
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of montbretin A analogues yields potent competitive inhibitors of human pancreatic α-amylase†
Christina R.
Tysoe
a,
Sami
Caner
b,
Matthew B.
Calvert
a,
Anna
Win-Mason
a,
Gary D.
Brayer
b and
Stephen G.
Withers
 *a
*a
aDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver BC, Canada V6T 1Z1. E-mail: withers@chem.ubc.ca
bDepartment of Biochemistry and Molecular Biology, University of British Columbia, 2350 Health Sciences Mall, Vancouver BC, Canada V6T 1Z3
First published on 18th October 2019
Abstract
Simplified analogues of the potent human amylase inhibitor montbretin A were synthesised and shown to bind tightly, KI = 60 and 70 nM, with improved specificity over medically relevant glycosidases, making them promising candidates for controlling blood glucose. Crystallographic analysis confirmed similar binding modes and identified new active site interactions.
The healthcare burden of diabetes mellitus has steadily risen over the past century, with an increase from 108 million adult cases worldwide in 1980 to 422 million cases in 2014.1 Strikingly, type II diabetes accounts for 90–95% of all cases.2 The inhibition of carbohydrate catabolism has been explored as a means of mediating post-prandial blood glucose levels for the treatment of type II diabetes.3 Targeting of carbohydrate catabolism provides two benefits: beyond directly modulating blood glucose levels it can also promote weight loss in pre-diabetic individuals to prevent the onset of diabetes. Miglitol, voglibose and acarbose are three inhibitors of carbohydrate catabolism that are currently in medical use. These compounds act as inhibitors of the mammalian gut α-glucosidases, thereby mediating post-prandial blood glucose levels.4 While effective in preventing hyperglycemia, the resulting displacement of di- and tri-saccharides into the lower gut produces unwanted side effects arising from their strong osmotic effects within the large intestine and their rapid processing by anaerobic bacteria, resulting in diarrhea, nausea, and abdominal discomfort.5
Human pancreatic α-amylase (HPA) catalyzes the endo-hydrolysis of ingested starches into maltose and maltotriose6 and its activity has been positively correlated with post-prandial blood glucose levels. Consequently the targeted inhibition of this enzyme has been highlighted as a means of managing post-prandial blood glucose levels while avoiding side effects associated with general α-glucosidase inhibition.7 Most designs of glycosidase inhibitors have been based upon azasugar scaffolds in which a nitrogen atom replaces O5 or C1 of the sugar ring in the −1 binding site.8 In this manuscript we explore a completely new pharmacophore for α-amylase inhibition involving stacked phenolic rings that interact closely with the conserved active site carboxylic acids. The parent molecule, montbretin A (MbA), is a complex flavonol glycoside produced by Crocosmia crocosmiiflora that acts as a potent and selective inhibitor of HPA (KI = 8 nM).9 This inhibitor has been tested in ZDF (Zucker diabetic fatty) rats and produced a significant decrease in blood glucose plasma levels, highlighting this compound as a promising lead for the therapeutic reduction of post-prandial blood glucose levels.10 However its large-scale extraction and purification from the corms of Crocosmia crocosmiiflora is a challenging multistep process and requires a substantial amount of biomass. Consequently the design and synthesis of novel inhibitors based upon MbA's structure has been proposed as a means of generating chemically accessible structures of similar potency and specificity.11
Montbretin A contains a myricetin flavonol core glycosylated at the 3- and 4′-positions (Fig. 1a). The 3-OH is adorned with an α-linked D-glucopyranosyl-(β-1 → 2)-D-glucopyranosyl-(β-1 → 2)-L-rhamnopyranoside trisaccharide to which a 6-O-caffeic ester is attached on the central D-glucosyl group. The 4′-OH of myricetin bears a β-linked L-rhamnopyranosyl-(α-1 → 4)-D-xyloside. X-ray crystallographic analysis of MbA in complex with HPA showed notable points of contact occurring between the enzyme's catalytic residues and MbA's caffeic ester and myricetin A-ring.11 Meanwhile, minimal interactions were observed with the myricetin B-ring or any of MbA's carbohydrate appendages. Step-wise chemoenzymatic degradation of MbA had yielded a ‘minimum inhibitory structure’ termed mini-MbA (Fig. 1b). This inhibitor contained only the myricetin flavonol and caffeic acid linked through a D-glucopyranosyl-(β-1 → 2)-L-rhamnopyranose disaccharide, and had a KI = 400 nM (ref. 12) towards HPA (lone myricetin and ethyl caffeate have KI = 100 μM and 1.3 mM, respectively9). The retained potency of the simplified mini-MbA structure suggested that a suitable synthetic analogue could achieve equivalent levels of potency, thus mini-MbA was used as inspiration for our synthesis. It was not clear however, whether such a compound would retain the required specificity for HPA over the gut alpha-glucosidases given the active site similarities.
