
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct functionalization of white phosphorus with anionic dicarbenes and mesoionic carbenes: facile access to 1,2,3-triphosphol-2-ides†
Dennis
Rottschäfer
 ,
Sebastian
Blomeyer
,
Beate
Neumann
,
Hans-Georg
Stammler
and
Rajendra S.
Ghadwal
*
,
Sebastian
Blomeyer
,
Beate
Neumann
,
Hans-Georg
Stammler
and
Rajendra S.
Ghadwal
*
Molecular Inorganic Chemistry and Catalysis, Inorganic and Structural Chemistry, Center for Molecular Materials, Faculty of Chemistry, Universität Bielefeld, Universitätsstr. 25, Bielefeld, D-33615, Germany. E-mail: rghadwal@uni-bielefeld.de; Web: http://https://www.ghadwalgroup.de
First published on 18th October 2019
Abstract
A series of unique C2P3-ring compounds [(ADCAr)P3] (ADCAr = ArC{(DippN)C}2; Dipp = 2,6-iPr2C6H3; Ar = Ph 4a, 3-MeC6H44b, 4-MeC6H44c, and 4-Me2NC6H44d) are readily accessible in an almost quantitative yield by the direct functionalization of white phosphorus (P4) with appropriate anionic dicarbenes [Li(ADCAr)]. The formation of 1,2,3-triphosphol-2-ides (4a–4d) suggests unprecedented [3 + 1] fragmentation of P4 into P3+ and P−. The P3+ cation is trapped by the (ADCAr)− to give 4, while the putative P− anion reacts with additional P4 to yield the Li3P7 species, a useful reagent in the synthesis of organophosphorus compounds. Remarkably, the P4 fragmentation is also viable with the related mesoionic carbenes (iMICsAr) (iMICAr = ArC{(DippN)2CCH}, i stands for imidazole-based) giving rise to 4. DFT calculations reveal that both the C3N2 and C2P3-rings of 4 are 6π-electron aromatic systems. The natural bonding orbital (NBO) analyses indicate that compounds 4 are mesoionic species featuring a negatively polarized C2P3-ring. The HOMO−3 of 4 is mainly the lone-pair at the central phosphorus atom that undergoes σ-bond formation with a variety of metal-electrophiles to yield complexes [{(ADCAr)P3}M(CO)n] (M = Fe, n = 4, Ar = Ph 5a or 4-Me-C6H45b; M = Mo, n = 5, Ar = Ph 6; M = W, n = 5, Ar = 4-Me2NC6H47).
Introduction
The direct conversion of white phosphorus (P4) into useful organophosphorus compounds (OPCs) is of significant interest because this excludes the involvement of corrosive Cl2 gas that is required to convert P4 into PCl3, a common starting material for OPCs, and thus minimizes the waste and energy consumption.1 The activation and subsequent functionalization of P4 has therefore become a topical objective.2 Both transition metal3 as well as main-group element4 compounds have been shown to activate or functionalize P4.5 In particular, compounds featuring a low-valent main-group element have made significant advances over the past years.6Among nonmetals, the use of singlet carbenes7 has given new impetus to the field of P4 activation as it leads to the direct C–P bond formation (Fig. 1).8 Several stable carbenes (L1–L7) undergo reactions with P4 and the fate of P4 fragmentation to give Pn (n = 1, 2, 4, 8 or 12) containing products II–IX depends on the relative σ-donor/π-acceptor (ambiphilic) property as well as the steric demand of carbenes.7 Weakly π-accepting NHCs such as IPr (IPr = C{(DippN)CH}2) do not react with P4, however, related derivatives containing the [P2] or [P3−] moiety are accessible by alternative methods.9 Sterically demanding 1,3-bis(tBu)imidazol-2-ylidene (IBut) activates P4 in combination with B(C6F5)3 to give X.6h This frustrated Lewis pair (FLP) type reactivity10 led to the transformation of the classical NHC (IBut) into the mesoionic carbene (iMIC) L8 based on an 1,3-imidazole framework.
 | ||
| Fig. 1 Singlet carbene-mediated P4 activation and fragmentation to II–X and a plausible intermediate I. Mesoionic carbenes (iMICsAr, XI) and anionic dicarbenes (XII) ([Li+] = solvated lithium ion) investigated in the current study. | ||
iMICs are very potent σ-donor ligands with almost negligible π-acceptor properties.11 Nonetheless, no reaction of an iMIC alone with P4 has been described so far. This is most likely due to their limited synthetic accessibility.11a We recently reported12 C5-protonated iMICsAr (XI) as well as C4/C5-ditopic anionic dicarbenes [Li(ADCAr)] XII (Fig. 1) by the deprotonation of C2-arylated 1,3-imidazolium salts.13 The dicarbenes XII feature two adjacent C4/C5-nucleophilic sites, and thus are well endowed to affect unique dual P4 functionalization.5i,14 Herein, we showcase the direct functionalization of P4via unprecedented [3 + 1] fragmentation with [Li(ADCAr] and iMICsAr to give the 1,2,3-triphosphol-2-ide derivatives [(ADCAr)P3] (ADCAr = ArC{NDipp)C}2; Dipp = 2,6-iPr2C6H3; Ar = C6H54a, 3-MeC6H44b, 4-MeC6H44c, and 4-Me2NC6H44d) (Scheme 1).
 | ||
| Scheme 1 Synthesis of 1,2,3-triphosphol-2-ide derivatives 4a–4d by the direct fragmentation of white phosphorus with [Li(ADCAr)] (2a–2d). Reaction of iMICsAr3a and 3c with P4 to form 4a and 4c. | ||
Results and discussion
Treatment of [Li(ADCAr)] (2a–2d),12 which are readily accessible by the double deprotonation of C2-arylated 1,3-imidazolium salts 1a–1d with n-BuLi, with P4 at room temperature afforded the 1,2,3-triphosphol-2-ides 4a–4d as crystalline solids in almost quantitative yields (Scheme 1). Compounds 4a–4d are indefinitely stable (as solids as well as in solutions) under an inert gas atmosphere. The formation of 4a–4d indicates formal [3 + 1] fragmentation of P4 into P3+ and P−. The cationic P3+ species is captured by the ADCs to give 4a–4d, whereas the P− nucleophile reacts with additional P4 to eventually form the phosphide (P7)3− anion, a very common species in metal mediated fragmentation of P4.15 Indeed, Li3P7 can be isolated as a red-brown solid,15,16 which was confirmed by its reaction with (IPr)HCl to give (IPr)PH, reported previously using Na3P7.17Interestingly, treatment of iMICsAr3a and 3c with P4 also afforded, albeit in a lower yield, the corresponding products 4a and 4c, respectively. 1H NMR analyses of the crude reaction product indicate the presence of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 4a:1a and 4c:1c, suggesting the reprotonation of iMICsAr3a and 3b to 1,3-imidazolium salts 1a and 1c. Pure 4a and 4c can be extracted from the mixture using toluene.
