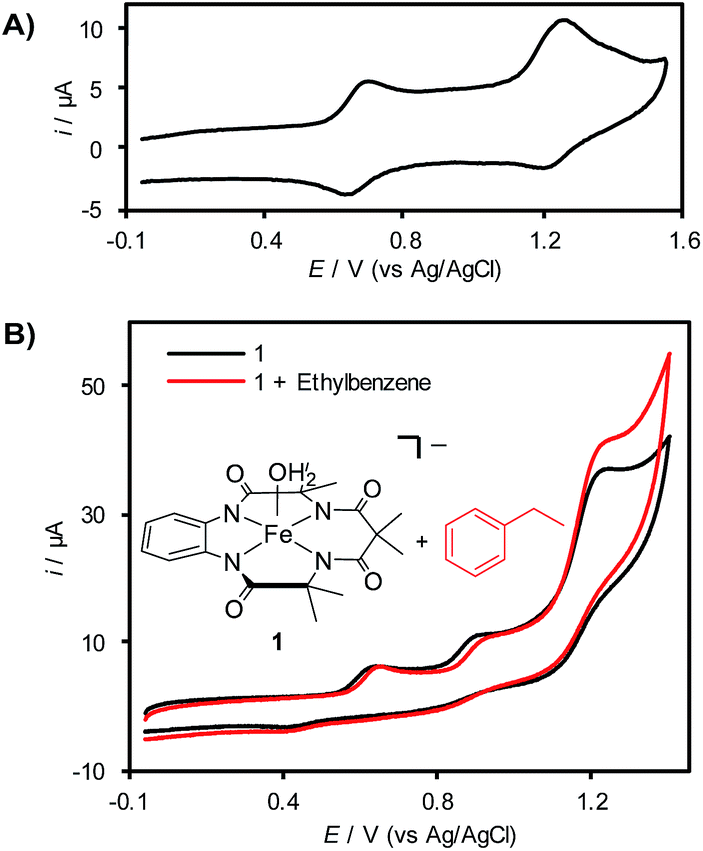 | ||
| Fig. 1 (A) CV of [(TAML)FeIII(OH2)]Na (1) in CH3CN (0.1 M LiClO4). (B) CVs of 0.5 mM 1 in the absence (black) and presence (red) of 20 mM ethylbenzene in 1:1 CH3CN:H2O and K2HPO4 (0.1 M). The increase in the current of the (TAML)Fe redox feature at 1.25 V (vs. Ag/AgCl) is attributed to catalytic ethylbenzene oxidation by a high-valent Fe-oxo species generated at this potential. Scan rate = 50 mV s−1. | ||
To gain further insights into the redox behavior of (TAML)Fe species under these conditions, the pH of the aqueous electrolyte was varied with different phosphate buffers while monitoring the redox potentials for the (TAML)Fe redox features by differential pulse voltammetry (DPV, Fig. 2A). Only the first redox feature (i) exhibits a significant dependence on the pH, shifting to more negative potentials with increasing pH. The other two features (ii and iii) are largely unaffected by changes in the pH. A plot of the pH dependence of redox feature (i) exhibits a slope of −59 mV pH−1, corresponding to a 1e−/1H+ stoichiometry and is consistent with proton-coupled oxidation of 1 to an [FeIV–OH] species (Fig. 2B). Control experiments show that redox feature (ii) disappears in the absence of the HPO42− buffer, suggesting that this redox step corresponds to an FeIII/IV process involving an Fe complex bearing HPO42− as an axial ligand (see the ESI for details†). Finally, the potential of feature (iii), assigned to the FeIV/V redox process, does not vary as the solution pH is changed (Fig. 2).