 | ||
| Fig. 1 Structures of montbretin A and mini-MbA. | ||
Quercetin was selected to replace myricetin as the flavonol core in our analogue synthesis since it, and its glycosylated derivatives, are substantially less expensive than other flavonols. Quercetin differs from myricetin only in hydroxylation of the B-ring and, as noted earlier, there are very few interactions between HPA and this ring.13 In mini-MbA, the caffeic acid and myricetin groups are connected by a disaccharide, which forms a bridge of 7 atoms. This region formed no direct interactions with the enzyme, though it likely provides some conformational constraints. As a result, a variety of different chemical structures could be explored for the linker region. Requirements for linker length and rigidity could be assessed, as well as the possibility of incorporating new functionalities to create favorable interactions with the enzyme.
Quercetin-3-O-rutinoside was used as a convenient and inexpensive starting material for the synthesis since the pre-existing sugar at C3 could be used to differentiate the hydroxyls in a protecting group strategy. Quercetin-3-O-rutinoside was thus reacted with benzyl bromide in the presence of K2CO3, followed by cleavage of the 3-O-glycoside by treatment with HCl/ethanol to yield benzyl quercetin 2, freeing the 3-OH for subsequent modification.14 Analogues with simple linkers were first synthesized to test the basic concept of linking the two phenolics (Fig. 2). Optimal linker length and polarity was then explored through a series containing three, five, seven and eight atoms. Analogues M01 to M03 contained simple alkyl linkers of three, five and seven atoms long while M04 contained a similarly flexible eight-atom triethylene glycol linker. Focus then changed to design of linkers that incorporate appropriate conformational constraints while seeking out favourable interactions of the side chains with active site residues. To that end analogues M05 to M11 were generated containing an amino acid linked to quercetin's 3-OH through a propyl chain (Scheme 1).