:1 mixture of 4a:1a and 4c:1c, suggesting the reprotonation of iMICsAr3a and 3b to 1,3-imidazolium salts 1a and 1c. Pure 4a and 4c can be extracted from the mixture using toluene.
The 1H NMR spectra of 4a–4d are very symmetric and show two doublets and one septet for the isopropyl groups along with the signals due to the aryl protons. The 13C{1H} NMR resonances for 4a–4d are fully consistent with their 1H NMR spectra. The 13C{1H} NMR spectrum of 4a–4d each exhibits a doublet at 167 ppm (JP–C ≈ 84 Hz) for the backbone carbon atoms due to coupling with the 31P nucleus. The 31P{1H} NMR spectrum of 4a–4d each shows a doublet at ∼73 ppm and a triplet at 325 ± 6 ppm in 2:1 ratio (JP–P ≈ 500 Hz), indicating the presence of an AB2 type system with unsaturated P–P bonds.18
Solid-state molecular structures19 of 4a (Fig. 2), 4b (Fig. S47†), and 4c (Fig. S48†) reveal the presence of a C2P3-ring that is coplanar with the imidazole C3N2-ring plane. The metrical parameters of 4a–4c are comparable (Table S1†) and hence, for brevity, only 4a is discussed here. The P1–P2 bond length of 4a (2.103(1) Å) is intermediate of the sum of covalent radii for P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P double (2.04 Å) and P–P single (2.22 Å) bond lengths,20 indicating a partial π-bond character. Similarly, the C1–P1 (1.757(3) Å) bond length of 4a is shorter compared to a classical C–P single bond length (1.85 Å)15 but compares well with CP bond lengths (ca. 1.75 Å) of inversely polarized phosphaalkenes.17 The C1–C1′ (1.395(5) Å) and C2–N1 (1.404(3) Å) bond lengths of 4a are elongated in comparison with those of 1a (1.350(2) and 1.344(2) Å, respectively).13 The C1–C1′, C1/C2–P1 and P1–P2 bond lengths of 4a–4c are comparable with the corresponding bond lengths of triphospholide anions [P3C2R2]− (R = H, C–P 1.726(2) and 1.781(3), and P–P 2.081(1) and 2.094(1) Å; R = Ph, C–P 1.760(2) and 1.762(2), and P–P 2.091(2), 2.098(2) Å).21 Thus, 4a–4d may be considered as the neutral analogues of the triphospholide anions.
P double (2.04 Å) and P–P single (2.22 Å) bond lengths,20 indicating a partial π-bond character. Similarly, the C1–P1 (1.757(3) Å) bond length of 4a is shorter compared to a classical C–P single bond length (1.85 Å)15 but compares well with CP bond lengths (ca. 1.75 Å) of inversely polarized phosphaalkenes.17 The C1–C1′ (1.395(5) Å) and C2–N1 (1.404(3) Å) bond lengths of 4a are elongated in comparison with those of 1a (1.350(2) and 1.344(2) Å, respectively).13 The C1–C1′, C1/C2–P1 and P1–P2 bond lengths of 4a–4c are comparable with the corresponding bond lengths of triphospholide anions [P3C2R2]− (R = H, C–P 1.726(2) and 1.781(3), and P–P 2.081(1) and 2.094(1) Å; R = Ph, C–P 1.760(2) and 1.762(2), and P–P 2.091(2), 2.098(2) Å).21 Thus, 4a–4d may be considered as the neutral analogues of the triphospholide anions.
 | ||
| Fig. 2 Solid-state molecular structure of 4a. Hydrogen atoms are omitted for clarity. Symmetry code: 1 − X, +Y, 3/2 − Z. Selected experimental and calculated [M06-2X/def2SVP] bond lengths (Å) and angles (°): C1–C1′ 1.395(5) [1.402], N1–C1 1.404(3) [1.399], P1–C1 1.757(3) [1.764], P1–P2 2.103(1) [2.112], C1–P1–P2 94.9(1) [94.8], and P1–P2–P1′ 104.0(1) [104.1]. | ||
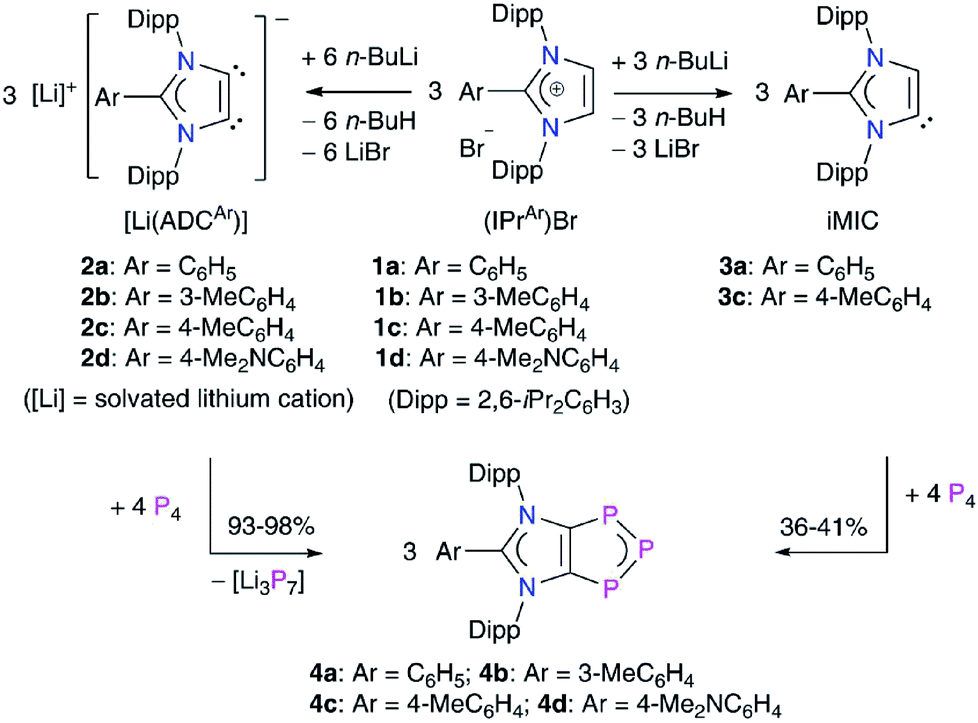
To gain further insight into the electronic structures of 4a–4d, we performed DFT calculations at the M06-2X/def2-TZVPP//M06-2X/def2-SVP level of theory. The computed NPA charges (Table S7†) at the P2 (−0.10e) and the C1/C2 (−0.24e) atoms are negative, whereas both the P1 atoms bear a positive charge (0.12e) (Scheme 2). The Wiberg Bond Indices (WBIs) of 1.40 (P–P), 1.18 (C–P), and 1.31 (C–C) indicate a partial double bond character. The WBI for the C1–C2 bond of 4a (1.31) is significantly smaller compared to that of the imidazolium salt 1a (WBI = 1.64). The WBIs for the C3–N1/2 bonds in 1 (1.28) and 4 (1.26) are, however, almost equal. Thus, compounds 4 may be described as mesoionic species with 6π-electron C2P3 and C3N2 aromatic systems (Scheme 2c). The nitrogen atoms contribute 4π-electrons to the C3N3-ring, whereas the P3 unit shares 4π-electrons with the C2P3-ring. The 2π-electrons of the C1C2 bond are pooled by both the ring systems. Indeed, calculated nucleus-independent chemical shift (NICS)22 values for 4a–4d (Table 1) suggest the aromaticity of the C3N2- and C2P3-rings. For comparison, we also calculated the NICS values for C6H6 and cyclobutadiene (CBD) molecules.