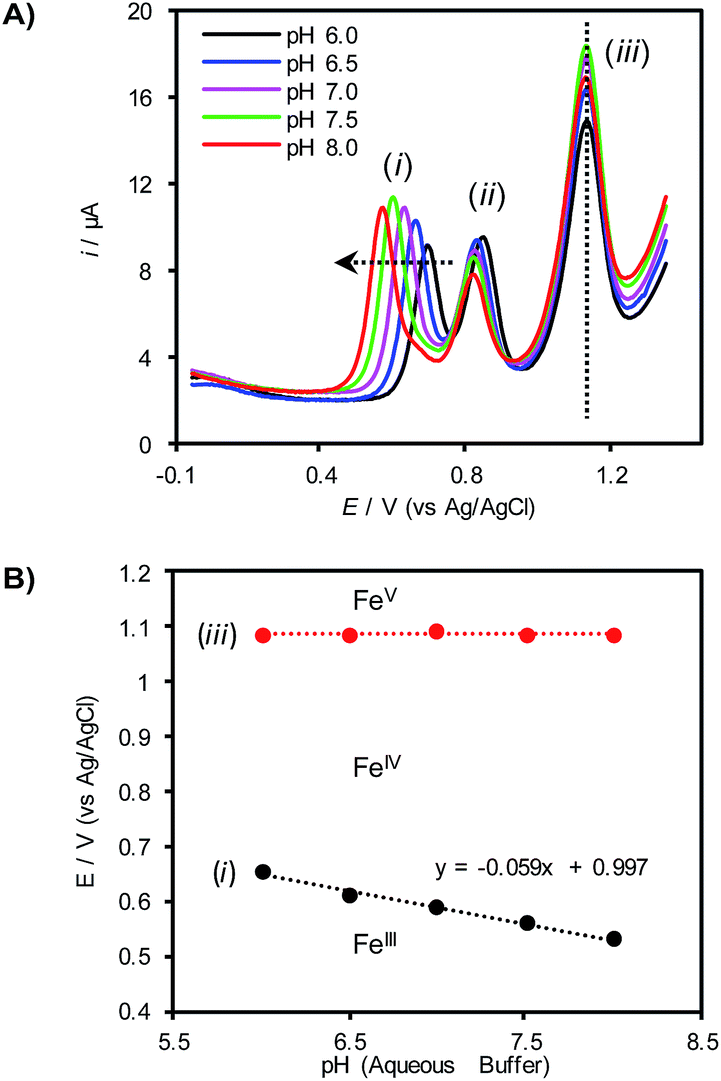 | ||
| Fig. 2 (A) Differential pulse voltammograms of 0.5 mM 1 in 1:1 CH3CN:buffer (0.1 M phosphate). (B) Pourbaix diagram derived from the peak potentials of redox features (i) and (iii) in (A). | ||
In anticipation of using (TAML)Fe complex 1 as a mediator for electrosynthetic applications, we investigated the behavior of 1 under bulk electrolysis conditions in a divided cell. A reticulated vitreous carbon (RVC) working electrode was paired with a Pt wire counter electrode, and a fiber-optic UV-visible dip probe was used to record optical spectra of the bulk solution during electrolysis. Bulk electrolysis was conducted with a solution of 1 in 1:1 CH3CN:H2O with K2HPO4 (0.1 M) at applied potentials of 800 and 1250 mV (Fig. 3). Electrolysis of the solution at 800 mV for 15 min led to clear changes in (TAML)Fe species in solution, and the spectral data reveal conversion of the FeIII species 1 into the dimeric oxo-bridged iron(IV) complex 3 (Fig. 3A).24 Electrolysis at an applied potential of 1250 mV led to the same spectral changes, albeit with more rapid formation of 3 (ca. 6 min) (Fig. 3B). Continued electrolysis at this potential leads to slow bleaching of the solution, indicating the onset of self-promoted decomposition of the high-valent (TAML)Fe species in solution (see the ESI for details†).17b,23
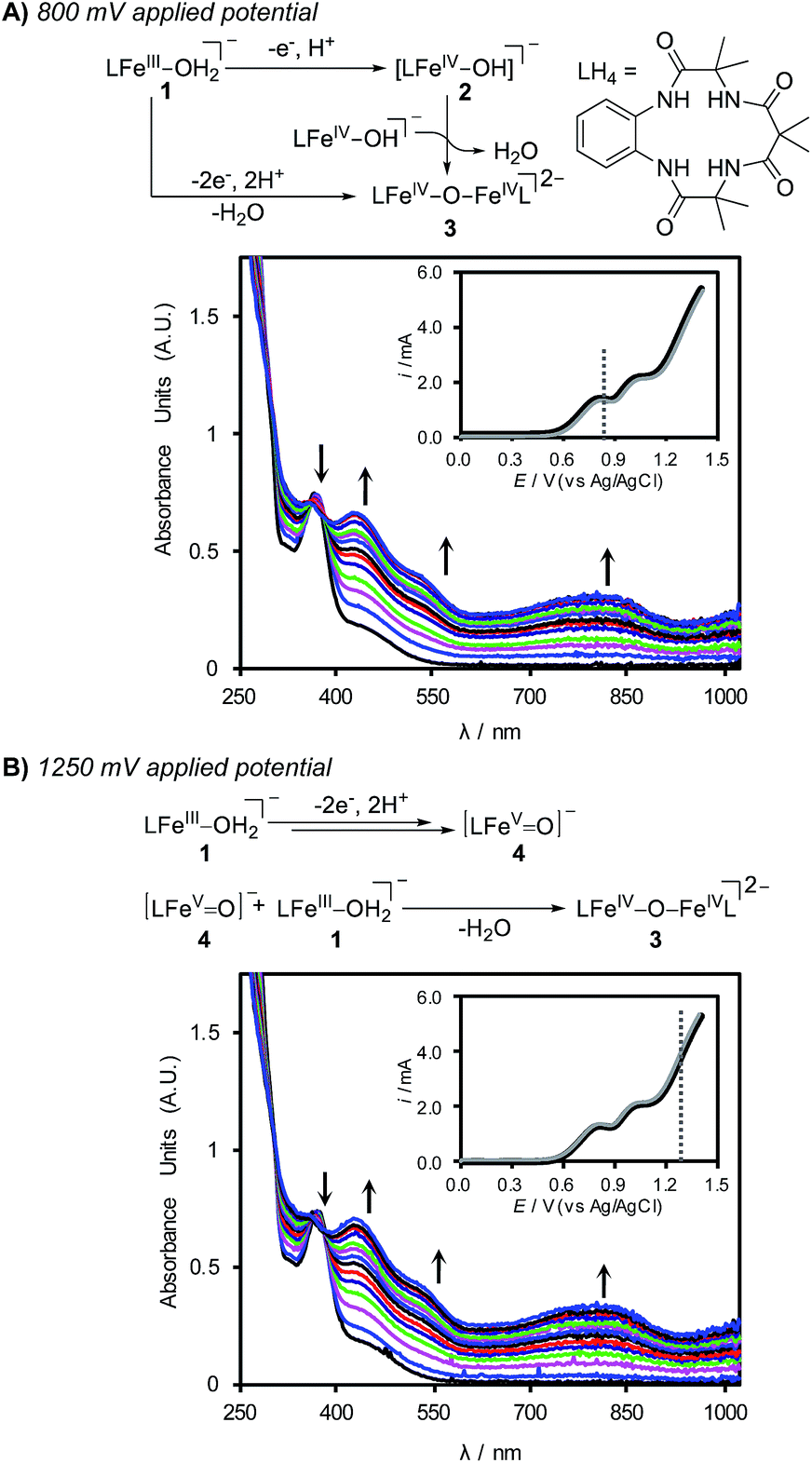 | ||
| Fig. 3 (A) UV-vis spectra recorded for 0.5 mM 1 undergoing bulk electrochemical oxidation at 800 mV (vs. Ag/AgCl) in 1:1 CH3CN:H2O with K2HPO4 (0.1 M) in a divided cell. Final spectra collected after 15.3 min of bulk electrolysis. (B) UV-vis spectra recorded for 0.5 mM (TAML)Fe undergoing bulk electrochemical oxidation at 1250 mV (vs. Ag/AgCl) in 1:1 CH3CN:H2O with K2HPO4 (0.1 M) in a divided cell at a RVC electrode. Final spectra collected after 6.7 min of bulk electrolysis. (Inset) LSV of bulk electrolysis solution at the RVC working electrode. The potential applied for each bulk electrolysis is indicated. Continuing electrochemical oxidation beyond the final spectra shown in (B) leads first to steady state absorption features followed by overall loss of all (TAML)Fe absorption features. | ||