 | ||
| Fig. 2 Structures of synthetic MbA analogues. | ||
 | ||
Scheme 1 Synthesis of analogues M01 to M04: (a) PFP-TFA (1.2 equiv.), pyr (1.2 equiv.), DMF, 93%; (b) BnBr (5 equiv.), K2CO3 (5 equiv.), DMF, 70 °C, 18 h; (c) EtOH/HCl (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 70 °C, 2 h 70% over two steps; (d) Br(CH2)nCl or Br(C2H4O)2 C2H4N3 (1.5 equiv.), K2CO3 (1.5 equiv.), DMF/THF, 61–90% (e) N(C4H9)4N3 (2 equiv.), DMF, 50 °C, 90–95%; (f) PMe3 (3 equiv.), THF/NaOH (0.05 M), 50–80%; (g) 1 (1 equiv.), pyr (1.5 equiv.), DCM/THF, 70%; (h) BBr3 (7 equiv.), DCM, −78 °C, 7–16%. :1), 70 °C, 2 h 70% over two steps; (d) Br(CH2)nCl or Br(C2H4O)2 C2H4N3 (1.5 equiv.), K2CO3 (1.5 equiv.), DMF/THF, 61–90% (e) N(C4H9)4N3 (2 equiv.), DMF, 50 °C, 90–95%; (f) PMe3 (3 equiv.), THF/NaOH (0.05 M), 50–80%; (g) 1 (1 equiv.), pyr (1.5 equiv.), DCM/THF, 70%; (h) BBr3 (7 equiv.), DCM, −78 °C, 7–16%. | ||
Tri-O-benzyl quercetin 2 was functionalized with 1-bromo-3-chloropropane, 1-bromo-5-chloropentane, 1-bromo-7-azidoheptane, or 1-bromo-8-azido-triethylene glycol depending on the analogue synthesized. Treatment with tetrabutylammonium azide produced a terminal azide, which was reduced with trimethylphosphine. In the cases of analogues M01 to M04, this could be coupled directly to caffeic acid through use of activated pentafluorophenyl caffeic ester 1. For analogues M05 to M11, the propylamine derivative 4 propyl was coupled to one of six chosen Fmoc-L-amino acids (with appropriate side chain protecting groups) (Scheme 2).15 The Fmoc group could be deprotected with 20% piperidine in dichloromethane followed by a second coupling with the pentafluorophenyl caffeic ester 1. Deprotection of the benzyl ethers and amino acid protecting groups was achieved through treatment with BBr3. While this synthesis allowed the construction of a small library of analogues, the final deprotection step in particular was prohibitively low yielding, posing a significant barrier to future applications. The continued study of any promising analogues required an improved synthesis, as will be explored below.
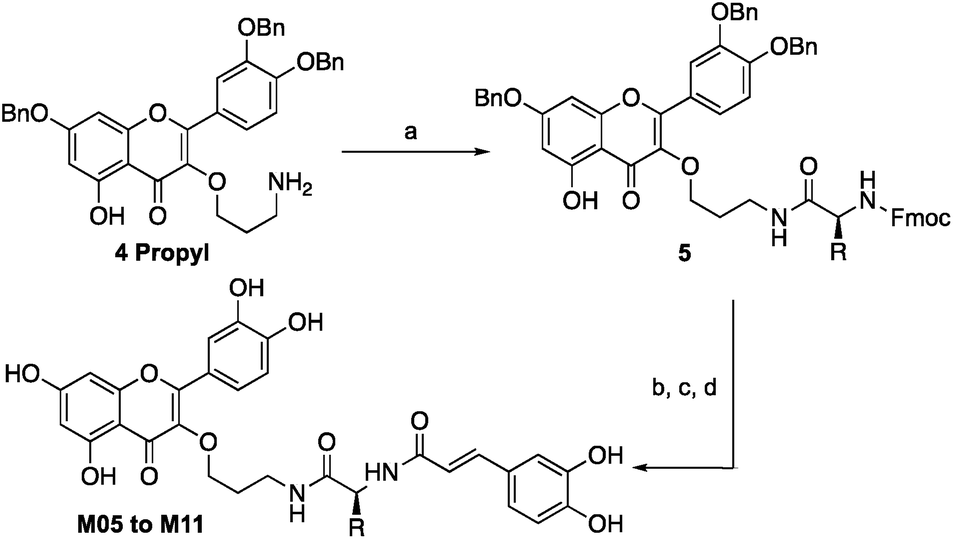 | ||
| Scheme 2 Synthesis of analogues M05 to M11: (a) Fmoc-L-Xaa-pfp (1 equiv.), pyr (1 equiv.), DCM, 75–90%; (b) 20% piperidine/DCM, 1 h; (c) 1 (1 equiv.), pyr (1.5 equiv.), THF/DCM, 50–75%; (d) BBr3 (7 equiv.), DCM, −78 °C, 5–25%. | ||
The eleven analogues were tested as inhibitors of HPA as shown in Table 1. Analogues M01 and M02 both had a KI = 4 μM, demonstrating that joining the flavonol and caffeic acid groups with a simple linker could increase potency by 25 times compared to the lone flavonol (KI ∼ 100 μM).9,13 However, these analogues were substantially less potent than mini-MbA, despite having the potential to form all the same interactions within the amylase active site. M03 was no more potent than the lone flavonol, suggesting that its three-atom linker was too short to allow for proper orientation of the phenolic moieties in the active site. M04, which contained an eight-atom triethylene glycol-based linker (TEG), also failed to offer a significant increase in potency over the lone flavonol. Despite having a linker of similar length and flexibility to those of M01 and M02, the incorporation of a triethylene glycol-based chain in place of a simple alkyl chain led to a 21-fold drop in potency, potentially due to the differences in polarity.