 | ||
| Scheme 2 (a) Calculated Wiberg Bond Indices (WBIs) and (b) NPA atomic charges of 1,2,3-triphosphol-1,2-ides 4. (c) Schematic representation of 4 with atom numberings. | ||
| C3N2/C2P3 | 4a | 4b | 4c | 4d | 5a | C6H6/CBDa |
|---|---|---|---|---|---|---|
| a CBD (cyclobutadiene). | ||||||
| NICS(0) | −7.08/−10.19 | −7.29/−10.31 | −7.29/−10.31 | −6.77/−10.37 | −7.57/−9.95 | −7.53/33.21 |
| NICS(1) | −5.94/−10.18 | −6.11/−10.28 | −6.11/−10.23 | −5.64/−10.21 | −6.29/−9.58 | −10.19/21.09 |
| NICS(2) | −2.43/−5.51 | −2.53/−5.21 | −2.53/−5.53 | −2.36/−5.52 | −2.52/−5.12 | −5.22/4.98 |
The anisotropy of current-induced density (AICD) has been used to study the aromatic behavior of several molecules.23 The AICD plots of 4a (Fig. 3) and 4b–4d (Fig. S62†) clearly show significant delocalization of the π-electrons of both the C3N2 and the C2P3 heterocycles, forming one coherent π-system.
 | ||
| Fig. 3 AICD plot (based on M06-2X/def2-TZVPP//def2-SVP calculations) of the C3N2P3 core of compound 4a. The isovalue was arbitrarily chosen to be 0.03, the magnetic field is orthogonal to the C2P3-plane and points towards the viewer, and thus clockwise ring currents represent aromatic systems, whereas counter-clockwise ring currents are indicative of antiaromatic systems. AICD plots of the complete molecules 4a–4d are given in the ESI.† | ||
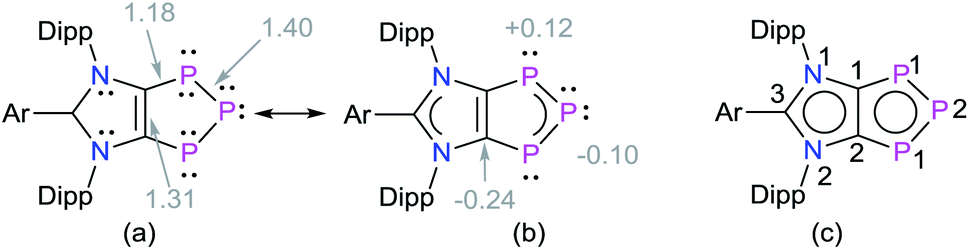
The HOMO of compounds 4a (Fig. 4) and 4b–4c (Fig. S58–S60†) corresponds to the π-orbitals of the C–P bonds with a small contribution from the lone pairs at the nitrogen atoms. The HOMO−1 corresponds mainly to the π-orbitals of the P3 and the C2 moieties of the C2P3-ring. Like in alkali metal 1,2,3-triphospholides,21b the analyses of frontier molecular orbitals, HOMO and HOMO−1 in particular, of 4a–4d reveal the mixing of phosphorus orbitals with lone-pair character amongst the π-manifold frontier orbitals. The HOMO−3 and HOMO−2 are the lone pairs on the central and neighbouring P atoms, respectively. The LUMO of 4a–4d corresponds to the π* orbital of the aryl group on the C3 carbon atom along with a p-orbital at the central phosphorus atom. The LUMO+2 corresponds mainly to the π*-orbitals of the C2P3 unit.