Formation of the oxo-bridged (TAML)FeIV dimer 3 at both electrolysis potentials was initially unexpected; however, these observations are readily rationalized from the CV data and previous studies of (TAML)Fe species with chemical oxidants.23b The pH-dependent DPV data in Fig. 2 suggest that electrochemical oxidation of 1 generates the [FeIV–OH] species 2 in a 1e−/1H+ process, and formation of 3 can arise from the dimerization of 2.25 The (TAML)FeV(O) species 4 has been shown to react rapidly with 1 to generate 3. For example, treatment of 1 with 0.5 equiv. of mCPBA affords oxo-bridged (TAML)FeIV dimer 3.23a,26 Complex 4 is not directly detected under the electrochemical conditions of Fig. 3B; however, it is expected to react rapidly with 1 in solution after being generated at the electrode to afford 3.
The identical (TAML)Fe speciation at the two electrolysis potentials is contrasted by different rates of substrate oxidation during electrochemical (TAML)Fe-catalyzed oxidation of organic molecules. Ethylbenzene and 1-phenylethanol were evaluated as substrates for electrochemical oxidation in the presence of 10 mol% 1, and constant potential electrolysis was conducted at 800 and 1250 mV. Both substrates generate acetophenone as the product; however, the rate of product formation varies with the substrate and applied potential. The formation of acetophenone from ethylbenzene is nearly 10-fold faster at 1250 mV relative to the rate at 800 mV, while oxidation of 1-phenylethanol exhibits a ca. five-fold increase in rate at the higher potential (Fig. 4). Small amounts of 1-phenylethanol are observed during the oxidation of ethylbenzene, but this intermediate does not build up to significant concentrations during the electrolysis, consistent with the much faster oxidation of 1-phenylethanol.