| MbA analogue | Linker | K I against HPA (μM) |
|---|---|---|
| Quercetin/myricetin | — | ∼100 |
| M01 | Heptylene | 4.2 ± 0.8 |
| M02 | Pentylene | 4.0 ± 0.4 |
| M03 | Propylene | 110 ± 10 |
| M04 | TEG | 90 ± 20 |
| M05 | Gly | 60 ± 10 |
| M06 | Pro | 1.0 ± 0.1 |
| M07 | Thr | 50 ± 10 |
| M08 | Glu | 34 ± 5 |
| M09 | Phe | 2.8 ± 0.4 |
| M10 | Tyr | 0.07 ± 0.01 |
| M11 | DOPA | 0.06 ± 0.02 |
While amino acid based analogues M05, M07, and M08 offered only meagre increases in potency compared to the lone flavonol, M06 fared better with a KI = 1 μM, perhaps due to the limited conformational range of its proline pre-organising the ligand for binding.
Introduction of aromatic residues into the linkers also afforded an increase in binding affinity. The phenylalanine-based analogue M09 bound 22-fold tighter than the un-functionalized glycine-based analogue M05, suggesting that its aromatic side chain likely creates favorable π stacking interactions in the active site, contributing 1.8 kcal mol−1 to binding affinity. Installation of hydroxyl groups onto the phenyl ring, as seen with the tyrosine-based analogue M10 and the DOPA-based analogue M11, produced a further 40 to 47-fold increase in potency relative to M09, equating to a further ∼2.5 kcal mol−1 contribution to the binding affinity. With inhibition constants of 70 and 60 nM, M10 and M11 bind approximately six times more tightly than the mini-MbA that inspired their synthesis. Thus not only have we replicated the affinity of the natural product with a simple, synthetically accessible analogue, but also we have substantially improved upon its binding affinity.
To gain insights into the binding modes of the best of these analogues, and particularly to understand the greatly improved affinity of M10 and M11, X-ray crystallographic analysis of analogues M06 and M10 in complex with HPA was pursued. The resulting crystal structures (PDBs 6OCN and 6OBX at 1.15 and 1.30 Å resolution) revealed the analogues in non-covalent complex within the enzyme active site. Importantly, an overlay of the M06/HPA and M10/HPA structures with that of mini-MbA/HPA showed essentially complete overlap of the flavonol and caffeic acyl moieties in each case, simplifying future design (Fig. 3a).