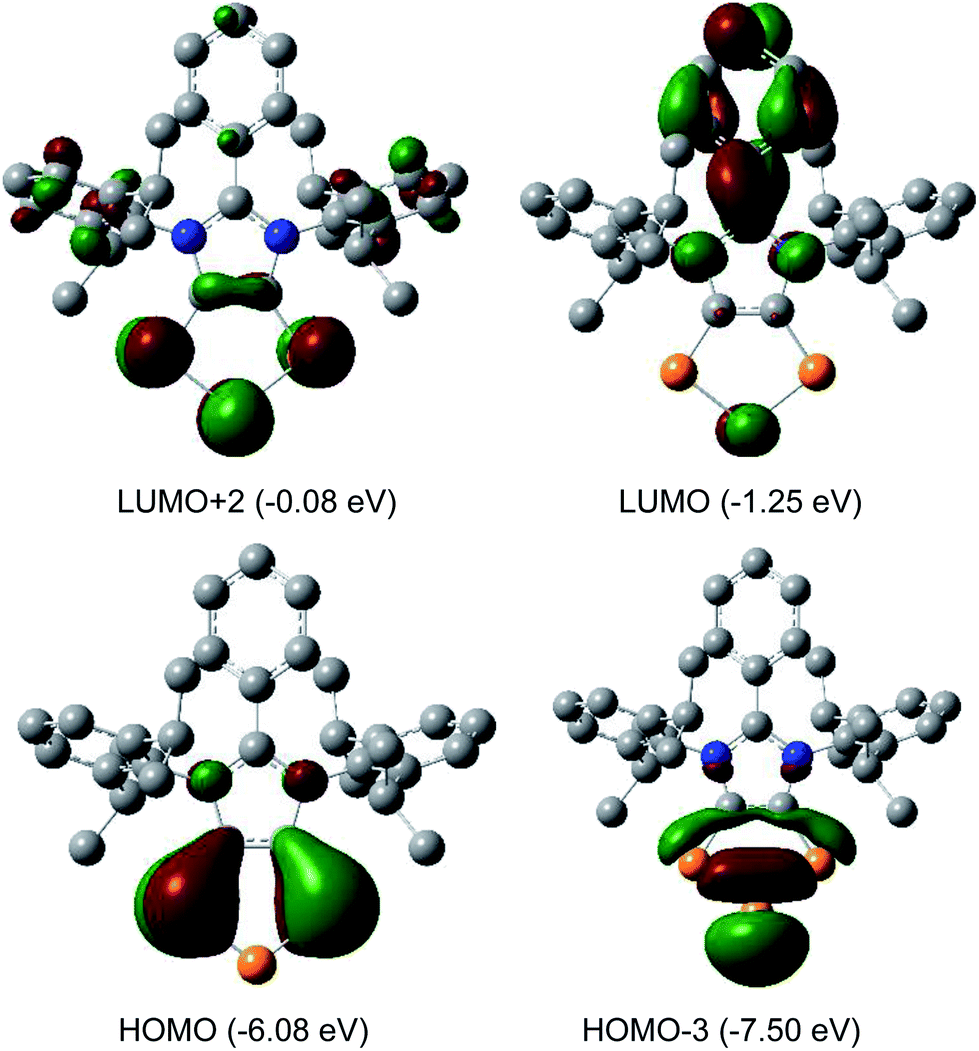 | ||
| Fig. 4 Selected MOs of 4a calculated at the M06-2X/def2-TZVPP//def2-SVP level of theory with an isovalue of 0.04. Hydrogen atoms were omitted for clarity. | ||
The intriguing electronic structures of 4 prompted us to investigate their ligand properties as they may function as neutral two electron σ-donors (via phosphorus atoms) and/or 6π-electron η5-donors (C2P3-ring) like triphospholide21 and cyclopentadienyl anions. Treatment of 4a, 4b, and 4c with Fe2(CO)9 or M(CO)5(THF) (M = Mo or W) led to the formation of related complexes 5a, 5b, 6, and 7 (Scheme 3). In all complexes, the central phosphorus atom functions as a two-electron σ-donor ligand to bind to the M(CO)n moiety. This is consistent with the NBO analysis, which suggests higher charge accumulation at the central phosphorus atom with respect to that of the neighbouring phosphorus atoms. The 31P{1H} NMR spectrum of 5a, 5b, 6, and 7 each exhibits one doublet (5a: 145; 5b: 145; 6: 160; 7: 157 ppm) and one triplet (5a: 316; 5b: 315; 6: 299; 7: 250 ppm), which have been upfield shifted with respect to that of 4a (173, 332 ppm), 4b (173, 331 ppm), and 4d (173, 319 ppm). In the 31P{1H} NMR spectrum of 7, the triplet at 250 ppm is accompanied by the 183W satellites (JP–W = 202 Hz).
 | ||
| Scheme 3 Synthesis of complexes [{(ADCAr)P3}M(CO)n] 5a, 5b, 6, and 7. | ||
The iron atom in 5a (Fig. 5) and 5b (Fig. S49†) each features a trigonal-bipyramidal geometry. Three equatorial positions are occupied by CO ligands, whereas one CO and one 4a or 4b are present at the axial positions. The P–Fe bond length of 5a (2.240(1) Å) compares well with that of triphosphaindane-derived P3Fe3 iron-carbonyl clusters (av. 2.244 Å).24 Interestingly, the metrical parameters of the C3N2- and C2P3-rings of 5a and 5b are very similar to those of the precursors 4a and 4b, respectively. This indicates that the aromatic π-systems remain virtually intact upon complexation of 4a and 4b with the Fe(CO)4 fragment. As expected, the molecular structures of 6 (Fig. S50†) and 7 (Fig. S51†) feature six-fold coordinated Mo and W atoms, respectively.
 | ||
| Fig. 5 Solid-state molecular structure of 5a. Hydrogen atoms and one solvent toluene molecule are omitted for clarity. Selected bond lengths (Å) and angles (°): C2–C3 1.394(2), C2–N1 1.399(1), C3–N2 1.403(1), C2–P1 1.756(1), C3–P3 1.764(1), P1–P2 2.081(1), P2–P3 2.089(1), P2–Fe1 2.240(1), Fe1–C34 1.791(1), Fe1–C35 1.797(2), Fe1–C36 1.810(1), Fe1–C37 1.783(1), P1–P2–P3 109.1(2), and P2–Fe1–C37 178.5(1). | ||
DFT calculations suggest that the HOMO of 5a (Fig. 6) is mainly located at the iron atom and has some contribution from the π-orbitals of the C–C and one P–P bond. The LUMO is comparable to that of 4a but is lower in energy by −0.26 eV, indicating metal-to-ligand π-back bonding. The aromaticity of the C2P3 moiety in 5a remains almost unchanged as indicated by NICS(0) = −9.95, NICS(1) = −9.58, and NICS(2) = −5.12 values. The aromaticity of 5a is also corroborated by the AICD plot (Fig. S62†).