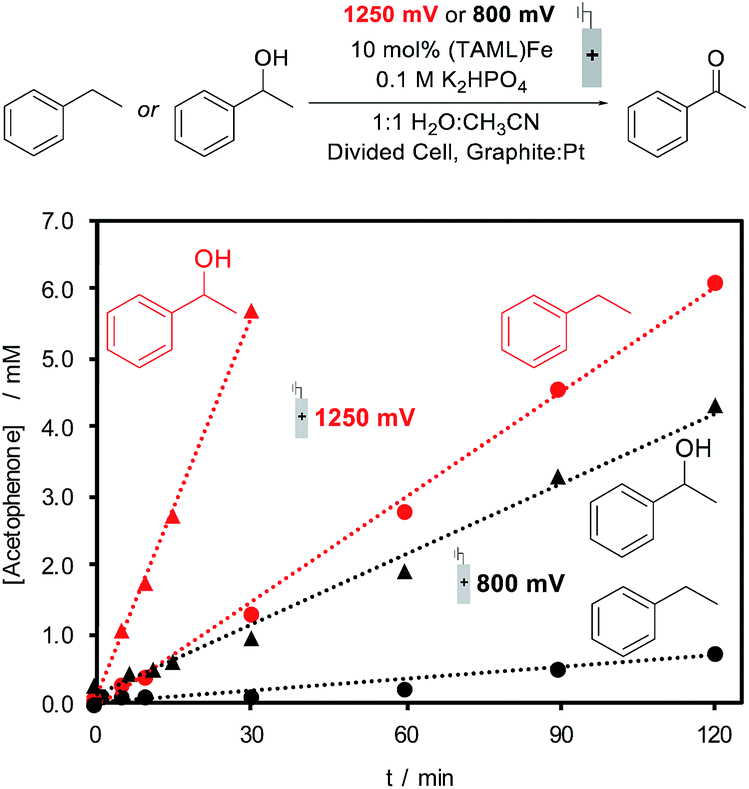 | ||
| Fig. 4 Formation of acetophenone during the first two hours of (TAML)Fe catalyzed electrochemical oxidation of ethylbenzene (circles, 20 mM) or 1-phenylethanol (triangles, 20 mM) at applied potentials of 1250 mV (red) and 800 mV (black) (vs. Ag/AgCl) in 1:1 CH3CN:H2O and 0.1 M K2HPO4 in a divided cell. | ||
The electrolysis results are consistent with previous observations that the FeIV dimer 3 is capable of oxidizing organic molecules;26 however, they also show that conditions capable of generating FeV (i.e., at 1250 mV) lead to even faster reactivity. The steady-state bulk concentration of FeIV dimer 3 is not affected by the electrolysis potential (see the ESI for details†). Therefore, the increased oxidation rate at 1250 mV is attributed to the generation of the highly reactive, but unobserved, FeV(O) species at the electrode, which will promote rapid reaction with the substrate.27
Electrochemical oxidation of ethylbenzene under constant potential electrolysis conditions at 1250 mV with 1 as the catalyst generates acetophenone in 79% yield. A 55% faradaic efficiency was observed during this reaction, probably reflecting that the background self-oxidation of (TAML)Fe species competes with productive reaction with the substrate.17b,23 A series of additional substrates containing benzylic C–H bonds was then tested under the electrochemical oxidation conditions (Fig. 5A; see the ESI for details†). Evaluation of modified ethylbenzene derivatives showed good performance with electron-rich and -neutral derivatives (5–7), while lower reactivity was observed with the very electron-deficient nitro-substituted derivative (8). The reaction with isobutylbenzene proceeds with high selectivity (10), arising exclusively from reaction at the benzylic position; no tertiary C–H oxidation product was detected. Similarly, no oxidation of the tertiary benzylic C–H position was observed in the reaction with the methyl ester of ibuprofen (11), probably reflecting both steric and electronic (i.e., from the electron-withdrawing ester) effects. The reaction also shows some tolerance to a pyridine substituent (12), but low conversion and yield are observed with the less reactive 2-ethylpyridine substrate (17). Pyridine coordination to Fe by these complexes could inhibit reactivity.25 We also examined the electrochemical oxidation of a small series of secondary alcohols (Fig. 5B). This reaction is more facile than benzylic C–H oxidation, and only 5 mol% catalyst was needed to observe good reactivity. In spite of the simplicity of cyclohexanol as a substrate, the good yield observed here (13, 85%) is noteworthy because a recent electrochemical oxidation of cyclohexanol with a different molecular Fe-based catalyst led to mineralization of this substrate (i.e., production of CO2).10 Good to excellent yields were observed with a series of more interesting alcohols under the electrolysis conditions (14–16). Many of the yields in Fig. 5 were determined by GC or 1H NMR analysis due to the volatility of the products; however, the steroidal ketone 15 was isolated in 97% yield. The ineffectiveness of electron-rich substrates, such as 18–20 in Fig. 5C, reflects the ability of these substrates to undergo direct electron-transfer at potentials below 1250 mV (see the ESI for details†).28 In general, this electrocatalytic approach does not yet achieve the breadth of reactivity demonstrated with (TAML)Fe and related catalysts that utilize chemical oxidants.17,18 The most significant constraint appears to be the oxidative self-decomposition of the (TAML)Fe catalyst under the bulk electrolysis conditions, which limits the catalyst lifetime (see Section 10 of the ESI†).