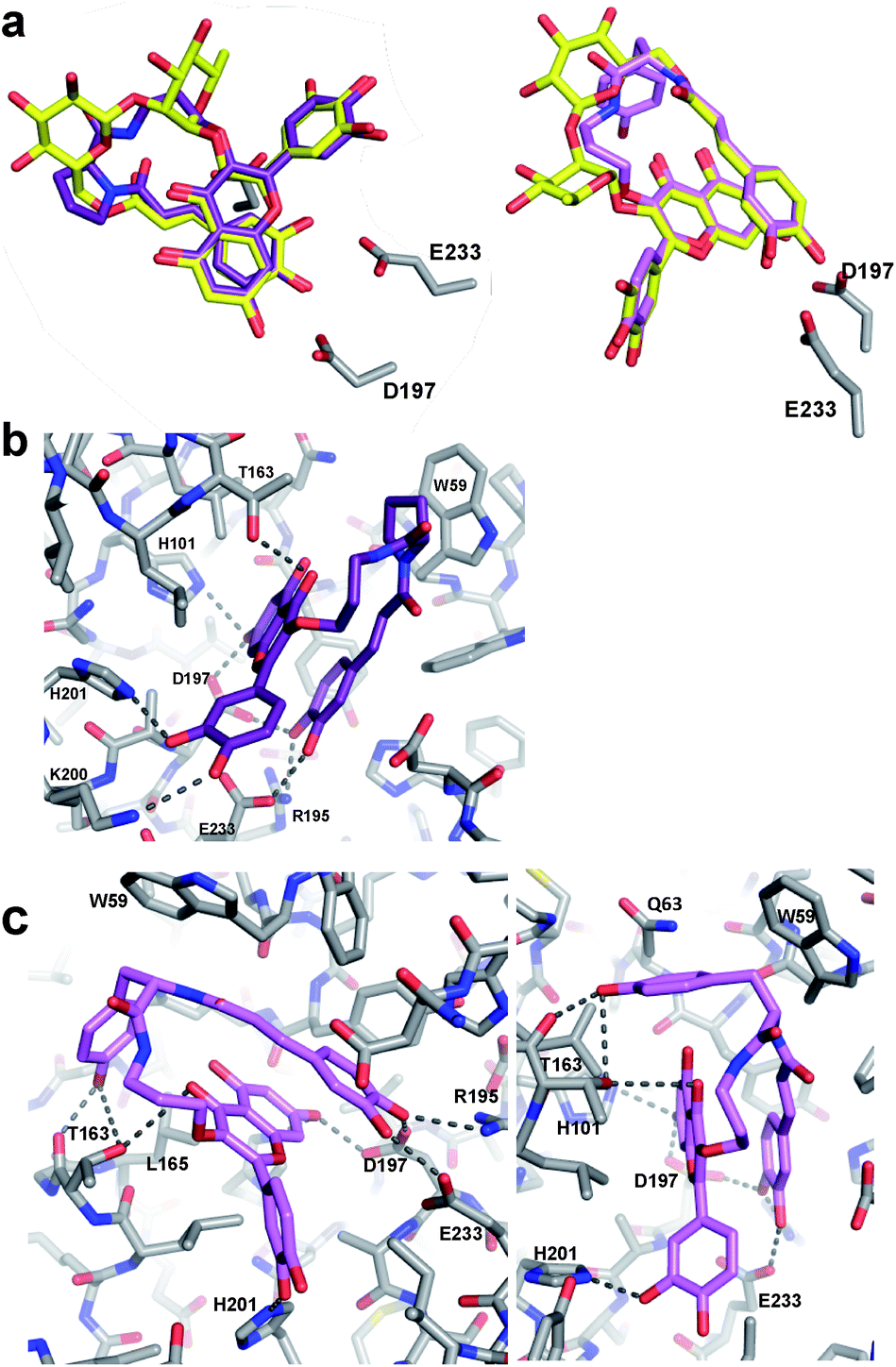 | ||
| Fig. 3 X-ray crystal structures of M06/HPA and M10/HPA complexes. (a) Overlays of M06 (purple) and M10 (pink) with mini-MbA (yellow) show very similar placement of flavonol and catechol groups. (b) Interactions between M06 (purple) and residues at the HPA active site (grey). Hydrogen bonds are shown as dashed lines. Polar interactions occurred between the flavonol and catechol of M06 and the enzyme. (c) Two different views of the interactions between M10 (pink) and residues of the HPA active site (grey). In addition to the polar contacts formed with the analogue flavonol and catechol, polar contacts occurred between the linker tyrosine and T163 of the enzyme. π-based interactions also occurred between the enzyme and aromatic tyrosine side chain. | ||
Proline-containing analogue M06 created all of the polar contacts within the HPA active site that were previously observed with mini-MbA (Fig. 3b). Hydrogen bonding was observed between the caffeic acid catechol and R195, D197 and E233 of the enzyme. The 7-OH of M06's flavonol formed hydrogen bonds with D197 and H201. A hydrogen bond was also observed between the C4 carbonyl of the flavonol and the side chain of T163. Meanwhile, the 3′-OH of the flavonol formed a hydrogen bond with the side chain of H201, as was also observed in the mini-MbA/HPA complex. This indicates that the more cost-effective quercetin was sufficient to form the necessary interactions within the active site. The absence of this 4′-O-disaccharide on mini-MbA opens this position up for hydrogen bonding with K200 in the active site, an interaction that is also seen with M06. In addition to these polar contacts, a π–CH stacking interaction appears to form between the proline side chain of M06 and W59 of the enzyme. Despite its high degree of alignment and reproducibility of polar contacts, M06 is still 2.5 times less potent than mini-MbA, highlighting the importance of MbA's glycosidic appendages in stabilization of a pre-stacked conformation.