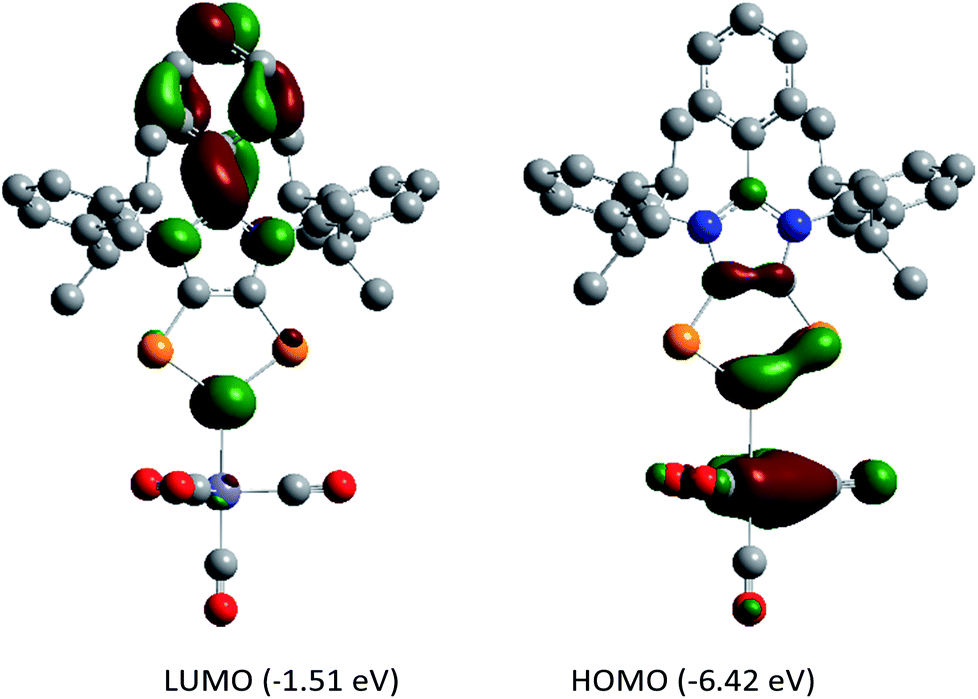 | ||
| Fig. 6 Frontier molecular orbitals of 5a calculated at the M06-2X/def2-TZVPP//def2-SVP level of theory. The isovalue was arbitrarily chosen to be 0.04. Hydrogen atoms were omitted for clarity. | ||
Experimental
All syntheses and manipulations were carried out under an inert gas atmosphere (Ar or N2) using standard Schlenk techniques or a glove box (MBraun LABMasterPro). Solvents were dried over appropriate drying agents, distilled, and stored over a 3 Å molecular sieve prior to use. Deuterated solvents were dried over appropriate drying agents, distilled, and stored inside a glove box. NMR spectra were recorded on a Bruker Avance III 500 or a Bruker Avance III 500 HD spectrometer. Chemical shifts (in δ, ppm) are referenced to the solvent residual signals of CD2Cl2: 1H 5.32; 13C 53.84 and C6D6: 1H 7.16; 13C 128.62 ppm. ESI mass spectra were recorded using an Esquire 3000 ion trap mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a nano-ESI source. Samples were dissolved in CH2Cl2 and introduced by static nano-ESI using in-house pulled glass emitters. Nitrogen served as a nebulizer gas as well as a dry gas and was generated by a Bruker nitrogen generator NGM 11. Helium served as a cooling gas for the ion trap. The mass axis was externally calibrated with ESI-L Tuning Mix (Agilent Technologies, Santa Clara, CA, USA) as the calibration standard. UV/vis spectra were recorded on a ThermoFisher Evolution 300 spectrophotometer. Infrared spectra were recorded using a Bruker Alpha-T FTIR spectrometer equipped with a Bruker Platinum diamond ATR unit. Melting points were measured with a Büchi B-545 melting point apparatus. (IPrAr)Br salts 1a–1d (Ar = Ph, 3-MeC6H4, 4-MeC6H4 or 4-Me2NC6H4) were synthesized following the reported method.13an-BuLi (2.5 M solution in hexanes, Sigma-Aldrich) was used as received. White phosphorus was sublimed and stored inside a glovebox. Commercially available Fe2(CO)9 (Sigma-Aldrich), Mo(CO)6 (Fluorochem), and W(CO)6 (Sigma-Aldrich) were used as supplied.Synthesis of compound (ADCPh)P3 (4a)
To a 15 mL THF suspension of 1a (0.88 g, 1.6 mmol), n-BuLi (2.5 M, 1.4 mL, 3.5 mmol) was added at −40 °C. The resulting reaction mixture was stirred at −20 °C for 1 h and then at room temperature (25 °C) for 15 minutes to obtain a clear light brown solution of 2a.12 To this solution, solid P4 (0.4 g, 3.2 mmol) was added in one portion and then stirred overnight at rt. The resulting dark suspension was refluxed for 2 h and the red insoluble material (probably a mixture of Li3P7 and other polyphosphides) was removed by filtration. The volatiles from the filtrate were removed under vacuum to give a brown residue, which was extracted with dichloromethane, dried under vacuum, washed with toluene (2 × 10 mL), and re-dried to obtain compound 4a as a yellow solid. Yield: 96% (0.86 g). Single crystals suitable for X-ray diffraction analysis were grown by storing a saturated toluene solution of 4a at −24 °C for three days. Mp: 343 °C. Elem. anal. (%), calcd for C33H39N2P3 (556.6): C, 71.21; H, 7.06; N, 5.03; found: C, 71.02; H, 6.84; N, 4.87. 1H NMR (500 MHz, CD2Cl2, 298 K): δ = 7.59 (t, J = 7.7 Hz, 2H, p-C6H3), 7.38 (d, J = 7.8 Hz, 4H, m-C6H3), 7.34 (t, J = 6.6 Hz, 1H, p-C6H5), 7.23–7.18 (m, 4H, o-, m-C6H5), 2.62 (sept, J = 6.6 Hz, 4H, CH(CH3)2), 1.26 (d, J = 6.6 Hz, 12H, CH(CH3)2), and 1.03 (d, J = 6.7 Hz, 12H, CH(CH3)2) ppm. 13C{1H} NMR (126 MHz, CD2Cl2, 298 K): δ = 167.6 (d, JP–C = 84.4 Hz, CP); 149.2 (NCN); 146.2, 133.5, 132.0, 131.8, 129.8, 129.1, 125.8, and 123.8 (C6H3, C6H5); 29.7 (CH(CH3)2); 26.1 and 23.5 (CH(CH3)2) ppm. 31P{1H} NMR (202 MHz, CD2Cl2, 298 K): δ = 332.3 (t, JP–P = 506 Hz) and 173.7 (d, JP–P = 506 Hz) ppm. MS (ESI, positive mode): m/z = 557.3 [4a + H]+. UV-vis (λ/nm ε (M−1 cm−1)): 282 (22336), 346 (31017), and 361 (31397).Compounds 4b–4d were prepared by employing a similar protocol to that described for 4a using the appropriate precursor 1b, 1c or 1d, n-BuLi, and P4.