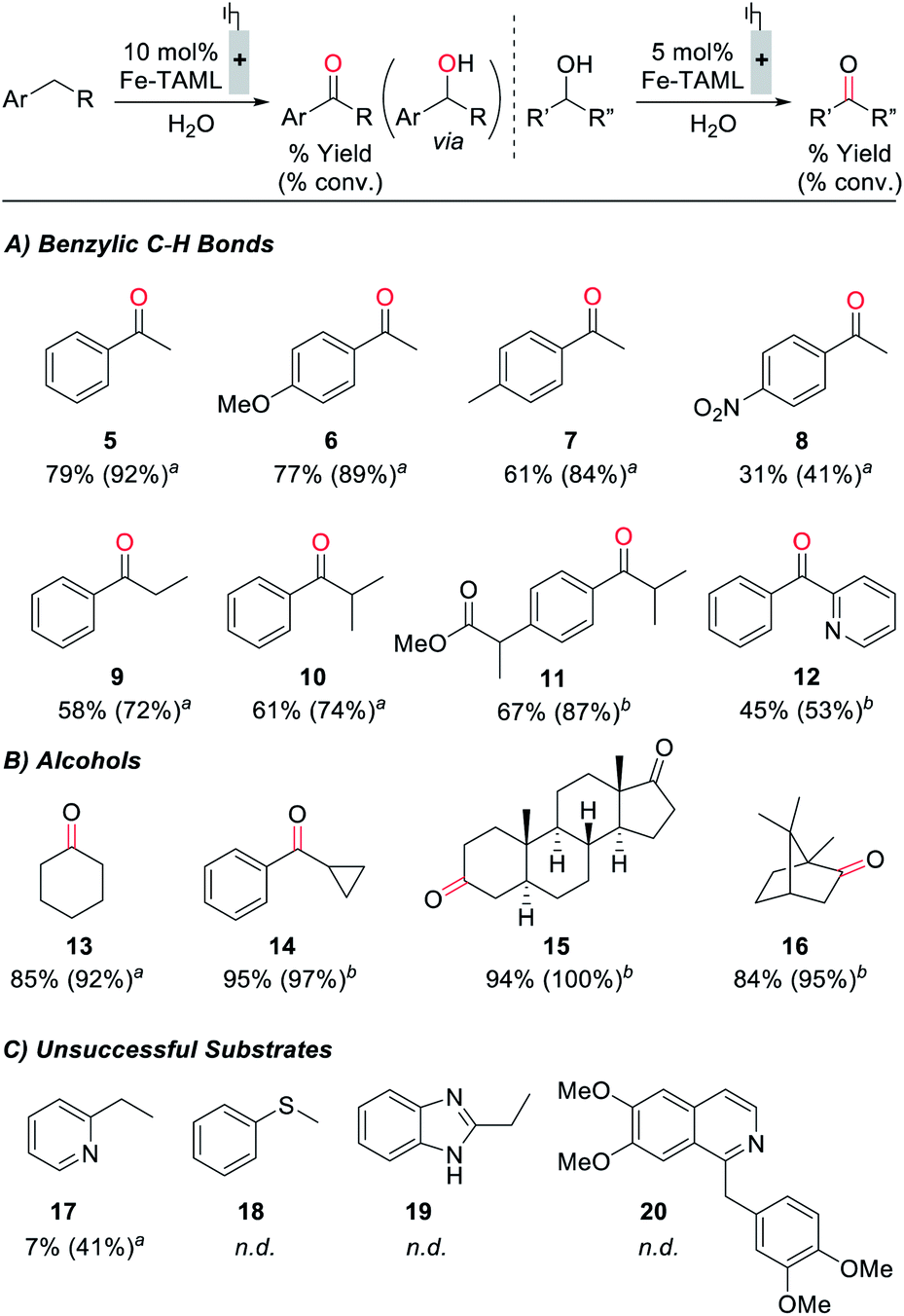 | ||
| Fig. 5 Assessment of (TAML)Fe-catalyzed electrochemical oxidation of substrates containing a benzylic C–H bond (A) or alcohol functional group (B), in addition to selected examples of unsuccessful substrates (C). Reactions performed in a divided H-cell in an N2 atmosphere with graphite:Pt electrodes at a constant applied potential of 1250 mV vs. Ag/AgCl. Anolyte: 0.1 mmol substrate, 0.1 M K2HPO4, 2.5 mL H2O, 2.5 mL CH3CN. Catholyte: 0.1 M K2HPO4, 2.5 mL H2O, 2.5 mL CH3CN. aYield% (conversion%) estimated by GC analysis with bromobenzene as the internal standard. bYield% (conversion%) estimated by 1H NMR analysis with 1,3,5-trimethoxybenzene as the external standard. n.d. = not determined. | ||
In spite of the existing limitations, the data in Fig. 5 establish an important benchmark for electrochemical oxidation of organic molecules. While electrochemical generation of transition-metal oxo species via proton-coupled oxidation of metal-aqua complexes is well established for water oxidation, analogous reactivity has not previously been demonstrated for preparative electrochemical oxidation of organic molecules. This approach warrants further development, and it would complement the application of organic hydrogen-atom transfer (HAT) mediators such as N-hydroxyphthalimide28a,29,30 and nitrogen-centered radicals28b,31 in electrochemical HAT-based oxidation of organic molecules.