Analogue M10 also formed all of the same hydrogen bonds within the amylase active site previously observed for mini-MbA and M06 (Fig. 3c). In addition, and presumably largely responsible for its affinity enhancement, the tyrosine of M10's linker formed hydrogen bonds with the main chain carbonyl and side chain hydroxyl of T163. These are accompanied by a number of CH–π–interactions between the tyrosine side chain and residues W59, Q63, and L165 of the enzyme.
To probe its specificity towards HPA, M10 was tested as an inhibitor of ten other glycosidases, including human intestinal maltase-glucoamylase and sucrase-isomaltase, and two intestinal bacterial α-amylases (Table 2). The analogue showed no inhibitory activity towards these α-glucosidases at a concentration over 100-fold higher than its KI value for HPA. M10 did show some inhibitory activity against the non-relevant S. cerevisiae α-glucosidase and Agrobacterium β-glucosidase. However, the binding affinity of M10 towards yeast α-glucosidase is actually lower than that of lone quercetin (IC50 = 7 μM).16 Comparison of these results with those of previous specificity analyses of mini-MbA and MbA indicated that M10 was in fact more selective for HPA over the intestinal glucosidases, since mini-MbA and MbA inhibited sucrase-isomaltase and B. fibrisolvens α-amylase, respectively.9,11 This bodes well for the use of M10 as a selective amylase inhibitor. It also implies that this pharmacophore may have restricted application to other glycosidases.
| Enzyme | M10 IC50 (μM) |
|---|---|
| Roseburia inulivorans amylase A | NI |
| Butyrivibrio fibrisolvens amylase B | NI |
| Homo sapiens maltase-glucoamylase | NI |
| H. sapiens C-term sucrase-isomaltase | NI |
| Saccharomyces cerevisiae α-glucosidase | 10.5 ± 0.7 |
| Agrobacterium β-glucosidase | 35.4 ± 5.7 |
| Green coffee bean α-galactosidase | NI |
| E. coli β-galactosidase | NI |
| Jack bean α-mannosidase | NI |
Inhibitor M10 demonstrates highly promising HPA inhibition, however the synthetic route that led to its discovery has a number of shortcomings that would hamper its development as a potential pharmaceutical. As such, an alternative approach was devised (Scheme 3). Selective azide reduction of 3 propyl could be carried out in the presence of 6 to give amide 7, which could then be globally deprotected to give amine 8. Treatment of crude amine 8 with 1 then delivered M10 in 49% yield over four steps, with no purification necessary between steps. This improved procedure delivers M10 in eight steps from rutin (24% overall yield), with further optimization certainly possible.