(ADC3-Tol)P3 (4b)
Yield: 98% (0.90 g). Mp: 338–341 °C. Elem. anal. (%), calcd for 4b, C34H41N2P3, (570.6): C, 71.56; H, 7.24; N 4.91; found C, 70.64; H, 7.33; N 4.68. 1H NMR (500 MHz, CD2Cl2, 298 K): δ = 7.57 (t, J = 7.8 Hz, 2H, p-C6H3), 7.35 (d, J = 7.8 Hz, 4H, m-C6H3), 7.14 (d, J = 7.6 Hz, 1H, o-C6H4), 7.08 (t, J = 7.8 Hz, 1H, m-C6H4), 7.02 (s, 1H, o-C6H4), 6.96 (d, J = 7.8 Hz, 1H, p-C6H4), 2.60 (sept, J = 6.7 Hz, 4H, CH(CH3)2), 2.10 (s, 3H, CH3), 1.24 (d, J = 6.7 Hz, 12H, CH(CH3)2), and 1.03 (d, J = 6.8 Hz, 12H, CH(CH3)2) ppm. 13C{1H} NMR (126 MHz, CD2Cl2, 298 K): δ = 167.5 (d, JP–C = 84.5 Hz, CP); 149.6 (NCN); 146.3, 139.3, and 133.6 (i-C6H4 and i-, m-C6H3); 132.7 (o-C6H4); 131.7 (p-C6H3); 130.5 (o-C6H4); 129.5 and 128.9 (m-C6H4); 126.9, 125.8, and 123.71 (m-C6H3); 29.7 (C(CH3)2); 26.2 and 23.5 (C(CH3)2); 21.2 (CH3) ppm. 31P{1H} NMR (202 MHz, CD2Cl2, 298 K): δ = 331.1 (t, JP–P = 506 Hz) and 173.6 (d, JP–P = 506 Hz) ppm. MS (ESI, positive mode): m/z = 571.3 [4b + H]+. UV-vis (λ/nm ε (M−1 cm−1)): 280 (25637), 345 (36323), and 361 (36539).(ADC4-Tol)P3 (4c)
Yield: 93% (0.85 g). Single crystals suitable for X-ray diffraction were obtained by storing a saturated toluene solution of 4c for three days at −24 °C. Mp: 339–343 °C. Elem. anal. (%), calcd for C34H41N2P3 (570.6): C, 71.56; H, 7.24; N, 4.91; found C, 71.11; H, 7.06; N, 4.65. 1H NMR (500 MHz, CD2Cl2, 298 K): δ = 7.59 (t, J = 7.8 Hz, 2H, p-C6H3), 7.38 (d, J = 7.8 Hz, 4H, m-C6H3), 7.05 (d, J = 8.4 Hz, 2H, C6H4), 7.02 (d, J = 8.3 Hz, 2H, C6H4), 2.61 (sept, J = 6.8 Hz, 4H, CH(CH3)2), 2.23 (s, 3H, CH3), 1.26 (d, J = 6.7 Hz, 12H, CH(CH3)2), and 1.03 (d, J = 6.9 Hz, 12H, CH(CH3)2) ppm. 13C{1H} NMR (126 MHz, CD2Cl2, 298 K): δ = 167.5 (d, JP–C = 84.1 Hz, CP); 146.2 (NCN); 143.1, 133.7, 131.7, 129.8, 129.7, 125.8, and 120.9 (C6H3 and C6H4); 29.7 (CH(CH3)2); 26.1 and 23.5 (CH(CH3)2); 21.7 (CH3) ppm. 31P{1H} NMR (202 MHz, CD2Cl2, 298 K): δ = 329.9 (t, JP–P = 506 Hz) and 173.6 (d, JP–P = 506 Hz) ppm. MS (ESI, positive mode): m/z = 571.3 [4c + H]+. UV-vis (λ/nm ε (M−1 cm−1)): 283 (23295), 336 (28771), 346 (29238), and 362 (29676).(ADC4-DMP)P3 (4d)
Yield: 94% (0.93 g). Mp: 270–273 °C (decomp.). Elem. anal. (%), calcd for C35H44N3P3 (599.3): C, 70.10; H, 7.40; N, 7.01; found: C, 69.66; H, 7.18; N 6.59. 1H NMR (500 MHz, CD2Cl2, 298 K): δ = 7.56 (t, J = 7.8 Hz, 2H, p-C6H3), 7.35 (d, J = 7.8 Hz, 4H, m-C6H3), 6.90 (d, J = 9.0 Hz, 2H, C6H4), 6.28 (d, J = 9.0 Hz, 2H, C6H4), 2.88 (s, 6H, N(CH3)2), 2.67 (sept, J = 6.8 Hz, 4H, CH(CH3)2), 1.24 (d, J = 6.7 Hz, 12H, CH(CH3)2), and 0.97 (d, J = 6.8 Hz, 12H, CH(CH3)2) ppm. 13C{1H} NMR (126 MHz, CD2Cl2, 298 K): δ = 167.0 (d, JP–C = 82.8 Hz, CP); 151.9 (NCN); 146.2, 138.4, 134.6, 131.4, 130.8, 126.3, 125.8, and 111.1 (C6H3 and C6H4); 40.0 (N(CH3)2); 29.6 (CH(CH3)2); 25.7 and 23.5 (CH(CH3)2) ppm. 31P{1H} NMR (202 MHz, CD2Cl2, 298 K): δ = 319.5 (t, JP–P = 504 Hz) and 173.3 (d, JP–P = 504 Hz) ppm. MS (ESI, positive mode): m/z = 600.3 [4d + H]+. UV-vis (λ/nm (ε M−1 cm−1)): 285 (37768), 322 (46655), 343 (47543), 366 (48310), and 398 (45288).PH17 by NMR spectroscopy. 1H NMR (500 MHz, C6D6, 298 K): δ = 7.23 (t, J = 7.7 Hz, 2H, p-C6H3), 7.14 (d, J = 7.6 Hz, 4H, m-C6H3), 6.18 (s, 2H, NCH), 3.06 (sept, J = 6.7 Hz, 4H, CH(CH3)2), 1.92 (d, JPH = 165.2 Hz, 1H, PH), 1.47 (d, J = 6.8 Hz, 12H, CH(CH3)2), and 1.15 (d, J = 6.9 Hz, 12H, CH(CH3)2) ppm. 31P NMR (C6D6, 298 K, 500 MHz): δ = −134.4 (d, JP–H = 165.2 Hz) ppm. 31P{1H} NMR (C6D6, 298 K, 500 MHz): δ = −134.4 ppm.