Conclusions
The results outlined herein demonstrate the electrochemical generation of high-valent iron species via proton-coupled oxidation of a TAML-ligated FeIII–OH2 complex, and the application of this reaction to preparative electrochemical oxidation of organic molecules. Voltammetry analysis reveals the sequential formation of FeIV and FeV species at increasing potentials (800 and 1250 mV, respectively). Electrolysis of [(TAML)FeIII(OH2)]− at the higher potential results in accumulation of the oxo-bridged, dimeric (TAML)FeIV species 3 in solution, which is attributed to the generation of FeV at the electrode followed by comproportionation with FeIII in solution. Sustained electrolysis at this potential provides the basis for selective electrochemical organic oxidation reactions, including oxygenation of benzylic C–H bonds and dehydrogenation of alcohols.These results establish an important foundation for future studies, such as those focused on the development of more robust metal complexes. The self-promoted decomposition of the high-valent intermediates generated under these reaction conditions limits the faradaic efficiency and the catalyst lifetime during electrolysis. Furthermore, complexes that exhibit stability under acidic conditions could support broader functional group compatibility by taking advantage of the ability to protect amines and other basic functional groups via protonation with a Brønsted acid.32
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was initiated with financial support from an NSF CCI grant (CHE-1305124) and completed with support from the DOE (DE-FG02-05ER15690). JEN acknowledges the NSF for a predoctoral fellowship (DGE-1747503). The spectrometers were purchased through NSF grant CHE-1048642 and by a generous gift from Paul J. and Margaret M. Bender, and the mass spectrometer was purchased through NIH grant NIH 1S10 OD020022-1.Notes and references
- (a) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2887 CrossRef CAS PubMed; (b) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed; (c) J. T. Groves, J. Inorg. Biochem., 2006, 100, 434–447 CrossRef CAS PubMed; (d) C. Krebs, D. G. Fujimori, C. T. Walsh and J. M. Bollinger Jr, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed; (e) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed; (f) P. R. Ortiz de Montellano, Cytochrome P450: Structure, Mechanism, and Biochemistry, Springer, New York, 4th edn, 2015 Search PubMed; (g) X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- (a) X. Shan and L. Que Jr, J. Inorg. Biochem., 2006, 100, 421–433 CrossRef CAS PubMed; (b) W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed; (c) L. Que Jr and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed; (d) X. Englemann, I. Monte-Pérez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef PubMed; (e) M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- (a) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC; (b) M. Costas, Coord. Chem. Rev., 2011, 255, 2912–2932 CrossRef CAS; (c) W. N. Oloo and L. Que Jr, Acc. Chem. Res., 2015, 48, 2612–2621 CrossRef CAS PubMed; (d) M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS PubMed.
- (a) F. Tiago de Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que Jr, E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835–838 CrossRef CAS PubMed; (b) P. Comba and S. Wunderlich, Chem.–Eur. J., 2010, 16, 7293–7299 CrossRef CAS PubMed; (c) M. S. Seo, N. H. Kim, K.-B. Cho, J. E. So, S. K. Park, M. Clémancey, R. Garcia-Serres, J.-M. Latour, S. Shaik and W. Nam, Chem. Sci., 2011, 2, 1039–1045 RSC; (d) I. Prat, J. S. Mathieson, M. Güell, X. Ribas, J. M. Luis, L. Cronin and M. Costas, Nat. Chem., 2011, 3, 788–793 CrossRef CAS PubMed; (e) S. A. Wilson, J. Chen, S. Hong, Y.-M. Lee, M. Clémancey, R. Garcia-Serres, T. Nomura, T. Ogura, J.-M. Latour, B. Hedman, K. O. Hodgson, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2012, 134, 11791–11806 CrossRef CAS PubMed; (f) J. England, J. O. Bigelow, K. M. Van Heuvelen, E. R. Farquhar, M. Martinho, K. K. Meier, J. R. Frisch, E. Münck and L. Que Jr, Chem. Sci., 2014, 5, 1204–1215 RSC; (g) M. Ghosh, K. K. Singh, C. Panda, A. Weitz, M. P. Hendrich, T. J. Collins, B. B. Dhar and S. S. Gupta, J. Am. Chem. Soc., 2014, 136, 9524–9527 CrossRef CAS PubMed; (h) A. N. Biswas, M. Puri, K. K. Meier, W. N. Oloo, G. T. Rohde, E. L. Bominaar, E. Münck and L. Que Jr, J. Am. Chem. Soc., 2015, 137, 2428–2431 CrossRef CAS PubMed; (i) J. Serrano-Plana, W. N. Oloo, L. Acosta-Rueda, K. K. Meier, B. Verdejo, E. García-España, M. G. Basallote, E. Münck, L. Que Jr, A. Company and M. Costas, J. Am. Chem. Soc., 2015, 137, 15833–15842 CrossRef CAS PubMed.
- (a) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed; (b) G. Renger and T. Renger, Photosynth. Res., 2008, 98, 53–80 CrossRef CAS PubMed; (c) R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319–5324 CrossRef CAS PubMed.