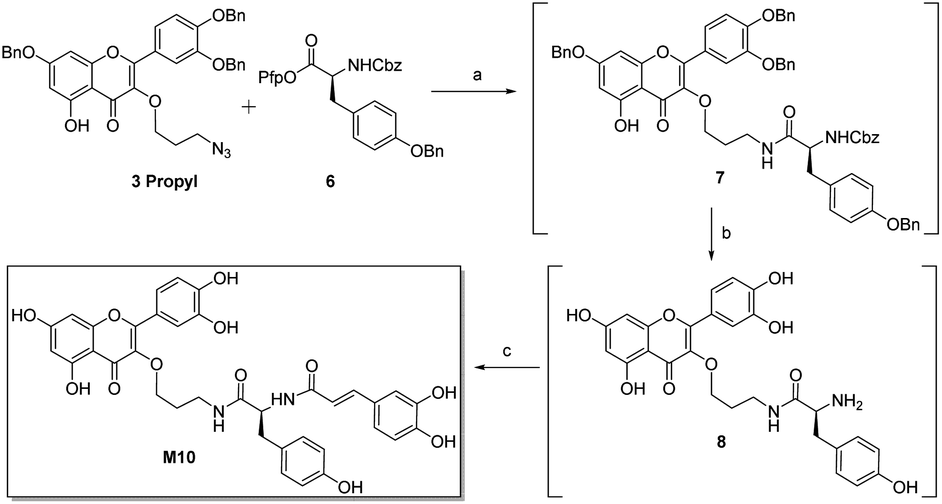 | ||
| Scheme 3 Improved synthesis of M10. (a) Lindlar catalyst (60% w/w), H2, DMF; (b) pentafluorophenol (4 equiv.), 5% Pd/Al2O3 (30% w/w), H2, DMF; (c) 1 (2 equiv.), Et3N (2.5 equiv.); 49%. | ||
Conclusions
The synthesis and kinetic analysis of MbA analogues M01 to M11 provides new insight into the structural requirements for HPA inhibition. The fact that most of the analogues bound more weakly than mini-MbA highlights the importance of MbA's constrained carbohydrate linker. Despite forming no direct interactions in the active site it appears to pre-stack the flavonol and caffeic acid moieties for optimal interaction. Initial exploration of the effect of incorporation of rigidity into the analogue linkers was achieved with the proline linker of M06. This increased potency some 60-fold compared to the non-functionalized glycine linker, with more possibly achievable by freezing out other motions. Affinity enhancement through the acquisition of stabilising interactions was explored through the incorporation of phenolic side chains to recruit some of the hydrophobic and hydrophilic interactions ordinarily developed by the starch substrate in the amylase active site.17 Indeed the phenolic linkers of M10 and M11 take advantage of both such features, rendering the inhibitors six times as potent as mini-MbA, and delivering a higher ligand efficiency than either MbA or mini-MbA (Table S1†). Further, and perhaps surprisingly, the specificity of inhibition is retained despite the reduction in complexity. Thus M10 and M11 represent amylase inhibitors that are both much more synthetically accessible and indeed more potent than the mini-MbA that inspired their development. With further optimization of their synthesis underway, pharmacokinetic and efficacy studies with these inhibitors are planned to determine their suitability for therapeutic usage.Experimental section
Please refer to ESI.†Conflicts of interest
The University of British Columbia has applied for patent protection on the inhibitors described.Acknowledgements
We thank Emily Kwan for purification of HPA and Ethan Goddard-Borger for useful discussions at the outset. We acknowledge funding from the Canadian Glycomics Network/Networks of Centres of Excellence (Project DO-2) DOI: 10.13039/501100009056. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.References
- World Health Organization, Global Report on Diabetes, 2006, vol. 6 Search PubMed.
- E. Adeghate, P. Schattner and E. Dunn, Ann. N. Y. Acad. Sci., 2006, 1084, 1 CrossRef CAS PubMed; K. G. Alberti and P. Z. Zimmet, Diabetic Med., 1998, 15(7), 539 CrossRef PubMed.
- A. D. Baron, Diabetes Res. Clin. Pract., 1998, 40, S51 CrossRef CAS PubMed.