Alternative synthesis of 4a and 4c from iMICsAr2a and 2c
To a 15 mL THF suspension of 1a (0.98 g, 1.8 mmol), n-BuLi (2.5 M, 0.8 mL, 2.0 mmol) was added at −40 °C. The resulting brown solution was stirred at −20 °C for 45 min and then for 15 min at rt. Subsequently, P4 (0.3 g, 2.4 mmol) was added in one portion and the resulting reaction mixture was stirred overnight at rt. The volatiles were removed under vacuum to obtain a dark residue, which was extracted with toluene (3 × 10 mL). The filtrate was dried in a vacuum to obtain 4a. Yield: 41% (0.4 g).Syntheses of complexes 5a, 5b, 6, and 7
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) /cm−1 = 2041, 1966, 1937, and 1919.
/cm−1 = 2039, 2007, 1962, and 1921.
/cm−1 = 2065, 2051, 1945, 1925, and 1911.
/cm−1 = 2063, 1978, 1925, and 1907.
/cm−1 = 2041, 1966, 1937, and 1919.
/cm−1 = 2039, 2007, 1962, and 1921.
/cm−1 = 2065, 2051, 1945, 1925, and 1911.
/cm−1 = 2063, 1978, 1925, and 1907.
Conclusions
In conclusion, the direct functionalization of white phosphorus (P4) with anionic dicarbenes (ADCs) (2a–2d) as well as with mesoionic carbenes (iMICsAr) (3a and 3c) that leads to the formation of unique 1,2,3-triphosphol-2-ide derivatives 4a–4d as crystalline solids up to 98% yield has been reported. The isolation of C2P3-heterocycles 4a–4d is unprecedented in the P4 activation by singlet carbenes and main-group compounds. The formation of 4a–4d suggests unique [3 + 1] fragmentation of P4 into P3+ and P−. The former species combines with an ADC to give 4a–4d, whereas the latter reacts with additional P4 to form (P7)3− that can be isolated as Li3P7. Electronic structures of 4a–4d have been analyzed by computational studies, which, along with the crystallographic data, show that both C3N2- and C2P3-rings of 4a–4d are 6π-electron aromatic systems. Thus, 4a–4d can be considered as neutral analogues of cyclopentadienyl anions. The C2P3-ring of 4a–4d is negatively polarized towards the central phosphorus atom, and hence 4a–4d may also function as potent two-electron σ-donor ligands. This feature has been demonstrated with the isolation of transition metal complexes 5a, 5b, 6, and 7. Consequently, 4a–4d have interesting perspectives as ligands in main-group element as well as transition-metal chemistry and catalysis. Further investigations in this direction are currently underway in this laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support from the Deutsche Forschungsgemeinschaft (DFG) and thank Professor Norbert W. Mitzel for his continuous support. The authors gratefully acknowledge the support by computing time provided by the Paderborn Center for Parallel Computing (PC2).References
- D. E. C. Corbridge, Phosphorus: Chemistry, Biochemistry and Technology, CRC Press, 6th edn, 2016 Search PubMed.
- J. M. Lynam, Angew. Chem., Int. Ed., 2008, 47, 831–833 CrossRef CAS PubMed.
- (a) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed; (b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed; (c) M. Peruzzini, L. Gonsalvi and A. Romerosa, Chem. Soc. Rev., 2005, 34, 1038–1047 RSC; (d) C. C. Cummins, Angew. Chem., Int. Ed., 2006, 45, 862–870 CrossRef CAS PubMed.
- (a) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed; (b) S. Khan, S. S. Sen and H. W. Roesky, Chem. Commun., 2012, 48, 2169–2179 RSC; (c) N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342–1359 CrossRef CAS; (d) M. H. Holthausen and J. J. Weigand, Chem. Soc. Rev., 2014, 43, 6639–6657 RSC.
- (a) S. Heinl, S. Reisinger, C. Schwarzmaier, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 7639–7642 CrossRef CAS PubMed; (b) W. W. Seidel, O. T. Summerscales, B. O. Patrick and M. D. Fryzuk, Angew. Chem., Int. Ed., 2009, 48, 115–117 CrossRef CAS PubMed; (c) B. M. Cossairt and C. C. Cummins, Angew. Chem., Int. Ed., 2008, 47, 169–172 CrossRef CAS PubMed; (d) B. M. Cossairt and C. C. Cummins, Angew. Chem., Int. Ed., 2008, 47, 8863–8866 CrossRef CAS PubMed; (e) N. A. Piro and C. C. Cummins, J. Am. Chem. Soc., 2008, 130, 9524–9535 CrossRef CAS PubMed; (f) A. R. Fox, C. R. Clough, N. A. Piro and C. C. Cummins, Angew. Chem., Int. Ed., 2007, 46, 973–976 CrossRef CAS PubMed; (g) N. A. Piro, J. S. Figueroa, J. T. McKellar and C. C. Cummins, Science, 2006, 313, 1276–1279 CrossRef CAS PubMed; (h) B. M. Cossairt, M.-C. Diawara and C. C. Cummins, Science, 2009, 323, 602 CrossRef CAS PubMed; (i) S. Du, J. Yin, Y. Chi, L. Xu and W.-X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 15886–15890 CrossRef CAS PubMed; (j) C. Schwarzmaier, A. Noor, G. Glatz, M. Zabel, A. Y. Timoshkin, B. M. Cossairt, C. C. Cummins, R. Kempe and M. Scheer, Angew. Chem., Int. Ed., 2011, 50, 7283–7286 CrossRef CAS PubMed; (k) C. Camp, L. Maron, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2014, 136, 17652–17661 CrossRef CAS PubMed; (l) S. Pelties, D. Herrmann, B. de Bruin, F. Hartl and R. Wolf, Chem. Commun., 2014, 50, 7014–7016 RSC; (m) S. L. Yao, N. Lindenmaier, Y. Xiong, S. Inoue, T. Szilvasi, M. Adelhardt, J. Sutter, K. Meyer and M. Driess, Angew. Chem., Int. Ed., 2015, 54, 1250–1254 CrossRef CAS PubMed; (n) F. Dielmann, A. Timoshkin, M. Piesch, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2017, 56, 1671–1675 CrossRef CAS PubMed; (o) F. Spitzer, C. Graßl, G. Balázs, E. M. Zolnhofer, K. Meyer and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 4340–4344 CrossRef CAS PubMed; (p) F. Spitzer, C. Graßl, G. Balázs, E. Mädl, M. Keilwerth, E. M. Zolnhofer, K. Meyer and M. Scheer, Chem.–Eur. J., 2017, 23, 2716–2721 CrossRef CAS PubMed; (q) B. L. Tran, M. Singhal, H. Park, O. P. Lam, M. Pink, J. Krzystek, A. Ozarowski, J. Telser, K. Meyer and D. J. Mindiola, Angew. Chem., Int. Ed., 2010, 49, 9871–9875 CrossRef CAS PubMed; (r) J. E. Borger, M. S. Bakker, A. W. Ehlers, M. Lutz, J. Chris Slootweg and K. Lammertsma, Chem. Commun., 2016, 52, 3284–3287 RSC.