- For electrochemical generation of high-valent iron-oxo complexes (i.e., non-catalytic examples), see: (a) W. A. Lee, T. S. Calderwood and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4301–4305 CrossRef CAS PubMed; (b) M. J. Collins, K. Ray and L. Que Jr, Inorg. Chem., 2006, 45, 8009–8011 CrossRef CAS PubMed; (c) H. Kotani, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 3249–3251 CrossRef CAS PubMed; (d) D. Wang, K. Ray, M. J. Collins, E. R. Farquhar, J. R. Frisch, L. Gómez, T. A. Jackson, M. Kerscher, A. Waleska, P. Comba, M. Costas and L. Que Jr, Chem. Sci., 2013, 4, 282–291 RSC.
- (a) M. K. Coggins, M.-T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531–5534 CrossRef CAS PubMed; (b) E. L. Demeter, S. L. Hilburg, N. R. Washburn, T. J. Collins and J. R. Kitchin, J. Am. Chem. Soc., 2014, 136, 5603–5606 CrossRef CAS PubMed; (c) M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed; (d) S. Pattanayak, D. R. Chowdhury, B. Garai, K. K. Singh, A. Paul, B. B. Dhar and S. S. Gupta, Chem.–Eur. J., 2017, 23, 3414–3424 CrossRef CAS PubMed; (e) K. G. Kottrup, S. D'Agostini, P. H. van Langevelde, M. A. Siegler and D. G. H. Hetterscheid, ACS Catal., 2018, 8, 1052–1061 CrossRef CAS PubMed.
- For general reviews of water oxidation with molecular electrocatalysts, see: (a) X. Sala, I. Romero, M. Rodriguez, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842–2852 CrossRef CAS PubMed; (b) M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åckermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed; (c) J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed; (d) B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- (a) D. L. Hickman and H. M. Goff, Inorg. Chem., 1983, 22, 2787–2789 CrossRef CAS; (b) J. T. Groves and J. A. Gilbert, Inorg. Chem., 1986, 25, 123–125 CrossRef CAS; (c) T.-s. Lee and Y. O. Su, J. Electroanal. Chem., 1996, 414, 69–73 CrossRef; (d) A. K. Vannucci, J. F. Hull, Z. Chen, R. A. Binstead, J. J. Concepcion and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 3972–3975 CrossRef CAS PubMed; (e) A. K. Vannucci, Z. Chen, J. J. Concepcion and T. J. Meyer, ACS Catal., 2012, 2, 716–719 CrossRef CAS.
- D. P. de Sousa, C. J. Miller, Y. Chang, T. D. Waite and C. J. McKenzie, Inorg. Chem., 2017, 56, 14936–14947 CrossRef CAS PubMed.
- A conceptually different approach to electrochemical generation of an Fe-oxo species relies on reduction of an FeIII complex, followed by activation of O2 by the reduced Fe species: M. Mukherjee and A. Dey, ACS Cent. Sci., 2019, 5, 671–682 CrossRef CAS PubMed.
- For a review see: S. Fukuzumi, T. Kojima, Y.-M. Lee and W. Nam, Coord. Chem. Rev., 2017, 333, 44–56 CrossRef CAS.
- For a selected recent example that shows considerable synthetic scope, see: E. McNeill and J. Du Bois, Chem. Sci., 2012, 3, 1810–1813 RSC.
- Y.-M. Lee, S. N. Dhuri, S. C. Sawant, J. Cho, M. Kubo, T. Ogura, S. Fukuzumi and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 1803–1806 CrossRef CAS PubMed.
- I. Garcia-Bosch, Z. Codolà, I. Prat, X. Ribas, J. Lloret-Fillol and M. Costas, Chem.–Eur. J., 2012, 18, 13269–13273 CrossRef CAS PubMed.
- (a) A. Company, G. Sabenya, M. González-Béjar, M. Gómez, M. Clémancy, G. Blondin, A. J. Jasniewski, M. Puri, W. R. Browne, J.-M. Latour, L. Que Jr, M. Costas, J. Pérez-Prieto and J. Lloret-Fillol, J. Am. Chem. Soc., 2014, 136, 4624–4633 CrossRef CAS PubMed; (b) T. Chantarojsiri, Y. Sun, J. R. Long and C. J. Chang, Inorg. Chem., 2015, 54, 5879–5887 CrossRef CAS PubMed; (c) B. Chandra, K. K. Singh and S. S. Gupta, Chem. Sci., 2017, 8, 7545–7551 RSC.
- (a) M. J. Bartos, S. W. Gordon-Wylie, B. G. Fox, L. J. Wright, S. T. Weintraub, K. E. Kauffmann, E. Münck, K. L. Kostka, E. S. Uffelman, C. E. F. Rickard, K. R. Noon and T. J. Collins, Coord. Chem. Rev., 1998, 174, 361–390 CrossRef CAS; (b) T. J. Collins, Acc. Chem. Res., 2002, 35, 782–790 CrossRef CAS PubMed; (c) T. J. Collins and A. D. Ryabov, Chem. Rev., 2017, 117, 9140–9162 CrossRef CAS PubMed.