- L. J. Scott and C. M. Spencer, Drugs, 2000, 59(3), 521 CrossRef CAS PubMed; K. Kaku, Expert Opin. Pharmacother., 2014, 15(8), 1181 CrossRef PubMed; M. Hanefeld, J. Diabetes Complicat., 1998, 12(4), 228 CrossRef PubMed; P. Phillips, J. Karrasch, R. Scott, D. Wilson and R. Moses, Diabetes Care, 2003, 26(2), 269 CrossRef PubMed.
- R. J. Andrade, M. Lucena, J. L. Vega, M. Torres, F. J. Salmerón, V. Bellot, M. D. García-Escaño and P. Moreno, Diabetes Care, 1998, 21(11), 2029 CrossRef CAS PubMed; G. C. Ezeji, T. Inoue, G. Bahtiyar and A. Sacerdote, BMJ Case Rep., 2015, 2015 Search PubMed; S. A. Cohen, Molecular and Cellular Pediatrics, 2016, 3(1), 5 CrossRef PubMed.
- G. D. Brayer, Y. Luo and S. G. Withers, Protein Sci., 1995, 4(9), 1730 CrossRef CAS PubMed; E. H. Rydberg, G. Sidhu, H. C. Vo, J. Hewitt, H. C. Côte, Y. Wang, S. Numao, R. T. MacGillivray, C. M. Overall, G. D. Brayer and S. G. Withers, Protein Sci., 1999, 8(3), 635 CrossRef PubMed.
- S. Numao, I. Damager, C. Li, T. M. Wrodnigg, A. Begum, C. M. Overall, G. D. Brayer and S. G. Withers, J. Biol. Chem., 2004, 279(46), 48282 CrossRef CAS PubMed.
- G. Horne, F. X. Wilson, J. Tinsley, D. H. Williams and R. Storer, Drug Discovery Today, 2011, 16(3–4), 107 CrossRef CAS PubMed.
- C. A. Tarling, K. Woods, R. Zhang, H. C. Brastianos, G. D. Brayer, R. J. Andersen and S. G. Withers, ChemBioChem, 2008, 9(3), 433 CrossRef CAS PubMed.
- V. G. Yuen, J. Coleman, S. G. Withers, R. J. Andersen, G. D. Brayer, S. Mustafa and J. H. McNeill, Mol. Cell. Biochem., 2016, 411(1–2), 373 CrossRef CAS PubMed.
- L. K. Williams, X. Zhang, S. Caner, C. Tysoe, N. T. Nguyen, J. Wicki, D. E. Williams, J. Coleman, J. H. McNeill, V. Yuen, R. J. Andersen, S. G. Withers and G. D. Brayer, Nat. Chem. Biol., 2015, 11(9), 691 CrossRef CAS PubMed.
- Revised KI for mini-MbA (Fig. S49†)..
- E. Lo Piparo, H. Scheib, N. Frei, G. Williamson, M. Grigorov and C. J. Chou, J. Med. Chem., 2008, 51(12), 3555 CrossRef CAS PubMed.
- H. Huang, Q. Jia, J. Ma, G. Qin, Y. Chen, Y. Xi, L. Lin, W. Zhu, J. Ding, H. Jiang and H. Liu, Eur. J. Med. Chem., 2009, 44(5), 1982 CrossRef CAS PubMed.
- M. Green and J. Berman, Tetrahedron Lett., 1990, 31(41), 2 CrossRef.
- K. Tadera, Y. Minami, K. Takamatsu and T. Matsuoka, J. Nutr. Sci. Vitaminol., 2006, 52(2), 149 CrossRef CAS PubMed.
- G. D. Brayer, G. Sidhu, R. Maurus, E. H. Rydberg, C. Braun, Y. Wang, N. T. Nguyen, C. M. Overall and S. G. Withers, Biochemistry, 2000, 39(16), 4778 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02610j |
| This journal is © The Royal Society of Chemistry 2019 |