- (a) S. Khan, R. Michel, J. M. Dieterich, R. A. Mata, H. W. Roesky, J.-P. Demers, A. Lange and D. Stalke, J. Am. Chem. Soc., 2011, 133, 17889–17894 CrossRef CAS PubMed; (b) J. W. Dube, C. M. E. Graham, C. L. B. Macdonald, Z. D. Brown, P. P. Power and P. J. Ragogna, Chem.–Eur. J., 2014, 20, 6739–6744 CrossRef CAS PubMed; (c) R. J. Schwamm, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2016, 55, 14798–14801 CrossRef CAS PubMed; (d) M. Arrowsmith, M. S. Hill, A. L. Johnson, G. Kociok-Köhn and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 7882–7885 CrossRef CAS PubMed; (e) K. X. Bhattacharyya, S. Dreyfuss, N. Saffon-Merceron and N. Mézailles, Chem. Commun., 2016, 52, 5179–5182 RSC; (f) S. Khan, R. Michel, S. S. Sen, H. W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786–11789 CrossRef CAS PubMed; (g) W. Lu, K. Xu, Y. Li, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2018, 57, 15691–15695 CrossRef CAS PubMed; (h) D. Holschumacher, T. Bannenberg, K. Ibrom, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2010, 39, 10590–10592 RSC; (i) J. E. Borger, A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2014, 53, 12836–12839 CrossRef CAS PubMed.
- (a) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530–5533 CrossRef CAS PubMed; (b) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052–7055 CrossRef CAS PubMed; (c) C. D. Martin, C. M. Weinstein, C. E. Moore, A. L. Rheingold and G. Bertrand, Chem. Commun., 2013, 49, 4486–4488 RSC; (d) C. L. Dorsey, B. M. Squires and T. W. Hudnall, Angew. Chem., Int. Ed., 2013, 52, 4462–4465 CrossRef CAS PubMed; (e) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180–14181 CrossRef CAS PubMed.
- (a) J. E. Borger, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, Chem.–Eur. J., 2017, 23, 11738–11746 CrossRef CAS PubMed; (b) D. Tofan and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 7516–7518 CrossRef CAS PubMed.
- (a) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 CrossRef CAS PubMed; (b) M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078–11082 CrossRef CAS PubMed.
- (a) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed; (c) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- (a) E. Aldeco-Perez, A. J. Rosenthal, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Science, 2009, 326, 556–559 CrossRef CAS PubMed; (b) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC; (c) G. Ung and G. Bertrand, Chem.–Eur. J., 2011, 17, 8269–8272 CrossRef CAS PubMed; (d) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759–4762 CrossRef CAS PubMed; (e) A. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef CAS PubMed.
- D. Rottschäfer, F. Ebeler, T. Strothmann, B. Neumann, H.-G. Stammler, A. Mix and R. S. Ghadwal, Chem.–Eur. J., 2018, 24, 3716–3720 CrossRef PubMed.
- (a) N. K. T. Ho, B. Neumann, H.-G. Stammler, V. H. Menezes da Silva, D. G. Watanabe, A. A. C. Braga and R. S. Ghadwal, Dalton Trans., 2017, 46, 12027–12031 RSC; (b) R. S. Ghadwal, S. O. Reichmann and R. Herbst-Irmer, Chem.–Eur. J., 2015, 21, 4247–4251 CrossRef CAS PubMed.
- (a) S. Du, J. Yang, J. Hu, Z. Chai, G. Luo, Y. Luo, W. X. Zhang and Z. Xi, J. Am. Chem. Soc., 2019, 141, 6843–6847 CrossRef CAS PubMed; (b) L. Xu, Y. Chi, S. Du, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2016, 55, 9187–9190 CrossRef CAS PubMed.
- M. Baudler and K. Glinka, Chem. Rev., 1993, 93, 1623–1667 CrossRef CAS.
- M. Baudler, Angew. Chem., Int. Ed., 1987, 26, 419–441 CrossRef.
- M. Cicač-Hudi, J. Bender, S. H. Schlindwein, M. Bispinghoff, M. Nieger, H. Grützmacher and D. Gudat, Eur. J. Inorg. Chem., 2016, 2016, 649–658 CrossRef.
- (a) C. L. Boulangé, in Encyclopedia of Analytical Science (Third Edition), ed. P. Worsfold, C. Poole, A. Townshend and M.Miró, Academic Press, Oxford, 2019, pp. 319–327 Search PubMed; (b) O. Kühl, Phosphorus-31 NMR Spectroscopy, Springer-Verlag Berlin Heidelberg, 2008 Search PubMed.
- CCDC 1939608–1939615 contain the supplementary crystallographic data for this paper.†.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- (a) R. S. P. Turbervill, A. R. Jupp, P. S. B. McCullough, D. Ergöçmen and J. M. Goicoechea, Organometallics, 2013, 32, 2234–2244 CrossRef CAS; (b) R. S. P. Turbervill and J. M. Goicoechea, Chem. Commun., 2012, 48, 6100–6102 RSC.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- E. P. Kyba, K. L. Hassett, B. Sheikh, J. S. McKennis, R. B. King and R. E. Davis, Organometallics, 1985, 4, 994–1001 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1939608–1939615. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04441h |
| This journal is © The Royal Society of Chemistry 2019 |