- S. Jana, M. Ghosh, M. Ambule and S. S. Gupta, Org. Lett., 2017, 19, 746–749 CrossRef CAS PubMed.
- (a) S. Pattanayak, A. J. Jasniewski, A. Rana, A. Draksharapu, K. K. Singh, A. Weitz, M. Hendrich, L. Que Jr, A. Dey and S. S. Gupta, Inorg. Chem., 2017, 56, 6352–6361 CrossRef CAS PubMed; (b) S. Jana, J. Thomas and S. S. Gupta, Inorg. Chim. Acta, 2019, 486, 476–482 CrossRef CAS.
- (TAML)Fe and related analogs have been used as catalysts for electrochemical water oxidation, as described in ref. 7b and d, respectively. However, we note that in ref. 7b, the electrochemical water oxidation studies were performed with (TAML)Fe immobilized on the electrode surface and conducted at low pH under conditions that lead to decomposition of the catalyst in solution. For non-electrochemical water oxidation by (TAML)Fe catalysts and related analogs, see: (a) W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef CAS PubMed; (b) C. Panda, J. Debgupta, D. D. Díaz, K. K. Singh, S. S. Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273–12282 CrossRef CAS PubMed.
- (a) A. Ghosh, F. Tiago de Oliveira, T. Yano, T. Nishioka, E. S. Beach, I. Kinoshita, E. Münck, A. D. Ryabov, C. P. Horwitz and T. J. Collins, J. Am. Chem. Soc., 2005, 127, 2505–2513 CrossRef CAS PubMed; (b) D.-L. Popescu, M. Vrabel, A. Brausam, P. Madsen, G. Lente, I. Fabian, A. D. Ryabov, R. van Eldik and T. J. Collins, Inorg. Chem., 2010, 49, 11439–11448 CrossRef CAS PubMed.
- See also: V. Polshin, D.-L. Popescu, A. Fischer, A. Chanda, D. C. Horner, E. S. Beach, J. Henry, Y.-L. Qian, C. P. Horwitz, G. Lente, I. Fabian, E. Münck, E. L. Bominaar, A. D. Ryabov and T. J. Collins, J. Am. Chem. Soc., 2008, 130, 4497–4506 CrossRef CAS PubMed.
- (a) S. Kundu, J. V. K. Thompson, A. D. Ryabov and T. J. Collins, J. Am. Chem. Soc., 2011, 133, 18546–18549 CrossRef CAS PubMed; (b) Q. Ren, Y. Guo, M. R. Mills, A. D. Ryabov and T. J. Collins, Eur. J. Inorg. Chem., 2015, 1445–1452 CrossRef CAS.
- A. Chanda, X. Shan, M. Chakrabarti, W. C. Ellis, D. L. Popescu, F. Tiago de Oliveira, D. Wang, L. Que Jr, T. J. Collins, E. Münck and E. L. Bominaar, Inorg. Chem., 2008, 47, 3669–3678 CrossRef CAS PubMed.
- A. Ghosh, D. A. Mitchell, A. Chanda, A. D. Ryabov, D. L. Popescu, E. C. Upham, G. J. Collins and T. J. Collins, J. Am. Chem. Soc., 2008, 130, 15116–15126 CrossRef CAS PubMed.
- S. Kundu, J. V. K. Thompson, L. Q. Shen, M. R. Mills, E. L. Bominaar, A. D. Ryabov and T. J. Collins, Chem.–Eur. J., 2015, 21, 1803–1810 CrossRef CAS PubMed.
- For comparative studies of FeIV and FeV species bearing TAML and closely related tetraanionic ligands, see ref. 4g, 19 and 26.
- The challenge of lowering the applied potential in electrochemical C–H oxidation reaction has been the specific focus of several recent studies: (a) M. Rafiee, F. Wang, D. P. Hruszkewycz and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 22–25 CrossRef CAS PubMed; (b) F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385–6390 CrossRef CAS PubMed.
- (a) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS PubMed; (b) D. P. Hruszkewycz, K. C. Miles, O. R. Thiel and S. S. Stahl, Chem. Sci., 2017, 8, 1282–1287 RSC.
- For a review of NHPI-mediated electrochemical reactions, see: J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS PubMed.
- (a) J. M. Howell, K. Feng, J. R. Clark, L. J. Trzepkowski and M. C. White, J. Am. Chem. Soc., 2015, 137, 14590–14593 CrossRef CAS PubMed; (b) A. M. Adams, J. Du Bois and H. A. Malik, Org. Lett., 2015, 17, 6066–6069 CrossRef CAS PubMed; (c) J. B. C. Mack, J. D. Gipson, J. Du Bois and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 9503–9506 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, procedures, and spectroscopic data. See DOI: 10.1039/c9sc02609f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |